Belara®
Ucrania
Contenido
INSTRUCCIONES para uso médico del medicamento BELARA®
Composición:
Principios activos: Cada tableta recubierta con película contiene: acetato de clormadinona 2 mg, etinilestradiol 0,03 mg.
Excipientes: lactosa monohidrato, almidón de maíz, povidona K-30, estearato de magnesio.
Recubrimiento: hipromelosa, lactosa monohidrato, dióxido de titanio (E 171), talco, polietilenglicol (macrogol 6000), propilenglicol, óxido de hierro rojo (E 172).
Forma farmacéutica: Tabletas recubiertas con película.
Propiedades fisicoquímicas principales: tabletas redondas, biconvexas, de color rosa claro, recubiertas con película.
Grupo farmacoterapéutico: Anticonceptivos hormonales para uso sistémico. Progestágenos y estrógenos, combinaciones fijas. Código ATC G03A A15.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
La administración continua del medicamento Belara® durante 21 días suprime la secreción de la hormona foliculoestimulante (FSH) y de la hormona luteinizante (LH) por la hipófisis y, como consecuencia, inhibe la ovulación. Se observa proliferación del endometrio y su transformación secretora. Asimismo, se modifica la consistencia del moco cervical, lo que dificulta el paso de los espermatozoides a través del canal cervical y altera su movilidad. También se producen cambios en el endometrio que hacen que este sea inadecuado para la implantación.
La dosis mínima de acetato de clormadinona que garantiza una supresión completa de la ovulación es de 1,7 mg. La dosis necesaria para la transformación del endometrio es de 25 mg por ciclo.
El acetato de clormadinona es un progestágeno con propiedades antiandrógenas. Su mecanismo de acción se basa en la capacidad de desplazar a los andrógenos de sus receptores específicos.
Eficacia clínica
En estudios clínicos en los que se evaluó el uso de tabletas que contenían etinilestradiol 0,03 mg y acetato de clormadinona 2 mg, con la participación de 1655 mujeres durante 2 años y más de 22000 ciclos, se registraron 12 embarazos. En 7 de estos casos, durante el período de fecundación del óvulo, las mujeres habían cometido errores en la toma del medicamento, padecían enfermedades concomitantes asociadas a náuseas o vómitos, o bien tomaban simultáneamente otros medicamentos que reducen el efecto contraceptivo de los preparados hormonales.
Tabla 1
| Uso habitual |
Cantidad de embarazos |
Índice de Pearl |
Intervalo de confianza del 95 % |
| Uso habitual |
12 |
0,698 |
[0,389; 1,183] |
| Uso correcto |
5 |
0,291 |
[0,115; 0,650] |
Farmacocinética.
Acetato de clormadinona (ACC)
Absorción
Tras la administración oral, el ACC se absorbe rápidamente y prácticamente por completo. La biodisponibilidad sistémica del ACC es alta, ya que no sufre metabolismo de primer paso en el hígado. La concentración máxima en plasma se alcanza entre 1 y 2 horas.
Distribución
Más del 95 % del ACC se une a las proteínas plasmáticas, principalmente a la albúmina. El ACC no se une a la globulina que transporta las hormonas sexuales ni a la globulina que une el cortisol. El ACC se acumula principalmente en el tejido adiposo.
Biocatransformación
Diversos procesos de reducción, oxidación y conjugación con glucurónidos y sulfatos conducen a la formación de múltiples metabolitos. Los metabolitos principales en plasma son el 3α- y el 3β-hidroxi-ACC, con un período de semidesaparición que no difiere significativamente del ACC no metabolizado. Los metabolitos 3-hidroxi presentan actividad antiandrógena similar a la del propio ACC. En la orina, los metabolitos se excretan principalmente en forma de conjugados. Tras escisión enzimática, el metabolito principal es el 2α-hidroxi-ACC; también se forman metabolitos 3-hidroxi y dihidroxi.
Eliminación
El período medio de semidesaparición del ACC en plasma es de aproximadamente 34 horas tras una dosis única y de unos 36-39 horas tras la administración múltiple. Tras la administración oral, el ACC y sus metabolitos se excretan en cantidades aproximadamente iguales tanto por vía renal como a través del intestino.
Ethinilestradiol (EE)
Absorción
El EE se absorbe rápidamente y prácticamente por completo tras la administración oral, alcanzando la concentración máxima en plasma a las 1,5 horas. Debido a la unión y metabolismo presistémicos en el hígado, la biodisponibilidad absoluta es solo de aproximadamente el 40 % y está sujeta a una marcada variabilidad individual (20-65 %).
Distribución
Los datos publicados disponibles sobre la concentración de EE en plasma muestran una gran variabilidad. Aproximadamente el 98 % del etinilestradiol se une a las proteínas plasmáticas, prácticamente de forma exclusiva con la albúmina.
Biocatransformación
Al igual que los estrógenos naturales, el EE sufre biotransformación mediante hidroxilación del anillo aromático (mediada por el sistema del citocromo P450). El metabolito principal es el 2-hidroxi-EE, que se transforma en otros metabolitos y conjugados. El etinilestradiol sufre unión presistémica tanto en la mucosa del intestino delgado como en el hígado. En la orina se encuentran principalmente glucurónidos, mientras que en la bilis y en plasma predominan los sulfatos.
Eliminación
El período medio de semidesaparición del EE en plasma es de aproximadamente 12-14 horas. El EE se elimina por vía renal y a través del intestino en una proporción de 2:3. El sulfato de EE, excretado en la bilis, tras la hidrólisis por bacterias intestinales, sufre recirculación enterohepática.
Datos de estudios preclínicos de seguridad
Los estrógenos presentan baja toxicidad aguda. Debido a las notables diferencias entre especies animales experimentales, así como entre animales y seres humanos, los resultados de los estudios con estrógenos en animales tienen un valor predictivo limitado para el ser humano. El etinilestradiol es un estrógeno sintético frecuentemente utilizado en anticonceptivos orales. Estudios de laboratorio en animales han demostrado que, incluso a dosis relativamente bajas, esta sustancia ejerce efectos embrioletales; en fetos masculinos se observaron anomalías en el desarrollo de los órganos del sistema urinario y genital, así como signos de feminización. Estos efectos se consideran específicos de la especie.
Se ha demostrado que el acetato de clormadinona ejerce efectos embrioletales en conejos, ratas y ratones. Además, se observó acción teratogénica tras la administración de dosis embriotóxicas en conejos y a la dosis más baja estudiada (1 mg/kg/día) en ratones. No se ha establecido la relevancia de estos datos respecto al uso del medicamento en humanos.
En el marco de estudios preclínicos de seguridad convencionales, que evaluaron la toxicidad crónica, genotoxicidad y potencial oncogénico del medicamento, no se identificaron riesgos particulares para el ser humano, salvo aquellos ya descritos en otras secciones del prospecto del medicamento.
Características clínicas.
Indicaciones.
Anticoncepción hormonal.
Antes de prescribir el medicamento Belara®, se debe evaluar la presencia de factores de riesgo individuales en la mujer, especialmente aquellos relacionados con el riesgo de tromboembolismo venoso (TEV), así como comparar el riesgo de TEV durante el uso de Belara® con el riesgo asociado con otros anticonceptivos hormonales combinados (AHC) (véase las secciones «Contraindicaciones» y «Precauciones y advertencias»).
Contraindicaciones.
No se debe utilizar AHC en caso de presencia de las enfermedades que se indican a continuación. Si se desarrolla uno de estos estados durante el tratamiento con Belara®, el medicamento debe suspenderse inmediatamente.
- Pérdida del control de la diabetes mellitus.
- Hipertensión arterial no controlada o marcado aumento de la presión arterial (valores que constantemente superan 140/90 mmHg).
- Presencia o riesgo de tromboembolismo venoso (TEV):
- tromboembolismo venoso actual (con anticoagulantes) o antecedentes (por ejemplo, trombosis venosa profunda (TVP), embolia pulmonar);
- predisposición hereditaria o adquirida conocida al desarrollo de TEV, tales como resistencia a la proteína C activada (incluyendo la mutación del factor V de Leiden), déficit de antitrombina III, déficit de proteína C, déficit de proteína S;
- intervenciones quirúrgicas mayores con inmovilización prolongada (véase la sección «Precauciones y advertencias»);
- alto riesgo de tromboembolismo venoso debido a la presencia de múltiples factores de riesgo (véase la sección «Precauciones y advertencias»).
- Presencia o riesgo de tromboembolismo arterial (TEA):
- tromboembolismo arterial actual o antecedentes (por ejemplo, infarto de miocardio) o estados precursores (angina de pecho);
- enfermedades cerebrovasculares − accidente cerebrovascular actual o antecedentes o estados precursores (ataque isquémico transitorio (AIT));
- predisposición hereditaria o adquirida conocida al desarrollo de TEA, en particular hiperhomocisteinemia, anticuerpos contra fosfolípidos (anticuerpos contra cardiolipina, anticoagulante lúpico);
- antecedentes de migraña con síntomas neurológicos focales;
- alto riesgo de tromboembolismo arterial debido a la presencia de múltiples factores de riesgo (véase la sección «Precauciones y advertencias») o presencia de factores de riesgo tales como diabetes mellitus con complicaciones vasculares; hipertensión arterial grave, dislipoproteinemia grave.
- Hepatitis, ictericia, alteraciones de la función hepática hasta la normalización de los parámetros hepáticos.
- Prurito generalizado, colestasis, especialmente durante embarazos previos o tratamiento con estrógenos.
- Síndrome de Dubin-Johnson, síndrome de Rotor, alteraciones en la excreción biliar.
- Meningioma o antecedentes de meningioma.
- Tumores hepáticos anteriores o actuales.
- Dolor intenso en epigastrio, aumento del tamaño del hígado o síntomas de hemorragia intraabdominal (véase la sección «Reacciones adversas»).
- Porfiria de aparición inicial o recurrente (las tres formas, especialmente porfiria adquirida).
- Tumores malignos dependientes de hormonas, anteriores o actuales, por ejemplo tumores de mama o útero.
- Alteraciones graves del metabolismo de lípidos.
- Pancreatitis anterior o actual, asociada con hipetrigliceridemia grave.
- Síntomas de migraña que aparecen por primera vez, así como cefalea más frecuente y extremadamente intensa.
- Trastornos sensoriales agudos, por ejemplo alteraciones visuales o auditivas.
- Trastornos motores (en particular parálisis).
- Aumento de la frecuencia de crisis epilépticas.
- Depresión grave.
- Otosclerosis que haya empeorado durante embarazos previos.
- Amenorrea de etiología desconocida.
- Hiperplasia endometrial.
- Hemorragia vaginal de etiología desconocida.
- Hipersensibilidad a los principios activos o a cualquier otro componente del medicamento.
- Contraindicación es la presencia de un factor de riesgo grave o varios factores de riesgo para el desarrollo de trombosis venosa o arterial (véase la sección «Precauciones y advertencias»).
El medicamento Belara® está contraindicado para su uso concomitante con medicamentos que contengan ombitasvir/paritaprevir/ritonavir y dasabuvir, o medicamentos que contengan glecaprevir/pibrentasvir o sofosbuvir/velpatasvir/voxilaprevir (véase la sección «Interacción con otros medicamentos y otras formas de interacción»).
Interacción con otros medicamentos y otras formas de interacción.
Nota: se debe consultar la información del medicamento que se administra simultáneamente para detectar posibles interacciones.
Interacciones farmacodinámicas
Durante estudios clínicos en pacientes que recibieron medicamentos para el tratamiento de la hepatitis vírica C (HCV) que contienen ombitasvir/paritaprevir/ritonavir y dasabuvir, con o sin ribavirina, se observó un aumento de las transaminasas (ALT) superior a 5 veces el límite superior normal (LSN). Esto ocurrió con mayor frecuencia en mujeres que utilizaban medicamentos que contienen etinilestradiol, incluyendo anticonceptivos hormonales combinados (AHC). Además, en pacientes que recibieron tratamiento con glecaprevir/pibrentasvir o sofosbuvir/velpatasvir/voxilaprevir, también se observó un aumento de ALT en mujeres que tomaban medicamentos que contenían etinilestradiol, tales como AHC (véase la sección «Contraindicaciones»).
Por lo tanto, a las pacientes que toman Belara® se les debe recomendar cambiar a un método anticonceptivo alternativo (por ejemplo, anticoncepción solo con progestágenos o métodos no hormonales) antes de iniciar el tratamiento con estos medicamentos combinados. El uso de Belara® puede reanudarse 2 semanas después de finalizar el tratamiento con estas combinaciones.
Interacciones farmacocinéticas
Efecto de otros medicamentos sobre Belara®
Puede producirse interacción con medicamentos que inducen enzimas microsomales, lo que puede aumentar el aclaramiento de hormonas sexuales, lo que a su vez puede provocar sangrado intermenstrual y/o pérdida de eficacia anticonceptiva.
Tratamiento
La inducción enzimática puede comenzar ya en unos pocos días de tratamiento. La máxima inducción enzimática generalmente se observa en varias semanas. Tras la interrupción del medicamento, la inducción enzimática puede persistir hasta 4 semanas.
Tratamiento a corto plazo
Las mujeres que toman medicamentos que inducen enzimas deben utilizar temporalmente un método barrera u otro método anticonceptivo adicional junto con los anticonceptivos orales combinados. El método barrera debe utilizarse durante todo el período de tratamiento con el medicamento correspondiente y durante 28 días adicionales tras finalizar dicho tratamiento.
Si el tratamiento con un medicamento inductor de enzimas comienza durante el período de toma de las últimas pastillas del anticonceptivo oral combinado (AOC) del envase actual, se debe comenzar inmediatamente con las pastillas del siguiente envase, sin hacer pausa entre envases.
Tratamiento a largo plazo
A las mujeres sometidas a tratamiento a largo plazo con sustancias que inducen enzimas hepáticas se recomienda utilizar un método anticonceptivo barrera u otro método anticonceptivo no hormonal adecuado.
Las siguientes interacciones se han documentado según datos científicos publicados.
Sustancias activas que aumentan el aclaramiento de AOC (disminución de la eficacia de AOC por inducción enzimática), por ejemplo: barbitúricos, bosentán, carbamazepina, barbexaclón, fenitoína, primidona, modafinilo, rifampicina, rifabutina y el medicamento para el VIH ritonavir, nevirapina y efavirenz, y posiblemente también felbamato, griseofulvina, oxcarbazepina, topiramato y preparaciones que contienen hipérico (Hypericum perforatum).
Medicamentos/sustancias activas que pueden reducir la concentración de etinilestradiol en suero:
- todos los medicamentos que aumentan la motilidad gastrointestinal (por ejemplo, metoclopramida) o interfieren con la absorción (por ejemplo, carbón activado).
Sustancias activas con efecto variable sobre el aclaramiento de AOC
Al administrarse conjuntamente con AOC, muchas combinaciones de inhibidores de la proteasa del VIH e inhibidores no nucleósidos de la transcriptasa inversa, incluyendo combinaciones con inhibidores de la proteasa del virus de la hepatitis C, pueden aumentar o disminuir la concentración de estrógenos o progestágenos en plasma. El efecto conjunto de estos cambios puede ser clínicamente relevante en algunos casos.
Por lo tanto, para detectar posibles interacciones y otras recomendaciones, se debe consultar la información del medicamento para el tratamiento del VIH/VHC. En caso de dudas, las mujeres deben utilizar adicionalmente un método barrera durante el tratamiento con inhibidores de la proteasa o inhibidores no nucleósidos de la transcriptasa inversa.
Medicamentos/sustancias activas que pueden aumentar la concentración de etinilestradiol en suero:
- sustancias activas que inhiben la sulfatación del etinilestradiol en la pared intestinal, por ejemplo ácido ascórbico o paracetamol;
- atorvastatina (aumenta el AUC del etinilestradiol en un 20 %);
- sustancias activas que inhiben la actividad de enzimas hepáticas, tales como antifúngicos derivados del imidazol (por ejemplo, fluconazol), indinavir o troleandomicina.
Efecto de Belara® sobre otros medicamentos:
- inhibición de la actividad de enzimas hepáticas y, por consiguiente, aumento de la concentración en suero de sustancias activas como diazepam (y otras benzodiazepinas cuyo metabolismo se realiza mediante hidroxilación), ciclosporina, teofilina y prednisolona;
- inducción de la glucuronidación en el hígado y, por consiguiente, disminución de la concentración en suero de sustancias como lamotrigina, clofibrato, paracetamol, morfina y lorazepam.
Puede cambiar la necesidad de insulina y medicamentos orales antidiabéticos, ya que el medicamento afecta la tolerancia a la glucosa (véase la sección «Precauciones y advertencias»).
Esto también puede aplicarse a medicamentos recientemente utilizados.
Se debe revisar el prospecto del medicamento recetado por el médico con el fin de detectar posibles interacciones con Belara®.
Estudios de laboratorio
La administración de anticonceptivos esteroides puede influir en los resultados de ciertos análisis de laboratorio, incluyendo parámetros bioquímicos de la función hepática, tiroides, glándulas suprarrenales y riñones; así como los niveles de proteínas plasmáticas transportadoras, tales como globulina fijadora de corticosteroides y fracciones de lípidos/lipoproteínas, así como parámetros del metabolismo de carbohidratos, coagulación y fibrinólisis. Los cambios suelen producirse dentro de los límites normales de los valores de laboratorio.
Características de uso.
Advertencias especiales.
Fumar aumenta el riesgo de reacciones adversas graves del sistema cardiovascular durante el uso de anticonceptivos hormonales combinados (AHC). Este riesgo aumenta con la edad, depende del número de cigarrillos fumados y es especialmente alto en mujeres de 35 años o más. Se recomienda a las mujeres de 35 años o más que fuman considerar la posibilidad de utilizar otros métodos anticonceptivos.
El uso de AHC se asocia con un mayor riesgo de enfermedades graves, tales como infarto de miocardio, tromboembolismo, accidente cerebrovascular o neoplasias hepáticas. Otros factores de riesgo, como hipertensión arterial, hiperlipidemia, obesidad y diabetes mellitus, aumentan significativamente el riesgo de complicaciones y mortalidad.
En presencia de cualquiera de las enfermedades o factores de riesgo mencionados a continuación, el uso del medicamento Belara® debe discutirse con la mujer.
Si estas enfermedades o factores de riesgo aparecen por primera vez o empeoran durante el uso del medicamento, se recomienda que la mujer consulte a su médico para determinar si debe interrumpir el uso de Belara®.
Tromboembolismo u otras enfermedades vasculares
Los resultados de estudios epidemiológicos indican que existe una relación entre el uso de anticonceptivos hormonales y el aumento del riesgo de enfermedades tromboembólicas venosas o arteriales, como infarto de miocardio, accidente cerebrovascular, trombosis venosa profunda y embolia de la arteria pulmonar. Estas enfermedades son raras.
Muy raramente se han notificado casos de trombosis en otros vasos sanguíneos, como venas hepáticas, mesentéricas, renales, venas de la retina y arterias, en mujeres que usaban AHC.
Riesgo de tromboembolismo venoso (TEV).
El uso de anticonceptivos hormonales combinados (AHC) incrementa el riesgo de tromboembolismo venoso (TEV) en las pacientes que los toman, en comparación con aquellas que no usan estos medicamentos. Los medicamentos que contienen levonorgestrel, norgestimato o noretisterona se asocian con un nivel bajo de riesgo de TEV. Otros AHC que contienen clormadinona/etinilestradiol, como Belara, pueden provocar un riesgo de TEV hasta 1,25 veces mayor en comparación con el levonorgestrel. La decisión de usar cualquier medicamento que no sea uno de bajo riesgo conocido debe tomarse únicamente tras una discusión con la mujer, asegurándose de que comprenda el riesgo de TEV asociado al uso de Belara®, cómo sus factores de riesgo individuales influyen en este riesgo, y que el riesgo de TEV es más alto durante el primer año de uso. Además, existen datos que indican que el riesgo aumenta al reanudar el uso de AHC tras una interrupción de 4 semanas o más.
En mujeres que no han usado AHC ni han estado embarazadas, aproximadamente dos casos de TEV por cada mil mujeres se desarrollan en un año. Sin embargo, en cualquier mujer concreta, el riesgo puede ser mucho mayor, dependiendo de sus factores de riesgo subyacentes (ver más abajo).
Estudios epidemiológicos en mujeres que usaban AHC de baja dosis (<50 mcg de etinilestradiol) mostraron que entre 6 y 12 mujeres de cada 10 000 desarrollaron TEV durante un año.
Se estima que entre 6 y 9 mujeres de cada 10 000 que usan AHC que contienen clormadinona desarrollarán TEV en un año; esto puede compararse con aproximadamente 6 mujeres que usan AHC que contienen levonorgestrel.
Número de casos de TEV por cada 10 000 mujeres en un año
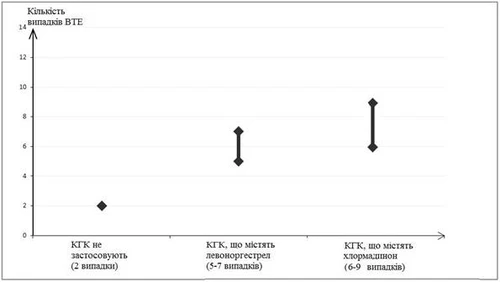
La frecuencia de TEV durante el uso de AHC de baja dosis es menor que la frecuencia en mujeres durante el embarazo o el período posparto.
El TEV puede provocar consecuencias fatales en el 1-2 % de los casos.
Factores de riesgo de TEV.
El riesgo de complicaciones tromboembólicas venosas en mujeres que usan AHC puede aumentar si existen factores de riesgo adicionales, especialmente si hay varios factores de riesgo como los indicados en la tabla 2.
Belara® está contraindicado si la mujer presenta varios factores de riesgo que la sitúan en el grupo de alto riesgo de trombosis venosa (ver sección «Contraindicaciones»). Si la mujer tiene más de un factor de riesgo para TEV, puede darse una situación en la que el riesgo aumente más que la suma de los factores individuales; en tales casos, debe considerarse el riesgo total de TEV.
Si, tras la evaluación, el balance beneficio/riesgo se considera negativo, no se debe recetar un AHC (ver sección «Contraindicaciones»).
Factores de riesgo de TEV. Tabla 2
| Factor de riesgo |
Explicación |
| Obesidad (índice de masa corporal superior a 30 kg/m²). |
Principalmente, el riesgo de desarrollo aumenta con el incremento del índice de masa corporal. Es especialmente importante tenerlo en cuenta si también están presentes otros factores de riesgo. |
| Inmovilización prolongada, intervención quirúrgica extensa, cualquier cirugía en las piernas o en la región pélvica, cirugías neurológicas o traumatismos graves. Nota: la inmovilización temporal, incluyendo vuelos aéreos de más de 4 horas de duración, también puede ser un factor de riesgo para la ETV, especialmente en mujeres con otros factores de riesgo de desarrollar ETV. |
En tales casos, se recomienda suspender el uso del parche/comprimidos/anillo vaginal (en caso de cirugía programada, al menos 4 semanas antes) y reiniciar su uso 2 semanas después de la completa removilización de la paciente. Se debe utilizar otro método anticonceptivo para evitar el embarazo. Debe considerarse la terapia anticoagulante si no se suspendió previamente el uso del medicamento Belara®. |
| Antecedentes familiares positivos (tromboembolismo venoso en algún momento en un hermano/a o padres, especialmente a edad relativamente temprana, por ejemplo antes de los 50 años). |
Si se sospecha una predisposición hereditaria, la mujer debe acudir a consulta con un especialista para tomar una decisión sobre el uso de ACO. |
| Otras afecciones médicas asociadas con ETV |
Cáncer, lupus eritematoso sistémico, síndrome hemolítico urémico, enfermedad inflamatoria intestinal crónica (enfermedad de Crohn o colitis ulcerosa) y anemia falciforme. |
| Edad |
Particularmente a partir de los 35 años. |
No existe un consenso unánime sobre si hay una relación entre la tromboflebitis superficial y la varicosis o la etiología de la tromboembolia venosa.
También debe tenerse en cuenta que el riesgo de complicaciones tromboembólicas aumenta durante el embarazo y especialmente durante las primeras 6 semanas después del parto (véase la sección «Uso durante el embarazo o la lactancia»).
Síntomas de la ETV (trombosis venosa profunda y embolia pulmonar)
Si aparecen síntomas, la mujer debe buscar atención médica de emergencia y debe informar al médico que está tomando un ACO.
Los síntomas de la trombosis venosa profunda (TVP) pueden incluir:
- hinchazón unilateral de la pierna y/o del pie o a lo largo de una vena de la pierna;
- dolor o sensibilidad dolorosa, que puede notarse solo al estar de pie o al caminar;
- sensación de calor aumentada en la pierna afectada; piel enrojecida o decolorada en la pierna.
Los síntomas de la embolia pulmonar (EP) pueden incluir:
- aparición repentina de disnea de causa desconocida o respiración acelerada;
- tos repentina, que puede ir acompañada de hemoptisis;
- dolor agudo en el pecho;
- fuerte dolor de cabeza o mareo;
- taquicardia o ritmo cardíaco irregular.
Algunos de estos síntomas (por ejemplo, disnea, tos) son inespecíficos y podrían interpretarse erróneamente como síntomas más comunes o menos graves (por ejemplo, infección de las vías respiratorias).
Otros signos de oclusión vascular pueden incluir dolor repentino, hinchazón y ligera coloración azulada de las extremidades.
Si la oclusión ocurre en los vasos sanguíneos del ojo, los síntomas pueden variar desde visión borrosa indolora, que puede progresar, hasta pérdida de la visión. A veces, la pérdida de la visión puede ocurrir de forma inmediata.
Riesgo de tromboembolia arterial (ATE).
Estudios epidemiológicos han mostrado que el uso de ACO aumenta el riesgo de desarrollar tromboembolia arterial o accidente cerebrovascular (por ejemplo, accidente isquémico transitorio, ictus). La tromboembolia arterial puede ser mortal.
Factores de riesgo para el desarrollo de ATE
El riesgo de complicaciones tromboembólicas arteriales (infarto de miocardio) o de accidente cerebrovascular agudo aumenta en mujeres que toman ACO y que presentan factores de riesgo descritos en la Tabla 3. El medicamento Belara® está contraindicado si la mujer presenta un factor de riesgo grave o varios factores de riesgo de ATE que la sitúan en un grupo de alto riesgo de trombosis arterial (véase la sección «Contraindicaciones»). Es muy probable que si una mujer presenta más de un factor de riesgo, el riesgo total de ATE aumente más que la suma de los riesgos individuales. Si el balance beneficio/riesgo se considera desfavorable, no se debe prescribir el ACO (véase la sección «Contraindicaciones»).
Factores de riesgo para el desarrollo de ATE. Tabla 3.
| Factor de riesgo |
Explicación |
| Edad |
Particularmente a partir de los 35 años. |
| Tabaquismo |
A las mujeres que utilizan anticonceptivos hormonales combinados se les debe recomendar encarecidamente que dejen de fumar. Las mujeres de 35 años o más que fuman deben considerar la posibilidad de utilizar otros métodos anticonceptivos. |
| Hipertensión arterial |
|
| Obesidad (índice de masa corporal superior a 30 kg/m²) |
Principalmente, el riesgo de desarrollo aumenta con el incremento del índice de masa corporal. Es especialmente importante tenerlo en cuenta si también están presentes otros factores de riesgo. |
| Antecedentes familiares positivos (tromboembolia arterial en hermanos, hermanas o padres en algún momento, especialmente a edades relativamente tempranas, por ejemplo antes de los 50 años) |
Si se sospecha de predisposición hereditaria, la mujer debe acudir a consulta con un especialista para una evaluación antes de decidir el uso de anticonceptivos hormonales combinados. |
| Migraña |
El aumento de la frecuencia o de la gravedad de la migraña durante el uso de anticonceptivos hormonales combinados (lo que podría ser un fenómeno prodromal o cerebrovascular) puede ser motivo para suspender inmediatamente el medicamento. |
| Otras afecciones médicas asociadas con eventos vasculares adversos |
Diabetes mellitus, hiperhomocisteinemia, enfermedades de las válvulas cardíacas y fibrilación auricular, displipoproteinemia y lupus eritematoso sistémico. |
Síntomas de ETA
En caso de presentación de síntomas, la mujer debe buscar atención médica de emergencia e informar al médico que está tomando ACO.
Los síntomas de un trastorno agudo de la circulación cerebral pueden incluir:
- Debilidad repentina u hormigueo en la cara, brazo o pierna, especialmente en un lado del cuerpo;
- Problemas repentinos al caminar, mareos, pérdida del equilibrio o de la coordinación;
- Confusión repentina, problemas para hablar o comprender;
- Problemas visuales repentinos en uno o ambos ojos;
- Dolor de cabeza intenso o prolongado sin causa aparente;
- Pérdida de conciencia o desmayo, con o sin convulsiones.
Síntomas transitorios indican un accidente isquémico transitorio (AIT).
Los síntomas de infarto de miocardio (IM) pueden incluir:
- Dolor, molestia, presión, pesadez, sensación de opresión o hinchazón en el pecho, brazo o detrás del esternón;
- Molestia que irradia hacia la espalda, mandíbula, garganta, brazo o abdomen;
- Sensación de plenitud, malestar estomacal o estreñimiento;
- Sudoración, náuseas, vómitos y mareos;
- Debilidad intensa, inquietud o dificultad para respirar;
- Palpitaciones rápidas o irregulares.
Las pacientes que toman ACO deben estar informadas de que, ante la aparición de posibles síntomas de trombosis, deben consultar al médico. En caso de sospecha o confirmación de trombosis, debe suspenderse el tratamiento con Belara®.
Neoplasias
Algunos estudios epidemiológicos indican que el uso prolongado de anticonceptivos orales es un factor de riesgo para el desarrollo de cáncer de cuello uterino en mujeres infectadas con el virus del papiloma humano (VPH). Sin embargo, este tema es controvertido, ya que no está claro hasta qué punto otros factores influyen en los resultados obtenidos (por ejemplo, diferencias en el número de parejas sexuales o uso de métodos anticonceptivos de barrera) (ver también la sección «Examen médico»).
Un metaanálisis de 54 estudios epidemiológicos mostró que el riesgo relativo de desarrollar cáncer de mama es ligeramente mayor en mujeres que toman ACO (RR = 1,24). Este riesgo aumentado disminuye gradualmente durante los 10 años posteriores a la suspensión del ACO. Sin embargo, estos estudios no han confirmado una relación causal entre la enfermedad y el uso del fármaco. El riesgo aumentado observado podría deberse a que en mujeres que toman ACO el cáncer de mama se diagnostica en una etapa más temprana que en aquellas que no los toman, así como a la acción biológica de los ACO o a una combinación de ambos factores.
Con el uso prolongado de anticonceptivos orales, se han observado raramente tumores benignos del hígado y, muy raramente, tumores malignos, que en algunos casos pueden provocar hemorragias intraabdominales potencialmente mortales. En caso de aparición de un dolor agudo intenso en la parte superior del abdomen que no desaparece espontáneamente, aumento del tamaño del hígado o signos de hemorragia intraperitoneal, debe considerarse la posibilidad de un tumor hepático y debe suspenderse el tratamiento con Belara®.
Meningioma.
Se han notificado casos de meningioma (solitarios y múltiples) asociados al uso de acetato de clormadinona, especialmente en dosis altas y durante períodos prolongados (varios años). Los pacientes deben ser vigilados según la práctica clínica habitual respecto a signos y síntomas de meningioma. Si a un paciente se le diagnostica meningioma, cualquier tratamiento que contenga acetato de clormadinona debe suspenderse como medida preventiva.
Existen algunas evidencias de que el riesgo de meningioma puede disminuir tras la suspensión del tratamiento con acetato de clormadinona.
Otras enfermedades
El estado de ánimo deprimido y la depresión son efectos adversos conocidos del uso de anticonceptivos hormonales (ver sección «Reacciones adversas»). La depresión puede ser grave y es un factor de riesgo conocido para la conducta suicida y el suicidio. Se debe aconsejar a las mujeres que consulten a su médico ante cambios de ánimo o síntomas depresivos, incluso poco después del inicio del tratamiento.
En muchas mujeres que toman anticonceptivos orales se observa un ligero aumento de la presión arterial. Un aumento clínicamente significativo de la presión arterial es raro. La relación entre el uso de anticonceptivos orales y la hipertensión arterial no está confirmada actualmente. Si durante el tratamiento con Belara® se observa un aumento clínicamente significativo de la presión arterial, debe suspenderse el tratamiento y debe iniciarse terapia antihipertensiva. Una vez que los valores de presión arterial se normalicen tras la terapia hipotensora, el tratamiento con Belara® puede reanudarse.
Los estrógenos exógenos pueden inducir o agravar los síntomas de angioedema hereditario o adquirido.
En mujeres con antecedentes de herpes gestacional, puede producirse un recidiva de esta enfermedad durante el uso de ACO.
En mujeres con antecedentes personales o familiares de hipertrigliceridemia durante el uso de ACO, aumenta el riesgo de desarrollar pancreatitis. En casos de alteraciones agudas o crónicas de la función hepática puede ser necesario suspender el ACO hasta la normalización de los parámetros funcionales hepáticos. En caso de recidiva de ictericia colestásica diagnosticada previamente durante el embarazo o el uso de hormonas sexuales, debe suspenderse el ACO.
El uso de ACO puede afectar la resistencia periférica a la insulina o la tolerancia a la glucosa. Por lo tanto, debe vigilarse cuidadosamente el estado de pacientes con diabetes mellitus que toman anticonceptivos orales.
En casos raros puede aparecer melasma, especialmente en mujeres con antecedentes de melasma gravídico. Las mujeres propensas al melasma deben evitar la exposición solar y la radiación ultravioleta durante el uso de anticonceptivos orales.
Precauciones
La administración de medicamentos que contienen estrógeno o estrógeno/progestágeno puede tener efectos negativos en ciertas enfermedades o estados. Situaciones que requieren una observación médica cuidadosa:
- Epilepsia;
- Esclerosis múltiple;
- Tetania;
- Migraña (ver sección «Contraindicaciones»);
- Asma;
- Insuficiencia cardíaca o renal;
- Corea menor;
- Diabetes mellitus (ver sección «Contraindicaciones»);
- Enfermedad hepática (ver sección «Contraindicaciones»);
- Dislipoproteinemia (ver sección «Contraindicaciones»);
- Enfermedades autoinmunes, incluyendo lupus eritematoso sistémico;
- Obesidad;
- Hipertensión arterial (ver sección «Contraindicaciones»);
- Endometriosis;
- Varices;
- Tromboflebitis (ver sección «Contraindicaciones»);
- Trastornos de la coagulación (ver sección «Contraindicaciones»);
- Mastopatía;
- Mioma uterino;
- Herpes gestacional;
- Depresión;
- Enfermedades inflamatorias intestinales crónicas (enfermedad de Crohn, colitis ulcerosa; ver sección «Reacciones adversas»).
Examen médico
Antes de iniciar o reanudar el tratamiento con Belara®, debe obtenerse un historial médico personal y familiar completo, realizarse un examen clínico y descartarse el embarazo. Debe medirse la presión arterial y realizarse un examen físico, prestando especial atención a las contraindicaciones (ver sección «Contraindicaciones») y advertencias descritas en esta sección.
Debe advertirse a la mujer sobre el riesgo de trombosis venosa y arterial, incluyendo el uso de Belara® en comparación con otros ACO, los síntomas de TVT y ETA, los factores de riesgo conocidos y qué hacer ante la sospecha de trombosis.
La mujer debe leer cuidadosamente el prospecto del medicamento y seguir las recomendaciones indicadas. La frecuencia y tipo de exámenes deben basarse en las recomendaciones clínicas establecidas, adaptadas a cada mujer individualmente.
Debe informarse a la mujer de que el uso de anticonceptivos orales no protege contra la infección por VIH (SIDA) ni contra otras enfermedades de transmisión sexual.
Reducción de la eficacia
La omisión de la toma de una tableta (ver sección «Toma irregular de las tabletas»), vómitos o trastornos intestinales, incluyendo diarrea, el uso prolongado de ciertos medicamentos concomitantes (ver sección «Interacción con otros medicamentos y otras formas de interacción») o, en casos muy raros, trastornos del metabolismo pueden reducir la eficacia anticonceptiva del fármaco.
Efecto sobre el control del ciclo menstrual
Sangrados de escape y manchado leve
El uso de todos los anticonceptivos orales puede provocar sangrados vaginales irregulares (sangrados de escape y manchado leve), especialmente durante los primeros ciclos de tratamiento. Por lo tanto, la evaluación médica de ciclos irregulares debe realizarse solo tras un período de adaptación de aproximadamente tres ciclos. Si durante el tratamiento con Belara® aparecen o persisten sangrados de escape, aunque previamente el ciclo fuera regular, debe realizarse una evaluación para descartar embarazo o enfermedades. Tras descartar embarazo o enfermedad, puede continuar el tratamiento con Belara® o cambiarse a otro fármaco.
Los sangrados intermenstruales pueden ser un signo de reducción de la eficacia anticonceptiva (ver secciones «Toma irregular de las tabletas», «Recomendaciones en caso de vómitos o diarrea», «Interacción con otros medicamentos y otras formas de interacción»).
Ausencia de sangrado de abstinencia
Generalmente, tras 21 días de tratamiento, aparece un sangrado de abstinencia. A veces, especialmente durante los primeros meses de tratamiento, puede no producirse sangrado de abstinencia. Sin embargo, esto no implica necesariamente una reducción del efecto anticonceptivo. Si no hay sangrado tras un ciclo completo en el que la paciente no olvidó tomar ninguna tableta, el período de siete días sin tomar tabletas no se prolongó y no hubo vómitos ni diarrea, la fecundación es improbable y puede continuar el tratamiento con Belara®. Si antes de la primera ausencia de sangrado de abstinencia el tratamiento con Belara® se realizó con incumplimiento de las instrucciones o si la ausencia de sangrado de abstinencia ocurre durante dos ciclos, debe descartarse el embarazo antes de continuar el tratamiento.
No deben tomarse simultáneamente con Belara® medicamentos de origen vegetal que contengan Hypericum perforatum (hipérico) (ver sección «Interacción con otros medicamentos y otras formas de interacción»).
Sustancias auxiliares
El medicamento contiene 69,5 mg de monohidrato de lactosa. No debe administrarse este medicamento a pacientes con intolerancia hereditaria rara a la galactosa, deficiencia total de lactasa o malabsorción de glucosa-galactosa.
Si tiene intolerancia a ciertos azúcares, consulte con su médico antes de tomar este medicamento.
Uso durante el embarazo o la lactancia.
Embarazo.
El medicamento está contraindicado durante el embarazo. Antes de iniciar el tratamiento debe descartarse el embarazo. Si se produce un embarazo durante el tratamiento con Belara®, debe suspenderse inmediatamente el medicamento. En estudios epidemiológicos amplios no se han obtenido pruebas de que la administración de estrógenos en combinación con otros progestágenos en dosis similares a las presentes en Belara®, cuando se toman accidentalmente anticonceptivos orales en las primeras etapas del embarazo, cause efectos teratogénicos o fetotóxicos. Aunque estudios en animales mostraron un efecto tóxico sobre la función reproductiva (ver sección «Datos de estudios preclínicos de seguridad»), los datos recopilados en 330 mujeres embarazadas no mostraron ninguna acción embriotóxica del acetato de clormadinona sobre el feto.
Debe tenerse en cuenta el riesgo aumentado de TVT durante el período posparto al reanudar el tratamiento con Belara® (ver secciones «Instrucciones de uso y dosis», «Precauciones especiales de uso»).
Lactancia.
Los estrógenos pueden afectar la lactancia, provocando una disminución en la cantidad y cambios en la composición de la leche materna. Pequeñas cantidades de esteroides anticonceptivos y/o sus metabolitos pueden pasar a la leche materna y afectar al niño, por lo tanto, no debe usarse Belara® durante la lactancia.
Capacidad para conducir y usar máquinas. No existen datos que indiquen que los anticonceptivos orales combinados afecten negativamente la capacidad para conducir automóviles u operar maquinaria.
Vía de administración y dosis.
Dosificación de los comprimidos recubiertos con película
Se debe tomar 1 comprimido recubierto con película cada día, a la misma hora (preferiblemente por la noche), durante 21 días consecutivos, seguido de un descanso de 7 días sin tomar comprimidos. Entre los días 2 y 4 después de tomar el último comprimido recubierto con película, aparecerá un sangrado de privación similar al sangrado menstrual. Tras finalizar el descanso de 7 días, se debe comenzar inmediatamente con el siguiente envase del medicamento Belara®, independientemente de si el sangrado ha cesado o no.
El comprimido recubierto con película, marcado en el blíster con el día correspondiente de la semana, debe extraerse del envase blíster, tragarse entero y, si es necesario, tomarse con un poco de agua. Los comprimidos recubiertos con película deben tomarse diariamente en el orden indicado en el envase mediante la flecha.
Inicio del tratamiento con comprimidos recubiertos con película
Si no se han utilizado previamente anticonceptivos hormonales (durante el último ciclo menstrual)
El primer comprimido recubierto con película debe tomarse el primer día del ciclo natural de la mujer, es decir, el primer día del siguiente sangrado menstrual. Si el primer comprimido se toma el primer día de la menstruación, el efecto anticonceptivo del medicamento comienza desde el primer día de toma y continúa durante la pausa de 7 días sin comprimidos.
El primer comprimido recubierto con película también puede tomarse entre el día 2 y el día 5 del sangrado menstrual, independientemente de si el sangrado ha cesado o no. En este caso, durante los primeros 7 días de tratamiento se debe utilizar un método anticonceptivo de barrera adicional.
Si la menstruación comenzó hace más de 5 días, se recomienda a la mujer esperar al inicio del siguiente ciclo menstrual para comenzar el tratamiento con Belara®.
Cambio desde otro anticonceptivo hormonal al tratamiento con Belara®
Cambio desde otro anticonceptivo oral combinado
La mujer debe comenzar el tratamiento con Belara® al día siguiente del descanso de 7 días o después de tomar el último comprimido placebo del envase del anticonceptivo oral combinado previamente utilizado.
Cambio desde preparados que contienen solo progestágeno («píldoras minipíldoras»)
El primer comprimido de Belara® debe tomarse al día siguiente de haber tomado el último comprimido que contiene solo progestágeno. Durante los primeros 7 días de tratamiento se debe utilizar un método anticonceptivo de barrera adicional.
Cambio desde inyecciones anticonceptivas hormonales o implante anticonceptivo
El tratamiento con Belara® puede comenzar el día de la extracción del implante o el día previamente programado para la inyección. Durante los primeros 7 días se debe utilizar un método anticonceptivo de barrera adicional.
Después de un aborto espontáneo o provocado en el primer trimestre
El tratamiento con Belara® puede comenzar inmediatamente después de un aborto espontáneo o provocado en el primer trimestre. En este caso, no es necesario utilizar medidas anticonceptivas adicionales.
Después del parto, aborto espontáneo o provocado en el segundo trimestre
Las mujeres que no amamantan pueden comenzar el tratamiento con Belara® entre los días 21 y 28 después del parto. En este caso, no es necesario utilizar métodos anticonceptivos de barrera adicionales.
Si el tratamiento comenzó más de 28 días después del parto, se debe utilizar un método anticonceptivo de barrera adicional durante los primeros 7 días.
Si la mujer ya ha tenido relaciones sexuales, se debe descartar un embarazo o esperar al inicio del siguiente ciclo menstrual antes de comenzar el tratamiento.
Lactancia (ver sección «Uso durante el embarazo o la lactancia»)
No se recomienda el uso de Belara® en mujeres que están amamantando.
Después de suspender el tratamiento con Belara®
Después de suspender Belara®, el ciclo menstrual actual puede prolongarse aproximadamente 1 semana.
Olvido de la toma de comprimidos
Si la paciente olvida tomar un comprimido, pero lo toma dentro de las siguientes 12 horas, no se requieren medidas anticonceptivas adicionales. La paciente debe continuar tomando el medicamento según lo habitual.
Si la paciente olvida tomar un comprimido y lo toma más de 12 horas después, la protección anticonceptiva puede reducirse. En caso de olvido, se deben seguir estas dos reglas principales:
- No se debe interrumpir la toma de comprimidos por más de 7 días.
- Es necesaria una pausa de 7 días sin comprimidos para lograr una supresión adecuada del eje hipotálamo-hipófisis-ovario.
El comprimido olvidado debe tomarse inmediatamente, incluso si esto implica tomar 2 comprimidos simultáneamente. Los siguientes comprimidos deben tomarse según lo habitual. Durante los siguientes 7 días se debe utilizar adicionalmente un método anticonceptivo de barrera, por ejemplo, preservativos. Si se olvidó tomar comprimidos durante la primera semana del ciclo y hubo relaciones sexuales en los 7 días previos al olvido (incluyendo la pausa de 7 días), se debe considerar la posibilidad de embarazo. Cuantos más comprimidos se hayan olvidado y más cerca estén del período habitual de descanso, mayor será el riesgo de embarazo.
Si quedan menos de 7 comprimidos en el envase, inmediatamente después de terminar los comprimidos del envase en uso, se debe comenzar con un nuevo envase de Belara® sin hacer pausa entre los dos envases. Es probable que no aparezca el sangrado de privación habitual hasta que se terminen los comprimidos del segundo envase; sin embargo, durante la toma de los comprimidos del nuevo envase puede aparecer un sangrado de escape o manchado. Si no aparece sangrado de privación tras finalizar el segundo envase, se debe realizar una prueba de embarazo.
Recomendaciones en caso de vómitos o diarrea
Si ocurren vómitos o diarrea severa dentro de las 4 horas posteriores a la toma del comprimido, la absorción del medicamento puede ser incompleta y, por lo tanto, la eficacia anticonceptiva no puede garantizarse. En este caso, se debe actuar según las recomendaciones indicadas en la sección «Olvido de la toma de comprimidos» (ver arriba). Se debe continuar tomando Belara®.
Cómo retrasar el sangrado de privación
Para retrasar el sangrado, la mujer debe continuar tomando comprimidos del siguiente envase de Belara® sin hacer pausa. Puede continuar tomando comprimidos según desee hasta terminar los comprimidos del segundo envase. Durante la toma de los comprimidos del segundo envase, puede aparecer un ligero sangrado o sangrado de escape. Tras la pausa habitual de 7 días sin comprimidos, se debe reanudar la toma regular de Belara®. Para posponer el inicio del sangrado a otro día de la semana distinto al del esquema actual, puede recomendarse a la mujer acortar la siguiente pausa de 7 días en el número deseado de días. Cuanto más corta sea la pausa sin comprimidos, mayor será el riesgo de ausencia del sangrado de privación y de sangrado de escape o manchado durante la toma de los comprimidos del siguiente envase (igual que en el caso de retrasar el sangrado).
Pacientes de edad avanzada. Belara® no está indicado para su uso tras la menopausia.
Niños. Belara® solo está indicado tras la menarquia. La seguridad y eficacia del acetato de clormadinona y etinilestradiol en adolescentes menores de 16 años no han sido establecidas. No existen datos.
El medicamento no debe administrarse a niños.
Sobredosis.
No hay información sobre efectos tóxicos graves del medicamento en caso de sobredosis. Pueden presentarse síntomas como náuseas, vómitos y, especialmente en jóvenes, ligeros sangrados vaginales. No existe antídoto específico; se debe realizar tratamiento sintomático. En casos raros, puede ser necesario controlar los parámetros del equilibrio hidroelectrolítico y la función hepática.
Reacciones adversas.
Durante los estudios clínicos del medicamento Belara®, se determinó que los efectos adversos más frecuentes del fármaco (> 20 %) fueron sangrado de escape, pequeños sangrados, cefalea y dolor en las mamas. La probabilidad de sangrados irregulares disminuye con el uso prolongado del medicamento Belara®.
En el marco de un estudio clínico realizado con participación de 1629 mujeres, se notificaron las siguientes reacciones adversas tras la administración del medicamento Belara®.
La frecuencia de las reacciones adversas se define de la siguiente manera:
muy frecuentes: ≥1/10; frecuentes: ≥1/100 a <1/10; infrecuentes: ≥1/1000 a <1/100; poco frecuentes: ≥1/10000 a <1/1000; raras: <1/10000; frecuencia desconocida: no se puede estimar a partir de los datos disponibles.
Infecciones e infestaciones:
infrecuentes: candidiasis vaginal;
poco frecuentes: vulvovaginitis.
Neoplasias benignas, malignas e inespecíficas (incluyendo quistes y pólipos):
infrecuentes: fibroadenoma de mama.
Alteraciones del sistema inmunitario:
infrecuentes: hipersensibilidad al medicamento, incluyendo reacciones alérgicas cutáneas;
frecuencia desconocida: empeoramiento de los síntomas de angioedema hereditario o adquirido.
Alteraciones del metabolismo y de la nutrición:
infrecuentes: alteraciones en los niveles de lípidos en sangre, incluyendo hipertigliceridemia;
poco frecuentes: aumento del apetito.
Trastornos psiquiátricos:
frecuentes: estado de ánimo deprimido, nerviosismo, irritabilidad;
infrecuentes: disminución del libido.
Alteraciones del sistema nervioso:
frecuentes: mareo, migraña (y/o empeoramiento de la migraña).
Alteraciones oculares:
frecuentes: trastornos visuales;
poco frecuentes: conjuntivitis, intolerancia a lentes de contacto.
Alteraciones del oído y del laberinto:
poco frecuentes: pérdida súbita de la audición, acúfenos.
Alteraciones vasculares:
poco frecuentes: hipertensión arterial, hipotensión arterial, colapso circulatorio, varices, trombosis venosa, tromboembolismo venoso (TEV) o tromboembolismo arterial (TEA)*.
Alteraciones gastrointestinales:
muy frecuentes: náuseas;
frecuentes: vómitos;
infrecuentes: dolor abdominal, distensión abdominal, diarrea.
Alteraciones de la piel y del tejido subcutáneo:
frecuentes: acné;
infrecuentes: alteraciones de la pigmentación, melasma, alopecia, sequedad de la piel, hiperhidrosis, caída del cabello;
poco frecuentes: erupciones urticariformes, eccema, eritema, prurito, empeoramiento del psoriasis, hirsutismo;
raras: eritema nodoso.
Alteraciones del sistema osteomuscular y del tejido conjuntivo:
frecuentes: sensación de pesadez;
infrecuentes: dolor de espalda, alteraciones musculares.
Alteraciones del sistema reproductor y de las mamas:
muy frecuentes: secreción vaginal, dismenorrea, amenorrea;
frecuentes: dolor abdominal bajo;
infrecuentes: galactorrea;
poco frecuentes: aumento de las mamas, menorragia, síndrome premenstrual.
Alteraciones generales y en el lugar de administración:
frecuentes: fatiga, edemas, aumento de peso.
Datos de laboratorio:
frecuentes: aumento de la presión arterial.
*Véase la sección «Descripción de reacciones adversas específicas».
Reacciones adversas relacionadas con la administración de etinilestradiol y clormadinona durante el período poscomercialización: astenia y reacciones alérgicas no relacionadas con alteraciones del sistema inmunitario.
Descripción de reacciones adversas específicas
Durante el uso de anticonceptivos orales combinados que contienen etinilestradiol 0,03 mg y acetato de clormadinona 2 mg, también se han notificado los siguientes efectos adversos:
- Se sabe que el uso de anticonceptivos hormonales combinados se asocia con un mayor riesgo de trombosis venosa y arterial y tromboembolismos, incluyendo infarto de miocardio, accidente cerebrovascular, ataque isquémico transitorio, trombosis venosa y embolia pulmonar. Este riesgo se describe con mayor detalle en la sección «Precauciones especiales de uso».
- Según datos de algunos estudios, el uso prolongado de AHC aumenta el riesgo de enfermedades de las vías biliares.
- En casos raros, tras la administración de anticonceptivos orales se han notificado tumores benignos del hígado, y aún más raramente tumores malignos; en algunos casos, estos tumores han provocado hemorragia intraabdominal con riesgo vital (véase la sección «Precauciones especiales de uso»).
- Empeoramiento de enfermedades inflamatorias intestinales crónicas (enfermedad de Crohn, colitis ulcerosa; véase la sección «Precauciones especiales de uso»).
Información sobre otros efectos adversos graves, como cáncer de útero o cáncer de mama, se proporciona en la sección «Precauciones especiales de uso».
Interacciones
Pueden producirse sangrados de escape y/o disminución del efecto anticonceptivo debido a interacciones entre otros medicamentos (inductores enzimáticos) y los anticonceptivos orales (véase la sección «Interacción con otros medicamentos y otras formas de interacción»).
Notificación de reacciones adversas sospechadas
Es importante notificar las reacciones adversas sospechosas durante el período de vigilancia poscomercialización. Esto permite realizar un seguimiento del balance beneficio-riesgo del medicamento. Los profesionales sanitarios deben informar sobre cualquier reacción adversa sospechosa.
Período de validez. 3 años.
Condiciones de conservación. Conservar a una temperatura no superior a 25 °C.
¡Mantener el medicamento fuera del alcance de los niños!
Envase. 21 comprimidos recubiertos con película en blíster; 1 o 3 blísteres por envase de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. Gedeon Richter S.A., Hungría.
Dirección del fabricante y lugar de actividad.
H-1103 Budapest, calle Demrédi, 19-21, Hungría.