Nuwiq®
Ucrania
Contenido
- INSTRUCCIONES para uso médico del medicamento NUWIQ®
- Composición:
- Propiedades farmacológicas.
- Características clínicas.
- Características de uso.
- Vía de administración y dosis.
- Reacciones adversas.
- La frecuencia se basa en estudios con todos los medicamentos del factor VIII que incluyeron pacientes con hemofilia A grave.
INSTRUCCIONES para uso médico del medicamento NUWIQ®
Composición:
Principio activo: simoctocog alfa (factor de coagulación sanguínea VIII recombinante);
1 frasco de polvo para solución inyectable contiene simoctocog alfa (factor de coagulación sanguínea VIII recombinante) 1500 UI, o 2500 UI, o 3000 UI, o 4000 UI;
Excipientes: cloruro de sodio; sacarosa; clorhidrato de L-arginina; cloruro de calcio dihidrato; poloxámero 188; citrato de sodio dihidrato.
Disolvente: agua para inyección.
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Principales propiedades físico-químicas:
Polvo: gránulo blanco. Puede haber una pequeña cantidad de polvo blanco.
Disolvente: líquido transparente, incoloro, sin partículas.
Grupo farmacoterapéutico. Agentes antihemorrágicos. Factor de coagulación sanguínea VIII.
Código ATC B02B D02.
Propiedades farmacológicas.
Farmacodinámica.
El factor de coagulación VIII se une al factor de von Willebrand en la circulación sanguínea del paciente. El factor VIII activado actúa como cofactor para el factor IX activado, reduciendo el tiempo de conversión del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Posteriormente, la trombina convierte el fibrinógeno en fibrina y se forma el coágulo.
La hemofilia A es una enfermedad hereditaria ligada al sexo, causada por un déficit congénito del factor de coagulación VIII:C, que conduce a hemorragias en articulaciones, músculos y órganos internos, que aparecen espontáneamente o como resultado de traumatismos accidentales o quirúrgicos. Con el tratamiento sustitutivo, los niveles plasmáticos del factor VIII aumentan, corrigiendo temporalmente el déficit del factor de coagulación sanguínea VIII y la tendencia a sangrar.
Población adulta y pacientes de 12 a 65 años.
Prevención: En un estudio clínico con 32 pacientes adultos con hemofilia A grave, la dosis profiláctica media del medicamento Nuwiq fue de 468,7 UI/kg/mes. Tratamiento de hemorragias: La dosis media para el tratamiento de hemorragias en pacientes que recibían profilaxis fue de 33,0 UI/kg. En otro estudio clínico, 22 pacientes adultos recibieron tratamiento a demanda. En un total de 986 episodios hemorrágicos se aplicó una dosis media de 30,9 UI/kg. En general, las hemorragias leves requirieron dosis medias menores, mientras que las hemorragias graves requirieron dosis medias tres veces mayores.
Profilaxis individualizada: La profilaxis individualizada basada en la farmacocinética (PK) se evaluó en 66 pacientes adultos con hemofilia A grave que habían recibido tratamiento previamente (PTPs). Tras un período estándar de 1-3 meses de profilaxis (con dosis administradas cada dos días o tres veces por semana), 44 pacientes (67 %) pasaron a un régimen de dosificación basado en la evaluación de su PK, y 40 pacientes completaron una profilaxis de 6 meses según las dosis y esquemas de tratamiento prescritos. De estos pacientes, 34 (85 %) recibieron tratamiento dos veces por semana o menos. 33 (82,5 %) no presentaron ninguna hemorragia y 36 (90,0 %) no tuvieron hemorragias espontáneas. La frecuencia anualizada media + DE (calculada sobre base anual) de hemorragias fue de 1,2 + 3,9, y la dosis media + DE fue de 52,2 + 12,2 UI/kg por inyección y de 99,7 + 25,6 UI/kg por semana.
Cabe señalar que la frecuencia anualizada de hemorragias (ABR) no es comparable entre diferentes concentrados de factor ni entre diferentes estudios clínicos.
Población pediátrica
Los datos se obtuvieron en 29 niños previamente tratados de 2 a 5 años de edad, 31 niños de 6 a 12 años y un adolescente de 14 años. La dosis media profiláctica por inyección fue de 37,8 UI/kg. Veinte pacientes recibieron dosis medias superiores a 45 UI/kg. La dosis media mensual del medicamento Nuwiq fue de 521,9 UI/kg. Para el tratamiento de hemorragias, a los niños se les administró una dosis media mayor del medicamento Nuwiq (43,9 UI/kg) que a los adultos (33,0 UI/kg), así como una dosis media mayor para el tratamiento de hemorragias tanto leves como graves (78,2 UI/kg frente a 41,7 UI/kg). En general, los niños más pequeños requirieron dosis medias mayores (6-12 años: 43,9 UI/kg; 2-5 años: 52,6 UI/kg). Estos datos fueron confirmados mediante un seguimiento a largo plazo de 49 de estos niños, tratados durante un período adicional medio de aproximadamente 30 meses (rango de 9,5 a 52 meses); durante este período, el 45 % de los niños no tuvo hemorragias espontáneas.
Los datos de 108 pacientes con hemofilia A grave (˂ 1 % de FVIII:C) que no habían recibido tratamiento previamente se obtuvieron en un estudio clínico prospectivo abierto. En la mayoría de los pacientes, el tratamiento profiláctico se inició tras la primera hemorragia que requirió tratamiento.
Farmacocinética.
Población adulta
Tabla 1.
Parámetros farmacocinéticos del medicamento Nuwiq (dosis de 50 UI/kg) en adultos de 18 a 65 años con hemofilia A grave que habían recibido tratamiento previamente (n=20).
| Parámetros farmacocinéticos |
Análisis cromogénico |
|
| Media ± DE |
Mediana (límites) |
|
| AUC (hora*MO/ml) |
22,6 ± 8,0 |
22,3 (8,4 – 38,1) |
| T 1/2 (hora) |
14,7 ± 10,4 |
12,5 (5,4 – 55,6) |
| IVR (%/MO/kg) |
2,5 ± 0,4 |
2,5 (1,7 – 3,2) |
| CL (ml/hora/kg) |
3,0 ± 1,2 |
2,7 (1,5 – 6,4) |
AUC - área bajo la curva (FVIII:C), T½ - período de semivida
IVR - recuperación de los parámetros in vivo, CL - aclaramiento, SD - desviación estándar
Tabla 2.
Parámetros farmacocinéticos del medicamento Nuwiq (dosis de 50 U.I./kg) en niños de 6 a 12 años de edad con hemofilia A grave, previamente tratados (n=12).
| Parámetros farmacocinéticos |
Análisis cromogénico |
|
| Media ± DE |
Mediana (límites) |
|
| AUC (h*MO/ml) |
13,2 ± 3,4 |
12,8 (7,8 – 19,1) |
| T 1/2 (h) |
10,0 ± 1,9 |
9,9 (7,6 – 14,1) |
| IVR (%/MO/kg) |
1,9 ± 0,4 |
1,9 (1,2 – 2,6) |
| CL (ml/h/kg) |
4,3 ± 1,2 |
4,2 (2,8 – 6,9) |
AUC - área bajo la curva (FVIII:C), T½ - período de semivida
IVR - recuperación de los parámetros in vivo, CL - aclaramiento, SD - desviación estándar
Tabla 3.
Parámetros farmacocinéticos del medicamento Nuwiq (dosis de 50 U.I./kg) en niños de 2 a 5 años de edad con hemofilia A grave, previamente tratados (n = 13).
| Parámetros farmacocinéticos |
Análisis cromogénico |
|
| Media ± DE |
Mediana (límites) |
|
| AUC (hora*MO/ml) |
11,7 ± 5,3 |
10,5 (4,9 – 23,8) |
| T 1/2 (horas) |
9,5 ± 3,3 |
8,2 (4,3 – 17,3) |
| IVR (%/MO/kg) |
1,9 ± 0,3 |
1,8 (1,5 – 2,4) |
| CL (ml/hora/kg) |
5,4 ± 2,4 |
5,1 (2,3 – 10,9) |
AUC - área bajo la curva (FVIII:C), T½ - período de semivida
IVR - recuperación de los parámetros in vivo, CL - aclaramiento, SD - desviación estándar
Población pediátrica
Según datos publicados, los índices de recuperación y el período de semivida fueron más bajos en niños pequeños que en adultos, mientras que el índice de eliminación fue más alto, lo cual podría explicarse parcialmente por un mayor volumen de plasma por kilogramo de peso corporal en pacientes de menor edad.
Subgrupos por peso corporal
Tabla 4.
Parámetros farmacocinéticos del medicamento Nuwiq (dosis de 50 U.I./kg) en adultos de 18 a 65 años con hemofilia A grave, previamente tratados (n = 20), según el peso corporal del paciente
| Parámetros farmacocinéticos |
Todos los pacientes (n =20) |
Peso corporal normal (n =14) |
Estado de preobesidad (n =4) |
Obesidad (n =2) |
| Análisis cromogénico, media ± DE |
||||
| AUC (h*MO/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
| T 1/2 (h) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
| IVR (%/MO/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
| CL (ml/h/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
| Análisis cromogénico, mediana (rangos) |
||||
| AUC (h*MO/ml) |
22,3 (8,4 – 38,1) |
21,2 (8,4– 32,6) |
23,3 (17,4 – 35,5) |
33,5 (28,9 – 38,1) |
| T 1/2 (h) |
12,5 (5,4 – 55,6) |
12,3 (5,4– 55,6) |
11,2 (9,3 – 22,0) |
17,2 (13,8 – 20,6) |
| IVR (%/MO/kg) |
2,5 (1,7 – 3,2) |
2,4 (1,7 – 3,1) |
2,8 (2,3 – 3,2) |
2,8 (2,6 – 3,0) |
| CL (ml/h/kg) |
2,7 (1,5 – 6,4) |
2,8 (1,7 – 6,4) |
2,5 (1,6 – 3,7) |
1,8 (1,5 – 2,0) |
Masa corporal normal IMC: 18,5 – 25 kg/m², Masa corporal aumentada: IMC 25 – 30 kg/m², Obesidad: IMC > 30 kg/m², DE - desviación estándar
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit congénito del factor VIII).
El medicamento Nuwiq puede utilizarse en pacientes de todas las edades.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento.
Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios sobre las interacciones del medicamento Nuwiq con otros medicamentos.
Características de uso.
Traza
Para mejorar el seguimiento de los medicamentos biológicos, se debe registrar claramente el nombre y el número de lote del medicamento administrado.
La actividad específica del medicamento Nuwiq es aproximadamente 9500 UI/mg de proteína.
La simoctocog alfa (factor de coagulación sanguínea VIII (ADNr)) es una proteína pura que contiene 1440 aminoácidos. La secuencia de aminoácidos es similar a la forma de 90+80 kDa del factor VIII de plasma humano (es decir, con el dominio B eliminado). Nuwiq se produce mediante tecnología de ADN recombinante a partir de células de riñón embrionario humano (HKE) 293F genéticamente modificadas. Durante el proceso de fabricación y en el medicamento final no se añaden materiales de origen animal o humano.
Hipersensibilidad
Como con cualquier medicamento proteínico administrado por vía intravenosa, existe la posibilidad de desarrollar reacciones alérgicas e hipersensibilidad. Nuwiq contiene trazas de proteínas celulares humanas que son diferentes del factor VIII. En caso de aparición de síntomas de hipersensibilidad, se debe interrumpir inmediatamente la administración del medicamento y buscar ayuda médica. Los pacientes deben ser informados sobre los signos tempranos de reacciones de hipersensibilidad, tales como urticaria, urticaria generalizada, sensación de opresión en el pecho, dificultad respiratoria, sibilancias, hipotensión arterial y anafilaxia.
En caso de shock, se debe aplicar el tratamiento médico estándar para el estado de shock.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida que puede ocurrir durante el tratamiento de personas con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG cuya acción está dirigida contra la actividad procoagulante del factor VIII, y se cuantifican en unidades de Bethesda (UB) por 1 ml de plasma, utilizando un ensayo modificado. El riesgo de formación de inhibidores está relacionado con la gravedad de la enfermedad y también depende de la exposición al factor VIII, siendo mayor durante los primeros 50 días de exposición, aunque continúa durante toda la vida, aunque el riesgo es raro.
Se han observado casos de reaparición de inhibidores (bajo título) al cambiar de un medicamento de factor VIII recombinante a otro en pacientes que previamente habían recibido tratamiento durante más de 100 días y que tenían antecedentes de formación de inhibidores. Por ello, se recomienda un monitoreo cuidadoso de todos los pacientes respecto a la formación de inhibidores tras cualquier cambio de medicamento.
La importancia clínica de la formación de inhibidores dependerá del título del inhibidor: los inhibidores con bajo título, que están presentes temporalmente o mantienen constantemente un bajo título, representan un menor riesgo de respuesta clínica insuficiente que los inhibidores con alto título.
Es necesario un monitoreo cuidadoso de todos los pacientes que reciben tratamiento con factores recombinantes de coagulación sanguínea VIII respecto a la formación de inhibidores, mediante observación clínica adecuada y análisis de laboratorio. Si no se alcanzan los niveles esperados de actividad del factor VIII en plasma o si la hemorragia no cesa con la dosis adecuada del medicamento, se deben realizar análisis para determinar la presencia de inhibidores del factor VIII. En pacientes con títulos altos de inhibidores, la terapia con factor VIII puede ser ineficaz, por lo que se deben considerar otras opciones terapéuticas, como la inducción de tolerancia inmunológica (ITI). El tratamiento de estos pacientes debe ser llevado a cabo por médicos con experiencia en el tratamiento de la hemofilia y en casos de formación de inhibidores del factor VIII.
Complicaciones cardiovasculares
En pacientes con factores de riesgo preexistentes para enfermedades cardiovasculares, la terapia sustitutiva con FVIII puede aumentar el riesgo de enfermedades cardiovasculares.
Complicaciones relacionadas con el uso del catéter
Si es necesario utilizar un dispositivo de acceso venoso central (DAVC), se deben considerar los riesgos de complicaciones asociadas con su uso, incluyendo infecciones locales, bacteriemia y trombosis en el sitio del catéter.
Se recomienda encarecidamente registrar, en cada administración del medicamento Nuwiq, el nombre y el número de lote (serie) del medicamento para poder establecer una relación entre el estado del paciente y el lote del medicamento administrado.
Todas las advertencias se aplican tanto a adultos como a niños.
Información sobre excipientes (contenido de sodio)
1 ml de solución diluida contiene 7,35 mg (18,4 mg de sodio por vial), es decir, prácticamente «sin sodio».
Sin embargo, dependiendo del peso corporal y la dosificación, el paciente puede recibir más de un vial. Esta información debe tenerse en cuenta en pacientes que siguen una dieta con control del contenido de sodio.
Uso durante el embarazo o la lactancia.
No se han realizado estudios sobre el efecto de Nuwiq en la función reproductiva en animales. Debido a que la hemofilia A es rara en mujeres, no existe experiencia en el uso de Nuwiq durante el embarazo y la lactancia. Por lo tanto, Nuwiq puede administrarse durante el embarazo y la lactancia cuando sea clínicamente necesario. No hay datos sobre el efecto sobre la fertilidad.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
No se ha observado ningún efecto sobre la capacidad para conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
El tratamiento debe realizarse bajo supervisión médica por un profesional con experiencia en el tratamiento de la hemofilia.
Monitorización del tratamiento
Durante el curso del tratamiento, se recomienda determinar adecuadamente los niveles de factor VIII para controlar la dosis necesaria a administrar y la frecuencia de las infusiones repetidas. Distintos pacientes pueden presentar respuestas diferentes al uso del factor VIII, mostrando diferentes periodos de semivida y recuperación (restablecimiento). La dosis ajustada según el peso corporal puede requerir ajustes (corrección) en caso de peso corporal insuficiente o excesivo. En caso de intervenciones quirúrgicas extensas, la monitorización precisa de la terapia sustitutiva mediante análisis de coagulación (actividad del factor VIII en plasma sanguíneo) es obligatoria.
Cuando se utiliza un análisis en una sola etapa de la actividad coagulante sanguínea basado en el tiempo de tromboplastina parcial activada (TTPa) in vitro para determinar la actividad del factor VIII en muestras de sangre de pacientes, tanto el tipo de reactivo TTPa como la muestra estándar utilizada en el análisis pueden influir significativamente en los resultados de la actividad del factor VIII en el plasma sanguíneo. Asimismo, pueden observarse discrepancias importantes entre los resultados obtenidos mediante el análisis en una sola etapa basado en el TTPa y los obtenidos mediante análisis cromogénico, según la Farmacopea Europea. Esto es especialmente relevante, particularmente al cambiar de laboratorio y/o de reactivos utilizados en el análisis.
Vía de administración
La dosificación y la duración de la terapia sustitutiva dependen de la gravedad del déficit del factor VIII, del lugar y grado de hemorragia, así como del estado clínico del paciente.
La cantidad de unidades del factor VIII administradas se expresa en unidades internacionales (UI), referidas al estándar activo de la OMS para medicamentos del factor VIII. La actividad del factor VIII en plasma se determina bien en porcentaje (en relación con el plasma normal humano), bien preferentemente en unidades internacionales (según el estándar internacional para el factor VIII en plasma).
1 unidad internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
Tratamiento según necesidad
Los cálculos de la dosis necesaria de factor VIII se basan en datos empíricos que indican que 1 unidad internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en plasma sanguíneo aproximadamente en un 2 % respecto a la actividad normal, o en 2 UI/dl. La dosis necesaria se determina mediante la siguiente fórmula:
I.
Unidades necesarias = peso corporal (kg) × incremento deseado del factor VIII (%) (UI/dl) × 0,5 (UI/kg por UI/dl)
II.
| Crecimiento esperado del factor VIII (% del normal) = |
2 × unidades infundidas |
| peso corporal (kg) |
II.
La dosis que debe administrarse y la frecuencia de administración siempre deben ajustarse según la eficacia clínica en cada caso individual.
En las situaciones hemorrágicas indicadas a continuación, la actividad del factor VIII no debe descender por debajo del nivel de actividad plasmática indicado (en % del valor normal o U/ml) durante el período correspondiente. La tabla 5 puede utilizarse para determinar la dosificación durante episodios de hemorragia y en intervenciones quirúrgicas.
Tabla 5
| Grado de hemorragia/ tipo de procedimiento quirúrgico |
Nivel necesario del factor VIII (%) (UI/dl) |
Frecuencia de la dosis (horas)/ duración del tratamiento (días) |
| Hemorragia Hemoartrosis precoz, hemorragia muscular u oral |
20–40 |
Repetir cada 12–24 horas durante al menos 1 día hasta que la hemorragia acompañada de dolor se detenga o hasta la curación completa |
| Hemoartrosis más extensa, hemorragia muscular o hematoma |
30–60 |
Repetir cada 12–24 horas durante 3–4 días o más hasta que desaparezca el dolor y las alteraciones agudas |
| Hemorragia potencialmente mortal |
60–100 |
Repetir las inyecciones cada 8–24 horas hasta que desaparezca el peligro |
| Intervención quirúrgica Intervención menor, incluyendo extracción dental Intervención mayor |
30–60 |
Cada 24 horas durante al menos 1 día hasta la curación completa |
| 80–100 (antes y después de la intervención quirúrgica) |
Repetir las inyecciones cada 8–24 horas hasta la cicatrización significativa de la herida; posteriormente, tratamiento durante al menos 7 días para mantener la actividad del factor VIII entre el 30% y el 60% (UI/dl) |
Prevención
Para la prevención prolongada del sangrado en pacientes con hemofilia A grave, la dosis habitual oscila entre 20 y 40 UI de factor VIII por kg de peso corporal cada 2-3 días. En algunos casos, especialmente en pacientes jóvenes, puede ser necesario aumentar la dosis o la frecuencia de administración del medicamento.
Se recomienda determinar los niveles de factor VIII durante el tratamiento para ajustar la dosis y la frecuencia de las infusiones repetidas. En caso de cirugía mayor, en particular, es necesario un control preciso de la terapia sustitutiva mediante la determinación de la actividad del factor VIII en plasma. Dependiendo del factor VIII, pacientes individuales pueden presentar diferentes periodos de semivida y recuperación. El régimen de dosificación puede ajustarse según la respuesta del paciente.
La vía de administración es la misma tanto para adultos como para niños y adolescentes; sin embargo, en niños y adolescentes pueden ser necesarios intervalos más cortos entre las administraciones o dosis más altas. No existen datos sobre la utilización en niños menores de 2 años.
Método de administración
Nuvig está indicado para administración intravenosa.
Se recomienda administrar no más de 4 ml por minuto.
Instrucciones para la reconstitución del medicamento antes de la administración.
El polvo debe reconstituirse únicamente con el diluyente suministrado (2,5 ml de agua para inyección), utilizando el conjunto para la preparación de la solución inyectable. El frasco debe agitarse suavemente hasta que el polvo se disuelva completamente. Tras la reconstitución, la solución debe aspirarse con la jeringa que contenía el diluyente.
La solución preparada debe examinarse visualmente para detectar partículas sólidas y cambios de color antes de la administración. La solución reconstituida debe ser transparente, incolora, sin partículas extrañas y con un pH entre 6,5 y 7,5. No se debe utilizar ninguna solución turbia o que contenga sedimentos.
Instrucciones para la preparación y administración del medicamento.
|
Fig. 1 |
|
Fig. 2 |
|
Fig. 3 |
|
Fig. 4 |
|
Fig. 5 |
|
Fig. 6 |
|
Fig. 7 |
|
Fig. 8 |
|
|
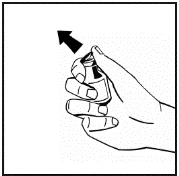
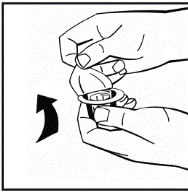
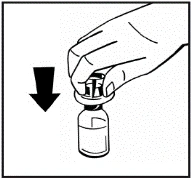
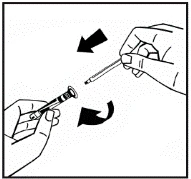
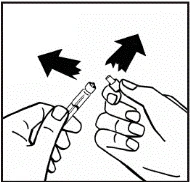
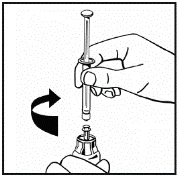
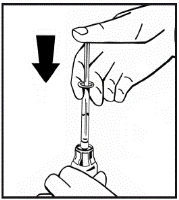
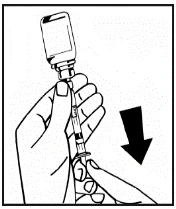
Si se utiliza más de un frasco de polvo para un mismo procedimiento, se puede volver a usar la misma aguja de inyección. El adaptador del frasco y la jeringa están destinados únicamente para uso único.
El medicamento no utilizado y los desechos deben eliminarse de acuerdo con los requisitos locales.
Niños.
Nuvic puede administrarse a niños de cualquier edad, pero no existen datos sobre su uso en niños menores de 2 años.
Sobredosis.
No se han notificado casos de sobredosis.
Reacciones adversas.
Resumen del perfil de seguridad
Se han observado reacciones de hipersensibilidad y alérgicas (que pueden incluir angioedema, sensación de ardor y hormigueo en el sitio de la infusión, escalofríos, hiperemia, urticaria generalizada, cefalea, urticaria, hipotensión arterial, somnolencia, náuseas, erupción cutánea, excitación, taquicardia, dificultad respiratoria, hormigueo, urticaria y urticaria generalizada, vómitos, sibilancias/ruidos respiratorios) raramente tras la administración de preparaciones del factor FVIII, pero en algunos casos pueden progresar hasta una anafilaxia grave (incluyendo shock).
En pacientes con hemofilia A tratados con factor VIII, así como con el medicamento Nuwiq, pueden desarrollarse anticuerpos neutralizantes (inhibidores) contra el factor VIII. Si se detectan inhibidores, la causa subyacente se considera una respuesta clínica insuficiente en forma de hemorragias persistentes o frecuentes a pesar de una profilaxis adecuada. En tales casos, se recomienda contactar con un centro especializado en hemofilia.
Lista de reacciones adversas en forma de tabla
La Tabla 6 que se presenta a continuación se clasifica según el sistema de órganos MedDRA (Diccionario Médico para la Actividad Regulatoria). La frecuencia se basa en informes de ensayos clínicos realizados con 355 pacientes con hemofilia A grave, de los cuales 247 eran pacientes previamente tratados (PTP) y 108 eran pacientes previamente no tratados (PUP).
La frecuencia se evaluó según los siguientes criterios: muy frecuentes (≥1/10); frecuentes (≥1/100 a <1/10); poco frecuentes (≥1/1000 a <1/100); raras (≥1/10000 a <1/1000); muy raras (<1/10000); desconocido (no puede determinarse con los datos disponibles).
Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 6.
| Clase de sistema de órganos MedDRA estándar |
Reacciones adversas |
Frecuencia |
| Alteraciones de la sangre y del sistema linfático |
Anemia Inhibición del factor VIII Anemia hemorrágica |
no frecuente* no frecuente (PTPs)# muy frecuente (PUPs)# no frecuente* |
| Alteraciones del sistema inmune |
Hipersensibilidad |
frecuente* |
| Alteraciones del sistema nervioso |
Vertigo Cefalea Parastesia (alteración de la sensibilidad) |
no frecuente* no frecuente* no frecuente* |
| Alteraciones del oído y del laberinto |
Vertigo |
no frecuente* |
| Alteraciones del aparato respiratorio, del tórax y del mediastino |
Disnea (dificultad respiratoria) |
no frecuente* |
| Alteraciones gastrointestinales |
Sequedad de boca |
no frecuente* |
| Alteraciones del músculo esquelético y del tejido conjuntivo |
Dolor de espalda |
no frecuente* |
| Alteraciones generales y en el lugar de administración |
Pirexia (fiebre) Dolor en el pecho Inflamación en el lugar de inyección Dolor en el lugar de inyección Malestar general |
frecuente* no frecuente* no frecuente* no frecuente* no frecuente* |
| Pruebas |
Presencia de anticuerpos positivos sin actividad neutralizante (en PTPs) |
no frecuente* |
*Calculado como el número de pacientes con reacciones adversas respecto al número total de 355 pacientes estudiados, de los cuales 247 pacientes habían recibido tratamiento previo (PTPs) y 108 pacientes no habían recibido tratamiento previo (PUPs).
La frecuencia se basa en estudios con todos los medicamentos del factor VIII que incluyeron pacientes con hemofilia A grave.
PTPs = pacientes previamente tratados, PUPs = pacientes previamente no tratados.
Descripción de reacciones adversas individuales
Se detectaron anticuerpos contra antígenos del factor VIII sin actividad neutralizante en un paciente adulto (ver tabla 6). El resultado fue positivo únicamente en la solución del factor 1, y el título de anticuerpos fue muy bajo. No se detectó actividad inhibitoria medida mediante la prueba modificada de Bethesda en este paciente. La eficacia clínica y la recuperación in vivo del medicamento Nuwiq no se vieron afectadas en este paciente.
Pediatría
Se espera que la frecuencia, tipo y gravedad de las reacciones adversas en niños sean similares a las de los adultos.
Notificación de reacciones adversas potenciales
La notificación de reacciones adversas potenciales tras la autorización del medicamento es importante. Permite continuar con el seguimiento del balance beneficio/riesgo del medicamento. Los profesionales sanitarios deben informar sobre cualquier reacción adversa potencial.
Periodo de validez.
Polvo: 2 años.
Solvente (agua para inyección): 5 años.
Durante todo el periodo de validez, el medicamento puede almacenarse hasta 1 mes a temperatura ambiente (hasta 25 °C). Si el medicamento ya ha sido retirado del refrigerador, no debe volver a colocarse allí.
Debe indicarse la fecha de inicio del periodo de almacenamiento a temperatura ambiente en el envase del medicamento.
Después de la reconstitución, la estabilidad química y física de la solución inyectable ha sido demostrada durante 24 horas cuando se almacena a temperatura ambiente.
Desde el punto de vista microbiológico, el medicamento debe usarse inmediatamente después de su preparación. Si no se utiliza inmediatamente, el usuario del medicamento asume la responsabilidad personal sobre el periodo de validez y las condiciones de uso del medicamento.
Condiciones de almacenamiento.
Polvo
Conservar entre 2 y 8 °C. No congelar.
Conservar en el embalaje exterior de cartón para protegerlo de la luz.
Solvente (agua para inyección)
Conservar entre 2 y 8 °C.
La solución reconstituida debe conservarse a temperatura ambiente.
No congelar la solución reconstituida.
Mantener fuera del alcance de los niños.
Incompatibilidades.
Debido a la falta de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Solo deben usarse los kits inyectables suministrados, ya que el tratamiento podría no ser eficaz debido a la adsorción del factor VIII de coagulación sanguínea en las superficies internas de ciertos dispositivos inyectables.
Envase. 1500 UI, 2500 UI, 3000 UI o 4000 UI de polvo en un frasco; 1 frasco con polvo, 1 jeringa precargada con 2,5 ml de solvente (agua para inyección) junto con un kit para la reconstitución y administración intravenosa (1 adaptador para frasco, 1 aguja-borla, 2 torundas impregnadas con alcohol) en una caja de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
Octapharma AB.
Dirección del fabricante y lugar de actividad.
Lars Forssellsgatan 23, Estocolmo, 11275, Suecia.
Fecha de la última revisión.