Nuvik / nuwiq®
Ukraine
Table of Contents
- INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT NUWIQ®
- Composition:
- Pharmacological properties.
- Clinical characteristics.
- Special precautions for use.
- Administration and Dosage
- Adverse reactions.
- Frequency is based on studies with all factor VIII products that included patients with severe hemophilia A.
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT NUWIQ®
Composition:
Active substance: simoctocog alfa (recombinant coagulation factor VIII);
1 vial of powder for solution for injection contains simoctocog alfa (recombinant coagulation factor VIII) 1500 IU, or 2500 IU, or 3000 IU, or 4000 IU;
Excipients: sodium chloride; sucrose; L-arginine hydrochloride; calcium chloride dihydrate; poloxamer 188; sodium citrate dihydrate.
Solvent: water for injections.
Pharmaceutical form. Powder and solvent for solution for injection.
Main physicochemical properties:
Powder: white solid. A small amount of white powder may be present.
Solvent: clear, colorless liquid, free from particles.
Pharmacotherapeutic group. Antihemorrhagics. Coagulation factor VIII.
ATC code B02BD02.
Pharmacological properties.
Pharmacodynamics.
Blood coagulation factor VIII binds to von Willebrand factor in the patient's circulation. Activated factor VIII acts as a cofactor for activated factor IX, reducing the conversion time of factor X to activated factor X. Activated factor X then converts prothrombin into thrombin. Thrombin subsequently converts fibrinogen into fibrin, leading to clot formation.
Hemophilia A is an inherited, sex-linked disorder caused by a congenital deficiency of coagulation factor VIII:C, resulting in bleeding into joints, muscles, and internal organs, occurring either spontaneously or following accidental or surgical trauma. With replacement therapy, plasma levels of factor VIII increase, thereby temporarily correcting the deficiency of blood coagulation factor VIII and reducing the tendency to bleed.
Adult population and patients aged 12–65 years.
Prophylaxis: In a clinical study involving 32 adult patients with severe hemophilia A, the mean prophylactic dose of Nuwiq was 468.7 IU/kg/month. Bleeding treatment: The mean dose for treating bleeding episodes in patients receiving prophylaxis was 33.0 IU/kg. In another clinical study, 22 adult patients received on-demand treatment. A mean dose of 30.9 IU/kg was administered in a total of 986 bleeding episodes. Overall, minor bleeds required lower doses, while major bleeds required approximately three times higher mean doses.
Individualized prophylaxis: Individualized prophylaxis based on pharmacokinetic (PK) assessment was evaluated in 66 previously treated adult patients (PTPs) with severe hemophilia A. After a 1- to 3-month standard prophylaxis phase (receiving doses every other day or three times weekly), 44 (67%) patients transitioned to a dosing regimen based on individual PK assessment, and 40 patients completed 6 months of prophylaxis according to the prescribed dosing regimen. Of these patients, 34 (85%) received treatment twice weekly or less. Bleeding episodes were absent in 33 (82.5%) patients, and 36 (90.0%) patients had no spontaneous bleeds. The mean + SD annualized (calculated on a yearly basis) bleeding rate (ABR) was 1.2 + 3.9, and the mean + SD dose was 52.2 + 12.2 IU/kg per injection and 99.7 + 25.6 IU/kg per week.
It should be noted that the annualized bleeding rate (ABR) is not directly comparable between different factor concentrates or across different clinical trials.
Pediatric population
Data were obtained from 29 previously treated children aged 2 to 5 years, 31 children aged 6 to 12 years, and one 14-year-old adolescent. The mean prophylactic injection dose was 37.8 IU/kg. Twenty patients received mean doses exceeding 45 IU/kg. The mean monthly prophylactic dose of Nuwiq was 521.9 IU/kg. Children received a higher mean dose of Nuwiq for bleeding treatment (43.9 IU/kg) compared to adults (33.0 IU/kg), as well as higher mean doses for treating both minor and major bleeds (78.2 IU/kg vs. 41.7 IU/kg). Younger children generally required higher mean doses (6–12 years: 43.9 IU/kg; 2–5 years: 52.6 IU/kg). These findings were confirmed during long-term follow-up of 49 such children treated over an additional mean period of approximately 30 months (range: 9.5 to 52 months); during this period, 45% of children had no spontaneous bleeding episodes.
Data from 108 previously untreated patients (PUPs) with severe hemophilia A (˂1% FVIII:C) were obtained in a prospective, open-label clinical trial. In most patients, prophylactic treatment was initiated after the occurrence of the first bleeding episode requiring treatment.
Pharmacokinetics.
Adult population
Table 1.
Pharmacokinetic parameters of Nuwiq (50 IU/kg dose) in adults aged 18 to 65 years with severe hemophilia A who were previously treated (n=20).
| Pharmacokinetic parameters |
Chromogenic assay |
|
| Mean ± SD |
Median (range) |
|
| AUC (hr*IU/mL) |
22.6 ± 8.0 |
22.3 (8.4 – 38.1) |
| T 1/2 (hr) |
14.7 ± 10.4 |
12.5 (5.4 – 55.6) |
| IVR (%/IU/kg) |
2.5 ± 0.4 |
2.5 (1.7 – 3.2) |
| CL (mL/hr/kg) |
3.0 ± 1.2 |
2.7 (1.5 – 6.4) |
AUC - area under the curve (FVIII:C), T½ - half-life
IVR - in vivo recovery, CL - clearance, SD - standard deviation
Table 2.
Pharmacokinetic parameters of Nuwiq administered at a dose of 50 IU/kg in children aged 6 to 12 years with severe hemophilia A who had been previously treated (n=12).
| Pharmacokinetic parameters |
Chromogenic assay |
|
| Mean ± SD |
Median (range) |
|
| AUC (hr*MU/mL) |
13.2 ± 3.4 |
12.8 (7.8 – 19.1) |
| T 1/2 (hr) |
10.0 ± 1.9 |
9.9 (7.6 – 14.1) |
| IVR (%/MU/kg) |
1.9 ± 0.4 |
1.9 (1.2 – 2.6) |
| CL (mL/hr/kg) |
4.3 ± 1.2 |
4.2 (2.8 – 6.9) |
AUC - area under the curve (FVIII:C), T½ - half-life
IVR - in vivo recovery, CL - clearance, SD - standard deviation
Table 3.
Pharmacokinetic parameters of Nuwiq after administration of 50 IU/kg in children aged 2 to 5 years with severe haemophilia A, who were previously treated (n=13).
| Pharmacokinetic parameters |
Chromogenic assay |
|
| Mean ± SD |
Median (range) |
|
| AUC (hr*OD/mL) |
11.7 ± 5.3 |
10.5 (4.9 – 23.8) |
| T 1/2 (hr) |
9.5 ± 3.3 |
8.2 (4.3 – 17.3) |
| IVR (%/OD/kg) |
1.9 ± 0.3 |
1.8 (1.5 – 2.4) |
| CL (mL/hr/kg) |
5.4 ± 2.4 |
5.1 (2.3 – 10.9) |
AUC - area under the curve (FVIII:C), T½ - half-life
IVR - in vivo recovery, CL - clearance, SD - standard deviation
Paediatric population
According to published data, recovery and half-life values were lower in younger children compared to adults, while clearance was higher, which may be partially explained by a larger plasma volume per kilogram of body weight in younger patients.
Subgroups by body weight
Table 4.
Pharmacokinetic parameters of Nuwiq (dose 50 IU/kg) in adults aged 18 to 65 years with severe haemophilia A who were previously treated (n=20), by patient body weight
| Pharmacokinetic parameters |
All patients (n =20) |
Normal body weight (n =14) |
Pre-obesity (n =4) |
Obesity (n =2) |
| Chromogenic assay, mean ± SD |
||||
| AUC (hr*IU/mL) |
22.6 ± 8.0 |
20.4 ± 6.9 |
24.9 ± 8.9 |
33.5 ± 6.5 |
| T 1/2 (hr) |
14.7 ± 10.4 |
14.7 ± 12.1 |
13.4 ± 5.9 |
17.2 ± 4.8 |
| IVR (%/IU/kg) |
2.5 ± 0.4 |
2.4 ± 0.4 |
2.7 ± 0.4 |
2.8 ± 0.3 |
| CL (mL/hr/kg) |
3.0 ± 1.2 |
3.2 ± 1.3 |
2.6 ± 1.0 |
1.8 ± 0.4 |
| Chromogenic assay, median (range) |
||||
| AUC (hr*IU/mL) |
22.3 (8.4 – 38.1) |
21.2 (8.4– 32.6) |
23.3 (17.4 – 35.5) |
33.5 (28.9 – 38.1) |
| T 1/2 (hr) |
12.5 (5.4 – 55.6) |
12.3 (5.4– 55.6) |
11.2 (9.3 – 22.0) |
17.2 (13.8 – 20.6) |
| IVR (%/IU/kg) |
2.5 (1.7 – 3.2) |
2.4 (1.7 – 3.1) |
2.8 (2.3 – 3.2) |
2.8 (2.6 – 3.0) |
| CL (mL/hr/kg) |
2.7 (1.5 – 6.4) |
2.8 (1.7 – 6.4) |
2.5 (1.6 – 3.7) |
1.8 (1.5 – 2.0) |
Normal body weight BMI - 18.5 – 25 kg/m², Increased body weight: BMI 25 – 30 kg/m², Obesity: BMI > 30 kg/m², SD - standard deviation
Clinical characteristics.
Indications.
Treatment and prophylaxis of bleeding in patients with hemophilia A (congenital factor VIII deficiency).
The Nuwiq medicinal product can be used in patients of all age groups.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients of the medicinal product.
Interaction with other medicinal products and other forms of interaction.
No studies on interactions between the Nuwiq medicinal product and other medicinal products have been conducted.
Special precautions for use.
Traceability
To improve traceability of biological medicinal products, the name and batch number of the administered product should be clearly recorded.
The specific activity of the medicinal product Nuwiq is approximately 9500 IU/mg protein.
Simoctocog alfa (recombinant DNA coagulation factor VIII) is a pure protein consisting of 1440 amino acids. The amino acid sequence is similar to the 90+80 kDa form of human plasma factor VIII (i.e., with the B-domain removed). Nuwiq is produced using recombinant DNA technology from genetically modified human embryonic kidney (HEK) 293F cells. No materials of animal or human origin are added during the manufacturing process or to the final medicinal product.
Hypersensitivity
As with any intravenous protein-containing product, there is a possibility of developing allergic reactions and hypersensitivity. Nuwiq contains trace amounts of human cell proteins that differ from factor VIII. If symptoms of hypersensitivity occur, administration of the medicinal product must be stopped immediately and medical attention sought. Patients should be informed about early signs of hypersensitivity reactions, such as urticaria, generalized urticaria, sensation of chest tightness, difficulty breathing, wheezing, hypotension, and anaphylaxis.
In the event of shock, standard medical treatment for shock should be initiated.
Inhibitors
The development of neutralizing antibodies (inhibitors) to factor VIII is a known complication in the treatment of patients with hemophilia A. These inhibitors are typically IgG immunoglobulins directed against the procoagulant activity of factor VIII and are quantified in Bethesda Units (BU) per 1 mL of plasma using a modified assay. The risk of inhibitor formation correlates with the severity of the disease and depends on exposure to factor VIII, being highest during the first 50 exposure days, but persists throughout life, although the risk is rare.
Cases of recurrent inhibitor formation (low titer) have been observed when switching from one recombinant factor VIII product to another in patients who had previously received treatment for more than 100 days and who had a history of inhibitor development. Therefore, careful monitoring of all patients for inhibitor formation is recommended following any change in medicinal product.
The clinical significance of inhibitor formation depends on the inhibitor titer: low-titer inhibitors, whether transient or persistently low, represent a lower risk of inadequate clinical response compared to high-titer inhibitors.
Careful monitoring of all patients receiving treatment with recombinant factor VIII is required for inhibitor development through appropriate clinical observation and laboratory testing. If expected plasma factor VIII activity levels are not achieved or if bleeding is not controlled with an appropriate dose, tests should be performed to determine the presence of factor VIII inhibitors. In patients with high-titer inhibitors, factor VIII therapy may be ineffective, and alternative therapeutic options such as immune tolerance induction (ITI) should be considered. Treatment of such patients should be managed by physicians experienced in the treatment of hemophilia and factor VIII inhibitor development.
Cardiovascular complications
In patients with existing risk factors for cardiovascular disease, replacement therapy with FVIII may increase the risk of cardiovascular events.
Complications associated with catheter use
If a central venous access device (CVAD) is required, the risks associated with its use, including local infections, bacteremia, and catheter-related thrombosis, should be considered.
It is strongly recommended that the name and batch (lot) number of the Nuwiq medicinal product be recorded at each administration to enable traceability between the patient's condition and the administered product batch.
All warnings apply to both adults and children.
Information on excipients (sodium content)
1 mL of reconstituted solution contains 7.35 mg (18.4 mg sodium per vial), which is essentially considered "sodium-free."
However, depending on body weight and dosage, a patient may receive more than one vial. This information should be taken into account by patients on a sodium-controlled diet.
Use during pregnancy or breastfeeding.
Studies on the effect of Nuwiq on reproductive function in animals have not been conducted. Since hemophilia A is rare in women, experience with the use of Nuwiq during pregnancy and breastfeeding is lacking. Therefore, Nuwiq may be prescribed during pregnancy and breastfeeding when clinically necessary. There are no data on the effect on fertility.
Ability to affect reaction speed when driving or operating machinery.
No effects on the ability to drive or operate machinery have been observed.
Administration and Dosage
Treatment should be administered under the supervision of a physician experienced in the management of hemophilia.
Monitoring of Therapy
During the course of treatment, appropriate monitoring of factor VIII levels is recommended to control the required dose and frequency of repeat infusions. Individual patients may exhibit varying responses to factor VIII administration, showing differences in half-life and recovery (restoration). The dose based on body weight may require adjustment in cases of significantly underweight or overweight patients. In the case of major surgical procedures, precise monitoring of replacement therapy using coagulation assays (plasma factor VIII activity) is mandatory.
When using a one-stage clotting activity assay based on activated partial thromboplastin time (aPTT) in vitro to determine factor VIII activity in patient plasma samples, both the type of aPTT reagent and the standard reference material used in the assay may significantly influence the measured factor VIII activity results. Considerable discrepancies may also occur between results obtained by the one-stage aPTT-based clotting assay and those obtained by chromogenic assay according to the European Pharmacopoeia. This is particularly important when changing laboratories and/or reagents used in testing.
Administration Method
The dosage and duration of replacement therapy depend on the severity of factor VIII deficiency, the site and extent of bleeding, and the patient's clinical condition.
The quantity of factor VIII administered is expressed in International Units (IU), calibrated against the WHO reference standard for factor VIII medicinal products. Factor VIII activity in plasma is measured either as a percentage (relative to normal human plasma) or preferably in International Units (according to the International Standard for factor VIII in plasma).
1 International Unit (IU) of factor VIII activity is equivalent to the amount of factor VIII present in 1 ml of normal human plasma.
On-demand Treatment
Dose calculations for factor VIII are based on empirical data indicating that 1 International Unit (IU) of factor VIII per kg of body weight raises plasma factor VIII activity by approximately 2% (or 2 IU/dL) from baseline. The required dose can be calculated using the following formula:
I.
Required units = body weight (kg) × desired factor VIII increase (%) (IU/dL) × 0.5 (IU/kg per IU/dL)
II.
| Expected rise in factor VIII (% of normal) = |
2 × administered IU |
| body weight (kg) |
II.
The dose to be administered and the frequency of administration must always be adjusted according to the clinical response in each individual case.
In the hemorrhagic situations listed below, factor VIII activity should not fall below the specified plasma activity level (% of normal or BU/dL) during the appropriate period. Table 5 may be used to determine dosing during bleeding episodes and surgery.
Table 5
| Severity of bleeding/ type of surgical procedure |
Required level of factor VIII (%) (IU/dl) |
Dosing frequency (hours)/ duration of therapy (days) |
| Bleeding Early haemarthrosis, muscle or oral bleeding |
20–40 |
Repeat every 12–24 hours for at least 1 day until bleeding accompanied by pain is controlled or until complete healing |
| More extensive haemarthrosis, muscle bleeding or haematoma |
30–60 |
Repeat every 12–24 hours for 3–4 days or more until pain and acute symptoms have resolved |
| Life-threatening bleeding |
60–100 |
Repeat injections every 8–24 hours until the threat has passed |
| Surgery Minor surgery, including tooth extraction Major surgery |
30–60 |
Every 24 hours for at least 1 day until complete healing |
| 80–100 (before and after surgery) |
Repeat injections every 8–24 hours until wound healing is well established, then continue therapy for at least 7 days to maintain factor VIII activity between 30% and 60% (IU/dl) |
Prophylaxis
For long-term prevention of bleeding in patients with severe hemophilia A, the usual dose is 20 to 40 IU of factor VIII per kg of body weight every 2 to 3 days. In some cases, particularly in younger patients, higher doses or more frequent administration may be required.
It is recommended to monitor factor VIII levels throughout the treatment course to adjust the dose and frequency of repeat infusions. Precise monitoring of replacement therapy by determining factor VIII activity in plasma is especially necessary during major surgical procedures. Depending on factor VIII, individual patients may exhibit different half-lives and recovery rates. The dosing regimen may be adjusted based on the patient's response.
The route of administration is the same for adults, children, and adolescents; however, shorter intervals between doses or higher doses may be required for children and adolescents. There is no data available on use in children under 2 years of age.
Method of administration
Nuvig is intended for intravenous use.
It is recommended to administer no more than 4 ml per minute.
Instructions for reconstitution of the medicinal product prior to administration.
The powder must be reconstituted only with the provided diluent (2.5 ml of water for injections), using the injection solution preparation kit. The vial should be gently swirled until the powder is completely dissolved. After reconstitution, the solution should be drawn into the syringe that contained the diluent.
The prepared solution should be visually inspected for particulate matter and discoloration prior to administration. The reconstituted solution should be clear, colorless, free of foreign particles, and have a pH between 6.5 and 7.5. Solutions that are cloudy or contain precipitate must not be used.
Instructions for preparation and administration of the medication.
|
Fig. 1 |
|
Fig. 2 |
|
Fig. 3 |
|
Fig. 4 |
|
Fig. 5 |
|
Fig. 6 |
|
Fig. 7 |
|
Fig. 8 |
|
|
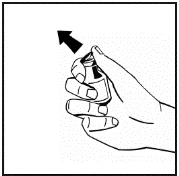
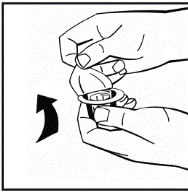
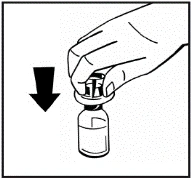
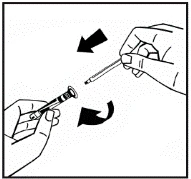
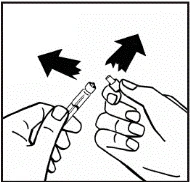
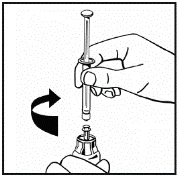
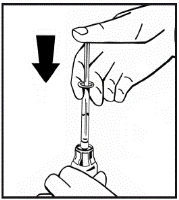
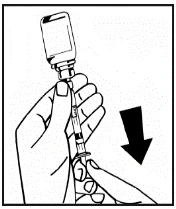
If more than one vial of powder is used for a single procedure, the same injection needle may be reused. The vial adapter and syringe are intended for single use only.
Any unused medicinal product and waste material must be disposed of in accordance with local requirements.
Children.
Novik can be used in children of any age, but there are no data on its use in children under 2 years of age.
Overdose.
There have been no reports of overdose.
Adverse reactions.
Summary of safety profile
Hypersensitivity and allergic reactions (which may include angioneurotic edema, burning and tingling at the infusion site, chills, flushing, generalized urticaria, headache, urticaria, arterial hypotension, somnolence, nausea, rash, restlessness, tachycardia, dyspnea, paresthesia, urticaria, as well as generalized urticaria, vomiting, wheezing/stridor) are rarely observed after administration of FVIII factor products, but may in some cases progress to severe anaphylaxis (including shock).
In patients with hemophilia A treated with factor VIII products, including Nuwiq, neutralizing antibodies (inhibitors) to factor VIII may develop. If inhibitors are detected, lack of expected clinical response in the form of persistent or frequent bleeding episodes despite appropriate prophylaxis is considered the cause of their occurrence. In such cases, consultation with a specialized hemophilia center is recommended.
List of adverse reactions in tabular form
Table 6 below is organized according to MedDRA (Medical Dictionary for Regulatory Activities) organ system class. The frequency is based on reports from clinical trials involving 355 patients with severe hemophilia A, including 247 previously treated patients (PTPs) and 108 previously untreated patients (PUPs).
Frequency was categorized according to the following criteria: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); not known (cannot be estimated from available data).
Within each frequency group, adverse reactions are listed in order of decreasing severity.
Table 6.
| MedDRA Standard System Organ Class |
Adverse Reactions |
Frequency |
| Blood and lymphatic system disorders |
Anaemia Inhibition of factor VIII Haemolytic anaemia |
uncommon* uncommon (PTPs)# very common (PUPs)# uncommon* |
| Immune system disorders |
Hypersensitivity |
common* |
| Nervous system disorders |
Dizziness Headache Paraesthesia (sensory disturbance) |
uncommon* uncommon* uncommon* |
| Ear and labyrinth disorders |
Dizziness |
uncommon* |
| Respiratory, thoracic and mediastinal disorders |
Dyspnoea (difficulty breathing) |
uncommon* |
| Gastrointestinal disorders |
Dry mouth |
uncommon* |
| Musculoskeletal and connective tissue disorders |
Back pain |
uncommon* |
| General disorders and administration site conditions |
Pyrexia (fever) Chest pain Injection site inflammation Injection site pain Malaise |
common* uncommon* uncommon* uncommon* uncommon* |
| Investigations |
Positive antibodies without neutralising activity (in PTPs) |
uncommon* |
*Calculated as the number of patients with adverse reactions divided by the total number of 355 investigated patients, of which 247 patients were previously treated patients (PTPs) and 108 patients were previously untreated patients (PUPs).
Frequency is based on studies with all factor VIII products that included patients with severe hemophilia A.
PTPs = previously treated patients, PUPs = previously untreated patients.
Description of individual adverse reactions
Antibodies to factor VIII antigens without neutralizing activity were detected in one adult patient (see Table 6). The result was positive only in dilution 1, and the antibody titer was very low. Inhibitor activity measured by a modified Bethesda test was not detected in this patient. The clinical efficacy and in vivo recovery of the medicinal product Nuwiq were not affected in this patient.
Children
The frequency, type, and severity of adverse reactions in children are expected to be the same as in adults.
Reporting of potential adverse reactions
Reporting of potential adverse reactions after marketing authorization of a medicinal product is important. This allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are required to report any potential adverse reactions.
Shelf life.
Powder: 2 years.
Solvent (water for injections): 5 years.
During the entire shelf life, the medicinal product may be stored for up to 1 month at room temperature (up to 25 °C). If the medicinal product has already been removed from the refrigerator, it must not be returned.
The date of the start of storage at room temperature must be indicated on the packaging of the medicinal product.
After reconstitution, chemical and physical stability of the solution for injection has been demonstrated for 24 hours when stored at room temperature.
From a microbiological standpoint, the medicinal product should be used immediately after reconstitution. If not used immediately, the user is personally responsible for the shelf life and conditions of use of the medicinal product.
Storage conditions.
Powder
Store at 2 to 8 °C. Do not freeze.
Store in the cardboard box to protect from light.
Solvent (water for injections)
Store at 2 to 8 °C.
The reconstituted solution should be stored at room temperature.
Do not freeze the reconstituted solution.
Keep out of the reach and sight of children.
Incompatibilities.
Due to lack of compatibility studies, this medicinal product must not be mixed with other medicinal products.
Only the provided infusion sets should be used, as treatment may fail due to adsorption of coagulation factor VIII onto the internal surfaces of certain infusion devices.
Packaging. 1500 IU, 2500 IU, 3000 IU, or 4000 IU powder in a vial; 1 vial of powder, 1 pre-filled syringe with 2.5 mL solvent (water for injections), together with a reconstitution and intravenous administration kit (1 vial adapter, 1 butterfly needle, 2 alcohol swabs) in a cardboard box.
Prescription status.
Prescription only.
Manufacturer.
Octapharma AB.
Manufacturer's address and location of its operations.
Lars Forssells gata 23, Stockholm, 11275, Sweden.
Date of last review.