Azacitidina Accord
UcraniaContenido
INSTRUCCIONES para uso médico del medicamento Azacitidina Accord (Azacitidine Accord)
Composición:
Principio activo: azacitidina;
1 frasco contiene 100 mg de azacitidina;
Sustancia auxiliar: manitol (E 421).
Forma farmacéutica. Polvo para suspensión inyectable.
Principales propiedades físico-químicas: polvo liofilizado blanco o masa en frasco de vidrio transparente.
Grupo farmacoterapéutico. Agentes antineoplásicos. Análogos de pirimidina.
Código ATC L01B C07.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
Se considera que la azacitidina ejerce su efecto antineoplásico mediante múltiples mecanismos, incluyendo la citotoxicidad sobre células hematopoyéticas anormales en la médula ósea y la hipometilación del ADN. El efecto citotóxico de la azacitidina puede deberse a múltiples mecanismos, incluyendo la inhibición de la síntesis de ADN, ARN y proteínas, la incorporación en ARN y ADN y la activación de las vías de daño del ADN. Las células no proliferativas son relativamente insensibles a la azacitidina. La incorporación de azacitidina en el ADN conduce a la inactivación de las ADN-metiltransferasas, lo que provoca hipometilación del ADN. La hipometilación del ADN de genes anormalmente metilados que participan en la regulación del ciclo celular normal, las vías de diferenciación y muerte celular puede conducir a la reexpresión de genes y restauración de funciones supresoras del cáncer en células tumorales. La importancia relativa de la hipometilación del ADN en comparación con la citotoxicidad u otra actividad de la azacitidina para lograr resultados clínicos no ha sido establecida.
Eficacia y seguridad clínicas
Población adulta (SMDS, LMMCr y LMA [20-30 % de blastas en médula ósea])
La eficacia y seguridad de la azacitidina se evaluaron en un estudio internacional, multicéntrico, controlado, abierto, aleatorizado, paralelo, de fase 3 (AZA PH GL 2003 CL 001) en adultos con: SMDS de riesgo intermedio 2 y alto según el Sistema Internacional de Puntuación Pronóstica (IPSS), anemia refractaria con exceso de blastas (AREB), anemia refractaria con exceso de blastas en transformación (AREB-T) y leucemia mielomonocítica crónica modificada (LMMCr) según la clasificación franco-estadounidense-británica (FAB). Los pacientes con AREB-T (21-30 % de blastas) ahora se consideran pacientes con LMA según la actual clasificación de la OMS. Se comparó el régimen de azacitidina más mejor tratamiento de apoyo (BSC) (n = 179) con regímenes terapéuticos convencionales (CCR). Los regímenes CCR incluían solo BSC (n = 105), citarabina en dosis bajas más BSC (n = 49) o quimioterapia de inducción estándar más BSC (n = 25). Los pacientes fueron previamente asignados por su médico a uno de los tres regímenes CCR antes de la aleatorización. Los pacientes recibieron este régimen previamente asignado si no fueron aleatorizados al grupo de azacitidina. Según los criterios de inclusión, los pacientes debían tener un estado funcional 0-2 según la clasificación del Grupo Cooperativo de Oncología del Este (ECOG). Se excluyeron pacientes con SMDS secundario del estudio. El punto final primario del estudio fue la supervivencia global. La azacitidina se administró por vía subcutánea a una dosis de 75 mg/m² diarios durante siete días, seguido de un período de descanso de 21 días (ciclo de tratamiento de 28 días); la mediana de ciclos fue de 9 (rango = 1-39) y el valor medio de ciclos fue de 10,2. En la población de intención de tratar (ITT), la edad media fue de 69 años (rango de 38 a 88 años).
En el análisis de la población ITT con 358 pacientes (179 en el grupo de azacitidina y 179 en el grupo CCR), el tratamiento con azacitidina se asoció con una supervivencia mediana de 24,46 meses frente a 15,02 meses en aquellos que recibieron tratamiento CCR; la diferencia fue de 9,4 meses con un valor p logarítmico estratificado de 0,0001. El coeficiente de riesgo (CR) para el efecto del tratamiento fue de 0,58 (IC del 95 %: 0,43, 0,77). Las tasas de supervivencia a dos años fueron del 50,8 % en pacientes que recibieron azacitidina frente al 26,2 % en pacientes que recibieron CCR (p < 0,0001).
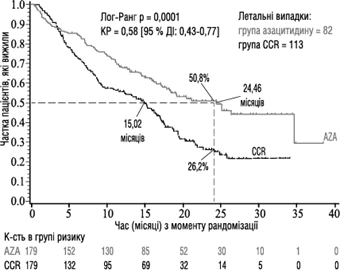
Abreviaturas: AZA = azacitidina; CCR = regímenes terapéuticos convencionales; IC = intervalo de confianza; CR = coeficiente de riesgo.
Se observó un aumento de la supervivencia en el grupo de azacitidina de forma constante independientemente del régimen CCR utilizado en el grupo control (solo BSC, dosis bajas de citarabina más BSC o quimioterapia de inducción estándar más BSC).
Durante el análisis de subgrupos citogenéticos según IPSS, se observaron resultados similares respecto a la supervivencia global mediana en todos los grupos (citogenética buena, intermedia y mala, incluyendo monosomía 7).
Durante el análisis de subgrupos por edad, se observó un aumento en la supervivencia global media en todos los grupos (< 65 años, ≥ 65 años y ≥ 75 años).
El tratamiento con azacitidina se asoció con un tiempo medio hasta muerte o transformación a LMA de 13,0 meses frente a 7,6 meses en aquellos que recibieron tratamiento CCR, lo que representa una mejora de 5,4 meses con un valor p estratificado logarítmico de rango de 0,0025.
El tratamiento con azacitidina también se asoció con una reducción de la citopenia y sus síntomas relacionados. El tratamiento con azacitidina condujo a una disminución en la necesidad de transfusiones de eritrocitos (glóbulos rojos) y plaquetas. Entre los pacientes en el grupo de azacitidina dependientes de transfusiones de eritrocitos al inicio, el 45,0 % se volvieron independientes de transfusiones de eritrocitos durante el período de tratamiento, en comparación con el 11,4 % de los pacientes en los grupos combinados de tratamiento CCR (diferencia estadísticamente significativa (p < 0,0001) del 33,6 % (IC del 95 %: 22,4, 44,6). En pacientes que eran dependientes de transfusiones de eritrocitos al inicio y que se volvieron independientes, la duración media del período de independencia de transfusiones de eritrocitos fue de 13 meses en el grupo de azacitidina.
La respuesta al tratamiento fue evaluada por el investigador o por un Comité Independiente de Revisión (IRC). La respuesta global (remisión completa [CR] + remisión parcial [PR]), determinada por el investigador, fue del 29 % en el grupo de azacitidina y del 12 % en el grupo combinado de tratamiento CCR (p = 0,0001). La respuesta global (CR + PR), determinada por el comité IRC en el estudio AZA PH GL 2003 CL 001, fue del 7 % (12/179) en el grupo de azacitidina frente al 1 % (2/179) en el grupo combinado de tratamiento CCR (p = 0,0113). Las diferencias entre las evaluaciones de respuesta del comité IRC y los investigadores fueron consecuencia de los criterios del Grupo de Trabajo Internacional (IWG), que requerían mejoría en los parámetros de sangre periférica y mantenimiento de estas mejorías durante al menos 56 días. La ventaja en supervivencia también se demostró en pacientes que no alcanzaron respuesta completa/parcial tras el tratamiento con azacitidina. La mejoría hematológica (importante o menor), según determinado por IRC, se logró en el 49 % de los pacientes que recibieron azacitidina frente al 29 % de los pacientes que recibieron CCR combinado (p < 0,0001).
En pacientes con una o más anomalías citogenéticas al inicio, el porcentaje de pacientes con respuesta citogenética significativa fue similar en los grupos de azacitidina y tratamiento CCR combinado. La respuesta citogenética menor fue estadísticamente significativa (p = 0,0015) más alta en el grupo de azacitidina (34 %) en comparación con el grupo de tratamiento CCR combinado (10 %).
Población adulta de 65 años o más con LMA con > 30 % de blastas en médula ósea
Los resultados que se indican a continuación se refieren a la población de tratamiento asignado estudiada en el ensayo AZA-AML-001 (indicaciones aprobadas se indican en la sección «Posología y forma de administración»).
La eficacia y seguridad de la azacitidina se evaluaron en un estudio internacional, multicéntrico, controlado, abierto, de fase 3, en grupos paralelos, con pacientes de 65 años o más con LMA de novo o secundaria diagnosticada recientemente con > 30 % de blastas en médula ósea según la clasificación de la OMS, que no cumplían los criterios para trasplante de células madre hematopoyéticas (TCMH). Se comparó azacitidina más BSC (n = 241) con un régimen CCR. El régimen CCR incluyó solo BSC (n = 45), dosis bajas de citarabina más BSC (n = 158) o quimioterapia intensiva estándar con citarabina y antraciclina más BSC (n = 44). Los pacientes fueron previamente asignados por su médico a uno de los tres regímenes de tratamiento CCR antes de la aleatorización. Los pacientes recibieron el régimen previamente seleccionado si no fueron aleatorizados al grupo de azacitidina. Como parte de los criterios de inclusión, los pacientes debían tener un estado funcional 0-2 según la escala ECOG y anomalías citogenéticas de riesgo intermedio o bajo. El punto final primario del estudio fue la supervivencia global.
La azacitidina se administró por vía subcutánea a una dosis de 75 mg/m²/día durante 7 días, seguido de un período de descanso de 21 días (ciclo de tratamiento de 28 días), con una mediana de 6 ciclos de tratamiento (rango: de 1 a 28): pacientes que recibieron solo BSC, con una mediana de 3 ciclos (rango: de 1 a 20); pacientes que recibieron dosis bajas de citarabina, con una mediana de 4 ciclos (rango de 1 a 25), y pacientes que recibieron quimioterapia intensiva estándar, con una mediana de 2 ciclos (rango: de 1 a 3, ciclo de inducción más 1 o 2 ciclos de consolidación).
Los parámetros basales individuales fueron comparables entre los grupos de azacitidina y CCR. La mediana de edad de los participantes fue de 75,0 años (rango: de 64 a 91 años), el 75,2 % eran de raza caucásica y el 59,0 % eran hombres. Al inicio, el 60,7 % se clasificaron como pacientes con LMA no especificada, el 32,4 % como pacientes con LMA con alteraciones relacionadas con displasia mieloide, el 4,1 % tenían neoplasias mieloides relacionadas con terapia y el 2,9 % tenían LMA con anomalías genéticas recurrentes según la clasificación de la OMS.
En el análisis de la población ITT con 488 pacientes (241 del grupo de azacitidina y 247 del grupo CCR), el tratamiento con azacitidina se asoció con una supervivencia mediana de 10,4 meses frente a 6,5 meses en aquellos que recibieron tratamiento CCR – la diferencia fue de 3,8 meses con un valor p logarítmico estratificado de rango de 0,1009 (bilateral). El coeficiente de riesgo para el efecto del tratamiento fue de 0,85 (IC del 95 % = 0,69, 1,03). La supervivencia a un año fue del 46,5 % en pacientes que recibieron azacitidina frente al 34,3 % en pacientes que recibieron el régimen CCR.
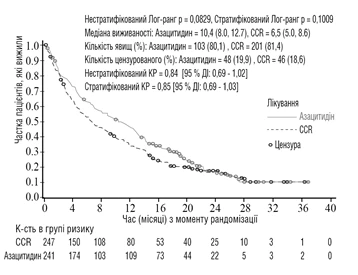
Abreviaturas: CCR = regímenes terapéuticos convencionales; IC = intervalo de confianza; CR = coeficiente de riesgo.
Según el modelo de riesgos proporcionales de Cox ajustado por factores pronósticos basales previamente definidos, se determinó un CR para azacitidina en comparación con el régimen CCR de 0,80 (IC del 95 % = 0,66, 0,99; p = 0,0355).
Además, aunque en el estudio no fue posible demostrar una diferencia estadísticamente significativa al comparar azacitidina con los regímenes CCR previamente seleccionados, la supervivencia de los pacientes que recibieron azacitidina fue más larga en comparación con estas opciones de tratamiento CCR, como solo BSC, dosis bajas de citarabina más BSC, y fue similar a la quimioterapia intensiva estándar más BSC.
En todos los subgrupos previamente definidos por edad [(< 75 años y ≥ 75 años), sexo, raza, estado funcional ECOG (0, o 1 y 2), riesgo citogenético basal (intermedio y bajo), región geográfica, clasificación OMS de LMA (incluyendo LMA con alteraciones relacionadas con displasia mieloide), recuento basal de leucocitos (≤ 5 x 10⁹/l y > 5 x 10⁹/l), recuento basal de blastas en médula ósea (≤ 50 % y > 50 %) y antecedentes previos de enfermedad de SMDS], se observó una tendencia a favor de la OS con azacitidina. En varios subgrupos previamente definidos, el CR de OS alcanzó significación estadística, incluyendo pacientes con riesgo citogenético bajo, pacientes con LMA con alteraciones relacionadas con displasia mieloide, pacientes menores de 75 años, pacientes de sexo femenino y pacientes de raza caucásica.
Las respuestas hematológicas y citogenéticas se evaluaron por el investigador y un comité de revisión institucional (IRC) en comparación con resultados similares. La frecuencia global de respuesta (remisión completa [CR] + remisión completa con recuperación hematológica incompleta [CRi]), según determinado por IRC, fue del 27,8 % en el grupo de azacitidina y del 25,1 % en el grupo combinado de tratamiento CCR (p = 0,5384). En pacientes que alcanzaron CR o CRi, la duración media de la remisión fue de 10,4 meses (IC del 95 % = 7,2, 15,2) para pacientes que recibieron azacitidina y de 12,3 meses (IC del 95 % = 9,0, 17,0) para pacientes que recibieron CCR. También se demostró una mejora en la supervivencia en pacientes que no alcanzaron respuesta completa con azacitidina en comparación con el régimen CCR.
El tratamiento con azacitidina mejoró los parámetros de sangre periférica y condujo a una reducción en la necesidad de transfusiones de eritrocitos y plaquetas. Un paciente se consideraba dependiente de transfusiones de eritrocitos o plaquetas al inicio si había recibido una o más transfusiones de eritrocitos o plaquetas durante los 56 días (8 semanas) previos al inicio o a la aleatorización, respectivamente. Un paciente se consideraba independiente de transfusiones de eritrocitos o plaquetas durante el período de tratamiento si no recibió transfusiones de eritrocitos o plaquetas durante ningún período consecutivo de 56 días durante el período de informe, respectivamente.
Entre los pacientes en el grupo de azacitidina dependientes de transfusiones de eritrocitos al inicio, el 38,5 % (IC del 95 % = 31,1, 46,2) de estos pacientes se volvieron independientes de transfusiones de eritrocitos durante el período de tratamiento, en comparación con el 27,6 % (IC del 95 % = 20,9, 35,1) de los pacientes en los grupos combinados de tratamiento CCR. En pacientes que eran dependientes de transfusiones de eritrocitos al inicio y alcanzaron independencia de transfusiones sanguíneas durante el tratamiento, la duración media de independencia de transfusiones de eritrocitos fue de 13,9 meses en el grupo de azacitidina; en el grupo CCR no se alcanzó dicha independencia.
Entre los pacientes en el grupo de azacitidina dependientes de transfusiones de plaquetas al inicio, el 40,6 % (IC del 95 % = 30,9, 50,8) de los pacientes se volvieron independientes de transfusiones de plaquetas durante el período de tratamiento, en comparación con el 29,3 % (IC del 95 % = 19,7, 40,4) de los pacientes en los grupos combinados de tratamiento CCR. En pacientes que eran dependientes de transfusiones de plaquetas al inicio y alcanzaron independencia de transfusiones sanguíneas tras el tratamiento, la duración media de independencia de transfusiones de plaquetas fue de 10,8 meses en el grupo de azacitidina y de 19,2 meses en el grupo CCR.
La calidad de vida relacionada con la salud (HRQoL) se evaluó mediante el Cuestionario Básico de Calidad de Vida de la Organización Europea para la Investigación y el Tratamiento del Cáncer (EORTC QLQ-C30). Los datos de HRQoL pueden analizarse para un subgrupo de la población total estudiada. A pesar de las limitaciones en el análisis, los datos obtenidos indican que los pacientes no experimentaron una disminución significativa en la calidad de vida durante el tratamiento con azacitidina.
Niños
El estudio AZA-JMML-001 fue un estudio internacional, multicéntrico, abierto, de fase 2, para evaluar la farmacocinética, farmacodinámica, seguridad y actividad de la azacitidina antes del TCMH en pacientes pediátricos con SMDS recientemente diagnosticado en progresión o LMMJu. El objetivo principal del estudio clínico fue evaluar el impacto de la azacitidina en la tasa de respuesta al tratamiento en el ciclo 3, día 28.
Los pacientes (con SMDS: n = 10; con LMMJu: n = 18, de 3 meses a 15 años; 71 % hombres) recibieron tratamiento intravenoso con azacitidina a una dosis de 75 mg/m² diarios del día 1 al 7 de un ciclo de 28 días durante un mínimo de tres ciclos y un máximo de seis ciclos.
La inclusión en el grupo de estudio de pacientes con SMDS se detuvo tras reclutar a 10 pacientes con SMDS debido a la eficacia insuficiente del tratamiento: no se registraron respuestas confirmadas en estos 10 pacientes.
Al grupo de estudio de LMMJu se incluyeron 18 pacientes (13 con mutaciones somáticas PTPN11, 3 con mutaciones NRAS, 1 con mutación KRAS y 1 con diagnóstico clínico de neurofibromatosis tipo 1 [NF-1]). Dieciséis pacientes completaron tres ciclos de terapia, y 5 de ellos completaron 6 ciclos. En total, 11 pacientes con LMMJu tuvieron respuesta clínica en el ciclo 3, día 28; de estos 11 participantes del estudio, 9 (50 %) tuvieron respuesta clínica confirmada (3 participantes con cCR y 6 participantes con cPR). Entre la cohorte de pacientes con LMMJu que recibieron azacitidina, 7 (43,8 %) pacientes tuvieron respuesta sostenida al tratamiento en el nivel de plaquetas (recuento ≥ 100 x 10⁹/l), y 7 (43,8 %) pacientes requirieron transfusión sanguínea durante el TCMH. Diecisiete de 18 pacientes pasaron al procedimiento de TCMH.
Debido al diseño del estudio (pequeño número de pacientes y diversos factores que dificultan el análisis), no es posible concluir, basándose en este estudio clínico, si el uso de azacitidina antes del procedimiento de TCMH mejora los resultados de supervivencia en pacientes con LMMJu.
El estudio AZA-AML-004 fue un estudio abierto, multicéntrico, de fase 2, para evaluar la seguridad, farmacodinámica y eficacia de la azacitidina en comparación con la ausencia de tratamiento antineoplásico en niños y adultos jóvenes con LMA en recaída molecular tras CR1.
Siete pacientes (mediana de edad de 6,7 años [rango de 2 a 12 años]; 71,4 % hombres) recibieron administración intravenosa de azacitidina a una dosis de 100 mg/m² diarios del día 1 al 7 de cada ciclo de 28 días durante un máximo de tres ciclos.
Cinco pacientes mostraron signos de enfermedad mínima residual (EMR) en el día 84, de los cuales 4 pacientes alcanzaron estabilidad molecular (n = 3) o mejora molecular (n = 1), y un paciente tuvo recaída clínica. Seis de los siete pacientes (90 % [IC del 95 % = 0,4, 1,0]) que recibieron azacitidina pasaron al TCMH.
Debido al pequeño tamaño de la muestra, no es posible establecer la eficacia de la azacitidina en niños con LMA. La información sobre seguridad se proporciona en la sección «Reacciones adversas».
Farmacocinética.
Absorción
Tras la administración subcutánea de una dosis única de 75 mg/m², la azacitidina fue rápidamente absorbida con una concentración máxima en plasma de 750 ± 403 ng/ml, observada a las 0,5 horas tras la administración de la dosis (primera toma de muestra).
La biodisponibilidad absoluta de la azacitidina tras la administración subcutánea en comparación con la vía intravenosa (dosis única de 75 mg/m²) fue aproximadamente del 89 %, basado en el valor del área bajo la curva farmacocinética (AUC).
El área bajo la curva y la concentración máxima en plasma (Cmax) tras la administración subcutánea de azacitidina fueron aproximadamente proporcionales en el rango de dosis de 25 a 100 mg/m².
Distribución
Tras la administración intravenosa, el volumen medio de distribución fue de 76 ± 26 l y el aclaramiento sistémico fue de 147 ± 47 l/h.
Biotransformación
Según datos obtenidos in vitro, el metabolismo de la azacitidina no está mediado por isoenzimas del citocromo P450 (CYP), UDP-glucuronosiltransferasas (UGT), sulfotransferasas (SULT) y glutatión transferasas (GST).
La azacitidina sufre hidrólisis espontánea y desaminación mediada por citidina desaminasa. En fracciones S9 de hígado humano, la formación de metabolitos no dependió del dinucleótido de nicotinamida-adenina fosfato reducido (NADPH), lo que indica que el metabolismo de la azacitidina no está mediado por isoenzimas del citocromo P450. Los datos de estudios in vitro con azacitidina en hepatocitos humanos cultivados indican que, en concentraciones de 1,0 µM a 100 µM (es decir, aproximadamente 30 veces más altas que las concentraciones clínicamente alcanzables), la azacitidina no induce CYP 1A2, 2C19 o 3A4 ni 3A5. En estudios de evaluación de inhibición de varias isoenzimas P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4), la azacitidina a dosis de hasta 100 µM no causó inhibición. Por lo tanto, la inducción o inhibición de la enzima CYP por azacitidina en concentraciones clínicamente alcanzables en plasma es poco probable.
Eliminación
La azacitidina se elimina rápidamente del plasma con una vida media (t1/2) media de 41 ± 8 minutos tras la administración subcutánea. Tras la administración subcutánea de 75 mg/m² de azacitidina una vez al día durante siete días, no se produce acumulación. La excreción urinaria es la vía principal de eliminación de la azacitidina y/o sus metabolitos. Tras la administración intravenosa y subcutánea de azacitidina marcada con 14C, el 85 % y el 50 % de la sustancia marcada administrada se detectó en la orina, respectivamente, mientras que < 1 % se detectó en las heces.
Poblaciones especiales
El impacto de la insuficiencia hepática (ver sección «Posología y forma de administración»), sexo, edad o raza sobre la farmacocinética de la azacitidina no ha sido estudiado oficialmente.
Niños
En el estudio AZA-JMML-001, el análisis farmacocinético se realizó en 10 pacientes con SMDS y 18 niños con LMMJu en el día 7 del ciclo 1 (ver sección «Propiedades farmacológicas»). La mediana de edad (rango) de los pacientes con SMDS fue de 13,3 (1,9-15) años y de 2,1 (0,2-6,9) años para pacientes con LMMJu.
Tras la administración intravenosa de una dosis de 75 mg/m², la azacitidina alcanzó rápidamente la Cmax en 0,083 horas tanto en las poblaciones de SMDS como de LMMJu. El valor medio geométrico de Cmax fue de 1797,5 y 1066,3 ng/ml, y el valor medio geométrico de AUC0-∞ fue de 606,9 y 240,2 ng*h/ml para pacientes con SMDS y LMMJu, respectivamente. El volumen medio geométrico de distribución en pacientes con SMDS y LMMJu fue de 103,9 y 61,1 l, respectivamente. Se encontró que la exposición total a la azacitidina en plasma fue más alta en pacientes con SMDS; sin embargo, se observó una variabilidad moderada o alta entre pacientes tanto para los parámetros AUC como para Cmax.
El tiempo medio geométrico de vida media t1/2 fue de 0,4 y 0,3 horas, y el aclaramiento medio geométrico fue de 166,4 y 148,3 l/h para SMDS y LMMJu, respectivamente.
Los datos farmacocinéticos del estudio AZA-JMML-001 se combinaron y compararon con los datos farmacocinéticos de seis pacientes adultos con SMDS que recibieron 75 mg/m² de azacitidina intravenosa en el estudio AZA-2002-BA-002. Los valores medios de Cmax y AUC0-t de azacitidina fueron similares en pacientes adultos y niños tras la administración intravenosa (2750 ng/ml frente a 2841 ng/ml y 1025 ng*h/ml frente a 882,1 ng*h/ml, respectivamente).
En el estudio AZA-AML-004, el análisis farmacocinético se realizó en 6 de 7 pacientes pediátricos que tuvieron al menos una concentración farmacocinética medida tras la administración de la dosis del fármaco (ver sección «Propiedades farmacológicas»). La mediana (rango) de edad de los pacientes con LMA fue de 6,7 (2-12) años.
Tras la administración de múltiples dosis del fármaco a 100 mg/m², las medias geométricas de Cmax y AUC0-t en el día 7 del ciclo 1 fueron de 1557 ng/ml y 899,6 ng*h/ml, respectivamente, con alta variabilidad interindividual (coeficiente de variación [CV] del 201,6 % y 87,8 %, respectivamente). La azacitidina alcanzó rápidamente el nivel de Cmax con una mediana de tiempo de 0,090 horas tras la administración intravenosa y disminuyó con un valor medio geométrico de vida media t1/2 de 0,380 horas. Las medias geométricas de aclaramiento y volumen de distribución fueron de 127,2 l/h y 70,2 l, respectivamente.
La exposición farmacocinética (azacitidina) observada en niños con LMA en recaída molecular tras CR1 fue comparable con la exposición obtenida a partir de datos combinados de 10 niños con SMDS y 18 niños con LMMJu, así como comparable con la exposición de azacitidina en adultos con SMDS.
Disfunción renal
La disfunción renal no tiene un impacto sustancial en la exposición farmacocinética de la azacitidina tras administración subcutánea única o múltiple. Tras la administración subcutánea de una dosis única de 75 mg/m², los valores medios de exposición (AUC y Cmax) en pacientes con disfunción renal leve, moderada y grave aumentaron en un 11-21 %, 15-27 % y 41-66 %, respectivamente, en comparación con pacientes con función renal normal. Sin embargo, la exposición se mantuvo dentro del mismo rango general de exposición observado en personas con función renal normal. La azacitidina puede administrarse a pacientes con disfunción renal sin ajuste de la dosis inicial, siempre que estos pacientes estén bajo vigilancia por signos de toxicidad, ya que la azacitidina y/o sus metabolitos se excretan principalmente por vía renal.
Farmacogenómica
El impacto de los polimorfismos conocidos de citidina desaminasa sobre el metabolismo de la azacitidina no ha sido estudiado oficialmente.
Características clínicas.
Indicaciones.
Azacitidina Accord está indicado para el tratamiento de pacientes adultos a los que no se adapta el trasplante de células madre hematopoyéticas (TCMH), con las siguientes enfermedades:
- Síndromes mielodisplásicos (SMD) de riesgo intermedio-2 y alto según el Sistema Internacional de Puntuación Pronóstica (IPSS),
- Leucemia mielomonocítica crónica (LMC) con 10-29 % de blastos en médula ósea sin trastorno mieloproliferativo,
- Leucemia mieloide aguda (LMA) con 20-30 % de blastos y displasia multilinaje, según la clasificación de la Organización Mundial de la Salud (OMS),
- LMA con > 30 % de blastos en médula ósea según la clasificación de la OMS.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Tumores malignos hepáticos en estadios avanzados (ver sección «Precauciones de uso»).
Lactancia (ver sección «Uso durante el embarazo o la lactancia»).
Precauciones especiales de seguridad.
Recomendaciones para una manipulación segura
Azacitidina Accord es un medicamento citotóxico; por tanto, como con otros compuestos potencialmente tóxicos, se debe tener precaución durante la manipulación del medicamento y la preparación de suspensiones de azacitidina. Se deben aplicar procedimientos adecuados de manipulación y eliminación de medicamentos antineoplásicos.
Si la azacitidina disuelta entra en contacto con la piel, debe lavarse inmediatamente con agua y jabón. Si entra en contacto con membranas mucosas, deben lavarse cuidadosamente con agua.
Procedimiento de dilución
Azacitidina Accord debe diluirse con agua para inyección. La vida útil del medicamento diluido puede prolongarse diluyéndolo con agua para inyección enfriada (entre 2 °C y 8 °C). La información detallada sobre el almacenamiento del producto diluido se proporciona más adelante.
Debe reunirse el siguiente material:
- Frasco(s) de azacitidina; frasco(s) de agua para inyección; guantes quirúrgicos no estériles; toallitas con alcohol; jeringa(s) de inyección de 5 ml con aguja(s).
- Debe aspirarse en la jeringa el volumen adecuado de agua para inyección (ver tabla más abajo) y debe eliminarse obligatoriamente el aire que haya entrado en la jeringa.
| Capacidad del frasco |
Volumen de agua para inyecciones |
Concentración final |
| 100 mg |
4 ml |
25 mg/ml |
- La aguja de la jeringa que contiene agua para inyección debe insertarse a través del tapón de goma del frasco de azacitidina y, a continuación, inyectar el agua para inyección en el frasco.
- Tras retirar la jeringa y la aguja, el frasco debe agitarse enérgicamente hasta obtener una suspensión homogénea turbia. Tras la reconstitución, cada ml de suspensión contendrá 25 mg de azacitidina (100 mg/4 ml o 150 mg/6 ml). El medicamento reconstituido es una suspensión homogénea turbia, sin aglomerados. El medicamento debe desecharse si contiene partículas de gran tamaño o aglomerados. No se debe filtrar la suspensión tras la reconstitución, ya que esto podría provocar la eliminación del principio activo. Téngase en cuenta que algunos conectores, conos y sistemas cerrados pueden contener filtros; por lo tanto, no deben utilizarse estos sistemas para la administración del medicamento tras la reconstitución.
- Debe desinfectarse el tapón de goma y se debe insertar una jeringa nueva con aguja en el frasco. A continuación, el frasco debe invertirse boca abajo, asegurándose de que la punta de la aguja quede por debajo del nivel del líquido. Después, se debe tirar suavemente del émbolo hacia atrás para extraer la cantidad de medicamento necesaria para la dosis adecuada, asegurándose de que el aire que haya entrado en la jeringa se haya eliminado. A continuación, se debe retirar la jeringa con la aguja del frasco y desechar la aguja.
- Debe acoplarse firmemente una nueva aguja para administración subcutánea (se recomienda calibre 25) a la jeringa. No debe purgarse la aguja con aire antes de la inyección, con el fin de reducir la frecuencia de reacciones locales en el sitio de inyección.
- Si se necesita administrar más de un frasco, deben repetirse todos los pasos descritos anteriormente para la preparación de la suspensión. En caso de dosis que requieran más de un frasco, la dosis debe distribuirse por igual, por ejemplo, una dosis de 150 mg = 6 ml, 2 jeringas con 3 ml cada una. Debido a la retención del medicamento en el frasco y en la aguja, puede no ser posible extraer toda la suspensión del frasco.
- El contenido de la jeringa precargada debe resuspenderse inmediatamente antes de la administración. La jeringa llena de suspensión reconstituida debe alcanzar una temperatura de aproximadamente 20–25 °C durante 30 minutos antes de la administración. Si el tiempo transcurrido supera los 30 minutos, la suspensión debe desecharse adecuadamente y prepararse una nueva dosis. Para resuspender el contenido, debe agitarse enérgicamente la jeringa entre las palmas de las manos hasta obtener una suspensión homogénea y turbia. El medicamento debe desecharse si contiene partículas de gran tamaño o aglomerados.
Conservación del medicamento reconstituido
Las condiciones de conservación tras la reconstitución del medicamento se indican en la sección «Período de validez».
Cálculo de la dosis individual
La dosis total según la superficie corporal (SC) puede calcularse de la siguiente manera:
Dosis total (mg) = dosis (mg/m²) × SC (m²)
La tabla siguiente se proporciona únicamente como ejemplo de cómo calcular las dosis individuales de azacitidina, basándose en un valor medio de superficie corporal de 1,8 m².
| Dosis, mg/m2 (% de la dosis recomendada inicial) |
Dosis total, basada en un APM de 1,8 m2 |
Cantidad necesaria de frascos |
Volumen total necesario de suspensión reconstituida |
| Frasco 100 mg |
|||
| 75 mg/m2 (100 %) |
135 mg |
2 frascos |
5,4 ml |
| 37,5 mg/m2 (50 %) |
67,5 mg |
1 frasco |
2,7 ml |
| 25 mg/m2 (33 %) |
45 mg |
1 frasco |
1,8 ml |
Vía de administración
El azacitidina Aco debe administrarse por vía subcutánea (introduciendo la aguja con un ángulo de 45-90°) mediante una aguja de calibre 25 en el hombro, muslo o abdomen.
Las dosis superiores a 4 ml deben administrarse en dos sitios diferentes.
Los sitios de inyección deben alternarse. Las nuevas inyecciones deben realizarse a una distancia mínima de 2,5 cm del sitio anterior y nunca en áreas dolorosas, con hematomas, enrojecidas o endurecidas.
Desecho
Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
Interacción con otros medicamentos y otras formas de interacción.
Según datos obtenidos in vitro, el metabolismo de la azacitidina no está mediado por las isoformas del citocromo P450 (CYP), UDP-glucuronosiltransferasas (UGT), sulfotransferasas (SULT) ni glutatión S-transferasas (GST); por lo tanto, se considera poco probable que se produzcan interacciones relacionadas con estas enzimas metabolizantes in vivo.
Es poco probable que la azacitidina tenga un efecto clínicamente significativo de inhibición o inducción sobre las enzimas del citocromo P450 (ver sección «Propiedades farmacológicas»).
No se han realizado estudios clínicos oficiales sobre interacciones con azacitidina.
Características de uso.
Toxicidad hematológica
El tratamiento con azacitidina se asocia con anemia, neutropenia y trombocitopenia, especialmente durante los dos primeros ciclos (véase la sección «Reacciones adversas»). Se debe realizar un hemograma completo para monitorizar la respuesta al tratamiento y la toxicidad según sea necesario, pero al menos antes de cada ciclo de tratamiento. Tras administrar la dosis recomendada en el primer ciclo, la dosis para los ciclos posteriores debe reducirse o la administración debe posponerse, según los datos sobre el recuento más bajo de células sanguíneas y la respuesta hematológica al tratamiento (véase la sección «Modo de administración y dosis»). Se debe recomendar a los pacientes que informen inmediatamente sobre cualquier episodio de fiebre. También se recomienda a los pacientes y médicos estar atentos a los signos y síntomas de hemorragia.
Alteración de la función hepática
No se han realizado estudios formales en pacientes con alteración de la función hepática. Se han notificado casos de coma hepático progresivo y desenlace fatal durante el tratamiento con azacitidina en pacientes con alta carga tumoral debido a enfermedad metastásica, especialmente en aquellos con un nivel basal de albúmina sérica < 30 g/l. La azacitidina está contraindicada en pacientes con tumores malignos hepáticos en estadios avanzados (véase la sección «Contraindicaciones»).
Alteración de la función renal
En pacientes que recibieron azacitidina por vía intravenosa en combinación con otros agentes quimioterapéuticos, se han notificado alteraciones de la función renal, desde el aumento de la creatinina sérica hasta insuficiencia renal y desenlace fatal. Además, cinco pacientes con leucemia mieloide crónica (LMC) que recibieron tratamiento con azacitidina y etopósido desarrollaron un síndrome de acidosis tubular renal, definido como disminución del bicarbonato sérico a < 20 mmol/l, combinado con alcalinidad urinaria aumentada e hipokalemia (nivel de potasio sérico < 3 mmol/l). En caso de disminución inexplicable del bicarbonato sérico (< 20 mmol/l) o aumento de la creatinina o de la TAS sérica, se debe reducir la dosis o posponer la administración del medicamento (véase la sección «Modo de administración y dosis»).
Se debe aconsejar a los pacientes que informen inmediatamente al médico sobre oliguria y anuria.
Aunque no se han observado diferencias clínicamente relevantes en la frecuencia de reacciones adversas entre personas con función renal normal y pacientes con insuficiencia renal, se debe observar cuidadosamente a los pacientes con insuficiencia renal por signos de toxicidad, ya que la azacitidina y/o sus metabolitos se excretan principalmente por vía renal (véase la sección «Modo de administración y dosis»).
Análisis de laboratorio
Antes del inicio del tratamiento y antes de cada ciclo de tratamiento, deben realizarse pruebas de función hepática, análisis del nivel de creatinina sérica y del bicarbonato sérico. El hemograma completo debe realizarse antes del inicio del tratamiento y según sea necesario para monitorizar la respuesta al tratamiento y la toxicidad, pero como mínimo antes de cada ciclo de tratamiento (véase también la sección «Reacciones adversas»).
Enfermedades cardíacas y pulmonares
Los pacientes con antecedentes de insuficiencia cardíaca congestiva grave, enfermedad cardíaca clínicamente inestable o enfermedad pulmonar fueron excluidos de los estudios registradores fundamentales (AZA PH GL 2003 CL 001 y AZA-AML-001), por lo que la seguridad y eficacia de la azacitidina en estos pacientes no han sido establecidas. Datos recientes de estudios clínicos en pacientes con antecedentes de enfermedad cardiovascular o pulmonar mostraron un aumento significativo en la frecuencia de complicaciones cardíacas con el uso de azacitidina (véase la sección «Reacciones adversas»). Por lo tanto, se recomienda tener precaución al prescribir azacitidina a estos pacientes. Antes del inicio y durante el tratamiento, se debe realizar una evaluación de la función cardíaca y pulmonar.
Fascitis necrotizante
En pacientes que recibieron azacitidina se han notificado casos de fascitis necrotizante, incluyendo desenlaces fatales. En los pacientes que desarrollen fascitis necrotizante, se debe interrumpir el tratamiento con azacitidina y comenzar inmediatamente el tratamiento adecuado.
Síndrome de lisis tumoral
Los pacientes con riesgo de desarrollar síndrome de lisis tumoral son aquellos con alta carga tumoral antes del tratamiento. Estos pacientes deben ser monitorizados cuidadosamente y se deben tomar medidas preventivas adecuadas.
Síndrome de diferenciación
Se han notificado casos de síndrome de diferenciación (también conocido como síndrome del ácido retinoico) entre pacientes que reciben azacitidina. El síndrome de diferenciación puede ser fatal, y sus síntomas y signos clínicos incluyen distress respiratorio, infiltrados pulmonares, fiebre, erupción cutánea, edema pulmonar, edema periférico, aumento rápido de peso corporal, derrame pleural, derrame pericárdico, hipotensión y alteración de la función renal (véase la sección «Reacciones adversas»). Se debe considerar el tratamiento con corticosteroides intravenosos en dosis altas y el monitoreo hemodinámico ante la primera aparición de síntomas u otros signos que sugieran síndrome de diferenciación. Se debe considerar la posibilidad de suspender temporalmente la administración de azacitidina inyectable hasta la desaparición de los síntomas, y si se reanuda el tratamiento, se debe proceder con precaución.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil/Control de la natalidad en hombres y mujeres
Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento y al menos 6 meses después de finalizarlo. Se recomienda a los hombres utilizar métodos anticonceptivos eficaces durante el tratamiento y durante 3 meses después de su finalización.
Embarazo
No existen datos adecuados sobre el uso de azacitidina en mujeres embarazadas. Los estudios en ratones mostraron toxicidad reproductiva del fármaco. El riesgo potencial de este medicamento para el ser humano es desconocido. En base a los resultados de estudios en animales y al mecanismo de acción, no se debe administrar azacitidina durante el embarazo, especialmente durante el primer trimestre, salvo que exista una necesidad absoluta. Se debe evaluar cuidadosamente el beneficio del tratamiento frente al riesgo potencial para el feto en cada caso individual.
Lactancia
No se sabe si la azacitidina/metabolitos se excretan en la leche materna. Debido al riesgo de reacciones adversas graves en el lactante, la lactancia está contraindicada durante el tratamiento con azacitidina.
Fertilidad
No existen datos sobre el efecto de la azacitidina sobre la fertilidad humana. En animales se han documentado efectos adversos sobre la fertilidad de los machos con el uso de azacitidina. Antes de iniciar el tratamiento, se debe recomendar a los pacientes varones que consideren la preservación del esperma.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
La azacitidina tiene un efecto leve o moderado sobre la capacidad para conducir vehículos o manejar maquinaria. Se ha notificado fatiga con el uso de azacitidina. Por lo tanto, se recomienda tener precaución al conducir vehículos o trabajar con maquinaria.
Vía de administración y dosis.
El tratamiento con Azacitidina Accord debe iniciarse y supervisarse bajo la dirección de un médico con experiencia en el uso de agentes quimioterapéuticos. A los pacientes se les debe administrar una premedicación con medicamentos antieméticos para prevenir náuseas y vómitos.
Dosis
La dosis inicial recomendada para el primer ciclo de tratamiento en todos los pacientes, independientemente de los valores hematológicos iniciales, es de 75 mg/m² de superficie corporal, administrada subcutáneamente una vez al día durante 7 días consecutivos, seguida de un período de descanso de 21 días (ciclo de tratamiento de 28 días).
Se recomienda que los pacientes reciban tratamiento durante un mínimo de 6 ciclos. El tratamiento debe continuar mientras el paciente obtenga beneficio clínico o hasta que la enfermedad progrese.
Debe vigilarse a los pacientes atentamente en busca de signos de respuesta hematológica/toxicidad y toxicidad renal (ver sección «Precauciones de uso»); puede ser necesario retrasar el inicio del siguiente ciclo o reducir la dosis, según se describe más adelante.
El medicamento Azacitidina Accord no debe utilizarse como intercambiable con la azacitidina oral. Debido a diferencias en la exposición, las recomendaciones de dosis y esquema de administración para la azacitidina oral son distintas de las recomendaciones para la azacitidina inyectable. Se recomienda a los profesionales sanitarios verificar cuidadosamente el nombre del medicamento, la dosis y la vía de administración.
Análisis de laboratorio
Antes del inicio de la terapia y antes de cada ciclo de tratamiento, deben realizarse pruebas de función hepática, así como medición del nivel de creatinina en suero y bicarbonato en suero. Se debe realizar un hemograma completo antes del inicio del tratamiento y, según sea necesario, para monitorear la respuesta al tratamiento y la toxicidad, pero como mínimo antes de cada ciclo de tratamiento.
Corrección de la dosis por toxicidad hematológica
La toxicidad hematológica se define como el valor más bajo alcanzado (nadir) en un ciclo determinado, si el recuento de plaquetas ≤ 50,0 x 10⁹/l y/o el recuento absoluto de neutrófilos (ANC) ≤ 1 x 10⁹/l.
La recuperación se define como el aumento de la línea celular (o líneas celulares) en la que se observó toxicidad hematológica, al menos hasta la mitad de la diferencia absoluta entre el valor más bajo y el valor inicial más el valor más bajo (es decir, el recuento celular en el momento de la recuperación ≥ valor más bajo + (0,5 x [valor inicial – valor más bajo])).
Pacientes sin alteraciones hematológicas iniciales (es decir, recuento de leucocitos (WBC) ≥ 3,0 x 10⁹/l y ANC ≥ 1,5 x 10⁹/l, y recuento de plaquetas ≥ 75,0 x 10⁹/l) antes del primer ciclo de tratamiento.
Si tras el tratamiento con Azacitidina Accord se observa toxicidad hematológica, el siguiente ciclo de tratamiento debe retrasarse hasta que se recupere el recuento de plaquetas y el ANC. Si la recuperación se alcanza dentro de los 14 días, no se requiere ajuste de dosis. Sin embargo, si la recuperación no se logra dentro de los 14 días, la dosis debe reducirse según la tabla que se indica a continuación. Tras cualquier cambio de dosis, la duración del ciclo debe volver a ser de 28 días.
| Nadir del ciclo |
Dosis en el siguiente ciclo si no se alcanza la recuperación* en 14 días (%) |
|
| ANC (x 109/l) |
Plaquetas (x 109/l) |
|
| ≤ 1,0 |
≤ 50,0 |
50 % |
| > 1,0 |
> 50,0 |
100 % |
*Recuperación = cantidad ≥ cantidad más baja + (0,5 x [cantidad inicial – cantidad más baja])
Pacientes con recuentos sanguíneos basales reducidos (es decir, leucocitos < 3,0 x 10⁹/l o ANC < 1,5 x 10⁹/l o plaquetas < 75,0 x 10⁹/l) antes del primer tratamiento
Si tras el tratamiento con azacitidina Accord la reducción del recuento de leucocitos, ANC o plaquetas en comparación con los niveles previos al tratamiento es ≤ 50 % o supera el 50 % pero existe mejoría en la diferenciación de cualquier línea celular, no se debe retrasar el siguiente ciclo ni ajustar la dosis.
Si la reducción del recuento de leucocitos, ANC o plaquetas supera el 50 % en comparación con los valores previos al tratamiento y no hay mejoría en la diferenciación de la línea celular, se debe retrasar el siguiente ciclo de tratamiento con azacitidina Accord hasta que se recupere el recuento de plaquetas y ANC. Si la recuperación se logra dentro de los 14 días, no se requiere ajuste de dosis. Sin embargo, si no se logra la recuperación dentro de los 14 días, se debe evaluar la celularidad de la médula ósea. Si la celularidad de la médula ósea es > 50 %, no se requiere ajuste de dosis. Si la celularidad de la médula ósea es ≤ 50 %, se debe retrasar el tratamiento y reducir la dosis según la tabla siguiente:
| Saturación de las células de la médula ósea |
Dosis en el siguiente ciclo si no se ha alcanzado la recuperación en 14 días (%) |
|
| Recuperación* ≤ 21 días |
Recuperación* > 21 días |
|
| 15-50 % |
100 % |
50 % |
| < 15 % |
100 % |
33 % |
*Recuperación = cantidad ≥ cantidad más baja + (0,5 x [cantidad inicial – cantidad más baja]).
Después de modificar la dosis, la duración del siguiente ciclo debe volver a ser de 28 días.
Poblaciones especiales
Pacientes de edad avanzada
No se recomienda un ajuste posológico especial para personas de edad avanzada. Dado que en los pacientes de edad avanzada es más frecuente la disminución de la función renal, puede ser útil el control de la función renal.
Pacientes con alteración de la función renal
Se puede administrar azacitidina a pacientes con alteración de la función renal sin ajuste de la dosis inicial (véase la sección «Propiedades farmacológicas»). Si se produce una disminución inexplicable del nivel de bicarbonato en suero por debajo de 20 mmol/l, la dosis debe reducirse en un 50 % durante el siguiente ciclo. En caso de aumento inexplicable del nivel de creatinina en suero o del nitrógeno ureico en sangre (NUS) ≥ al doble del valor inicial y por encima del límite superior normal (LSN), el siguiente ciclo debe posponerse hasta que los valores regresen a la normalidad o al nivel inicial, y la dosis debe reducirse en un 50 % durante el siguiente ciclo de tratamiento (véase la sección «Precauciones de uso»).
Pacientes con alteración de la función hepática
No se han realizado estudios formales en pacientes con alteración de la función hepática (véase la sección «Precauciones de uso»). Los pacientes con insuficiencia hepática grave deben estar bajo estrecha vigilancia respecto a eventos adversos. Para los pacientes con alteración de la función hepática no se recomienda modificar la dosis inicial antes del inicio del tratamiento; los ajustes posteriores de la dosis deben basarse en los parámetros hematológicos de laboratorio. Azacitidina Accord está contraindicada en pacientes con tumores hepáticos malignos en estadios avanzados (véanse las secciones «Contraindicaciones» y «Precauciones de uso»).
Niños
La seguridad y eficacia de azacitidina en niños de 0 a 17 años aún no han sido establecidas. Los datos disponibles se describen en las secciones «Propiedades farmacológicas» y «Reacciones adversas», pero no es posible proporcionar recomendaciones sobre la dosificación.
Vía de administración
Después de la reconstitución, cada ml de suspensión contendrá 25 mg de azacitidina. La azacitidina reconstituida debe administrarse por vía subcutánea en el hombro, muslo o abdomen. Los sitios de inyección deben alternarse. Las nuevas inyecciones deben realizarse a una distancia mínima de 2,5 cm del lugar anterior y en ningún caso en áreas dolorosas, con equimosis, enrojecidas o endurecidas.
Después de la reconstitución, la suspensión no debe filtrarse. Las instrucciones para la reconstitución del medicamento antes de su uso se proporcionan en la sección «Precauciones especiales de seguridad».
Niños.
No debe administrarse a niños (menores de 18 años), ya que la seguridad y eficacia del medicamento en esta categoría de pacientes no han sido establecidas.
Sobredosis.
Durante los estudios clínicos se notificó un caso de sobredosis con azacitidina. Un paciente presentó diarrea, náuseas y vómitos tras la administración intravenosa única de una dosis del medicamento de aproximadamente 290 mg/m², lo que casi cuadruplica la dosis inicial recomendada.
En caso de sobredosis, el estado del paciente debe vigilarse mediante análisis de sangre y, si es necesario, debe administrarse tratamiento de soporte. No existe un antídoto específico conocido para la sobredosis de azacitidina.
Reacciones adversas.
Resumen del perfil de seguridad
Población adulta con síndrome mielodisplásico (SMD), leucemia mielomonocítica crónica (LMC) y leucemia mielóide aguda (LMA) (20-30 % de blastos en médula ósea)
Se observaron reacciones adversas probablemente o posiblemente relacionadas con el uso de azacitidina en el 97 % de los pacientes.
Las reacciones adversas graves más frecuentes notificadas en el estudio principal (AZA PH GL 2003 CL 001) incluyeron neutropenia febril (8,0 %) y anemia (2,3 %), también comunicadas en estudios complementarios (CALGB 9221 y CALGB 8921). Otras reacciones adversas graves en estos tres estudios incluyeron infecciones, tales como sepsis neutropénica (0,8 %) y neumonía (2,5 %) (algunas con desenlace fatal), trombocitopenia (3,5 %), reacciones de hipersensibilidad (0,25 %) y manifestaciones hemorrágicas (por ejemplo, hemorragia cerebral [0,5 %], hemorragia gastrointestinal [0,8 %] y hemorragias intracraneales [0,5 %]).
Las reacciones adversas más frecuentes durante el tratamiento con azacitidina fueron reacciones hematológicas (71,4 %), incluyendo trombocitopenia, neutropenia y leucopenia (generalmente grados 3-4), trastornos gastrointestinales (60,6 %), incluyendo náuseas, vómitos (generalmente grados 1-2) o reacciones en el sitio de administración (77,1 %; generalmente grados 1-2).
Población adulta de 65 años o más con LMA y > 30 % de blastos en médula ósea
Las reacciones adversas graves más frecuentes (≥ 10 %) observadas en el estudio AZA-AML-001 en el grupo tratado con azacitidina fueron neutropenia febril (25,0 %), neumonía (20,3 %) y fiebre (10,6 %). Otras reacciones adversas graves menos frecuentes en el grupo tratado con azacitidina incluyeron sepsis (5,1 %), anemia (4,2 %), sepsis neutropénica (3,0 %), infección del tracto urinario (3,0 %), trombocitopenia (2,5 %), neutropenia (2,1 %), celulitis (2,1 %), vértigo (2,1 %) y disnea (2,1 %).
Las reacciones adversas más frecuentemente notificadas (≥ 30 %) durante el tratamiento con azacitidina fueron trastornos gastrointestinales, incluyendo estreñimiento (41,9 %), náuseas (39,8 %) y diarrea (36,9 %; generalmente grados 1-2); trastornos generales y reacciones en el sitio de administración, incluyendo fiebre (37,7 %; generalmente grados 1-2); y manifestaciones hematológicas, incluyendo neutropenia febril (32,2 %) y neutropenia (30,1 %; generalmente grados 3-4).
Lista tabulada de reacciones adversas
La Tabla 1, mostrada a continuación, contiene las reacciones adversas relacionadas con el tratamiento con azacitidina, obtenidas durante los principales estudios clínicos en pacientes con SMD y LMA, así como durante el período poscomercialización.
La frecuencia de las reacciones adversas se definió de la siguiente manera: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras (< 1/10.000) y frecuencia desconocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, los efectos adversos se presentan en orden decreciente de gravedad.
Las reacciones adversas se presentan en la tabla siguiente según la frecuencia más alta observada en cualquiera de los estudios clínicos principales.
Reacciones adversas notificadas en pacientes con SMD o LMA que recibieron azacitidina (estudios clínicos y período poscomercialización)
Tabla 1
| Clase de órganos y sistemas |
Muy frecuentes |
Frecuentes |
Infrecuentes |
Aislados |
Frecuencia desconocida |
| Infecciones e infestaciones |
neumonía* (incluida bacteriana, vírica y fúngica), nasofaringitis |
sepsis* (incluida bacteriana, vírica y fúngica), sepsis neutrópénica*, infección de las vías respiratorias (incluida infección de las vías respiratorias altas y bronquitis), infección del tracto urinario, celulitis, diverticulitis, infección fúngica de la cavidad oral, sinusitis, faringitis, rinitis, herpes simple, infección cutánea |
fascitis necrotizante* |
||
| Neoplasias benignas, malignas e inespecíficas (incluidos quistes y pólipos) |
síndrome de diferenciación*, a |
||||
| Trastornos de la sangre y del sistema linfático |
neutropenia febril*, neutropenia, leucopenia, trombocitopenia, anemia |
pancitopenia*, insuficiencia medular |
|||
| Trastornos del sistema inmunitario |
reacciones de hipersensibilidad |
||||
| Trastornos del metabolismo y de la nutrición |
anorexia, disminución del apetito, hipokalemia |
deshidratación |
síndrome de lisis tumoral |
||
| Trastornos psiquiátricos |
insomnio |
confusión, ansiedad |
|||
| Trastornos del sistema nervioso |
mareo, dolor de cabeza |
hemorragia intracraneal*, síncope, somnolencia, letargo |
|||
| Trastornos oculares |
hemorragia ocular, hemorragia conjuntival |
||||
| Trastornos cardiacos |
derrame pericárdico |
pericarditis |
|||
| Trastornos vasculares |
hipotensión*, hipertensión, hipotensión ortostática, hematoma |
||||
| Trastornos respiratorios, torácicos y mediastínicos |
disnea, epistaxis |
derrame pleural, disnea con esfuerzo, dolor faringo-laríngeo |
enfermedad pulmonar intersticial |
||
| Trastornos gastrointestinales |
diarrea, vómitos, estreñimiento, náuseas, dolor abdominal (incluido malestar en la parte superior del abdomen y en el abdomen) |
hemorragia gastrointestinal* (incluida hemorragia oral), hemorragias hemorroidales, estomatitis, sangrado de encías, dispepsia |
|||
| Trastornos hepáticos y biliares |
insuficiencia hepática*, coma hepático progresivo |
||||
| Trastornos de la piel y del tejido subcutáneo |
pétquias, prurito (incluido generalizado), erupción cutánea, equimosis |
púrpura, alopecia, urticaria, eritema, erupción macular |
dermatosis neutrofílica febril aguda, pioderma gangrenoso |
vasculitis cutánea |
|
| Trastornos del sistema músculo-esquelético y del tejido conjuntivo |
artralgia, dolor del sistema músculo-esquelético (incluido dolor de espalda, huesos y extremidades) |
espasmos musculares, mialgia |
|||
| Trastornos renales y urinarios |
insuficiencia renal*, hematuria, aumento de la creatinina en suero |
acidosis tubular renal |
|||
| Trastornos generales y condiciones en el sitio de administración |
fiebre*, fatiga, astenia, dolor en el pecho, eritema en el sitio de inyección, dolor en el sitio de inyección, reacción en el sitio de inyección (no especificada) |
equimosis, hematoma, induración, erupción, prurito, inflamación, cambio de color, nódulos y hemorragia (en el sitio de inyección), malestar general, escalofríos, hemorragia en el sitio del catéter |
necrosis del sitio de administración (en el sitio de administración) |
||
| Análisis de laboratorio |
disminución de peso corporal |
* – en casos raros se han notificado consecuencias fatales.
a – véase la sección «Propiedades especiales de utilización».
Descripción de reacciones adversas individuales
Reacciones adversas hematológicas
Las reacciones adversas hematológicas más frecuentemente notificadas (≥ 10 %), relacionadas con el tratamiento con azacitidina, incluyeron anemia, trombocitopenia, neutropenia, neutropenia febril y leucopenia, y generalmente fueron de grado 3 o 4. Existe un mayor riesgo de aparición de estos eventos durante los dos primeros ciclos de tratamiento, tras los cuales ocurren con menor frecuencia en pacientes con recuperación de la función hematológica. La mayoría de las reacciones adversas hematológicas se controlaron mediante monitoreo regular del hemograma completo, aplazamiento de la administración de azacitidina en el siguiente ciclo, tratamiento profiláctico con antibióticos y/o apoyo con factores de crecimiento (por ejemplo, G-CSF (factor estimulante de colonias de granulocitos)) para tratar la neutropenia, y transfusiones de sangre para controlar la anemia o la trombocitopenia, si fuera necesario.
Infecciones
La mielosupresión puede provocar neutropenia y un mayor riesgo de infección. Se han notificado reacciones adversas graves, como sepsis, incluyendo sepsis neutropénica, y neumonía en pacientes que recibieron azacitidina, algunas con resultado fatal. Las infecciones pueden tratarse con agentes antiinfecciosos y apoyo con factores de crecimiento (por ejemplo, G-CSF) para tratar la neutropenia.
Sangrado
Puede ocurrir sangrado en pacientes que reciben azacitidina. Se han notificado reacciones adversas graves, tales como hemorragias gastrointestinales y hemorragias intracraneales. Los pacientes deben ser vigilados en busca de signos y síntomas de sangrado, especialmente aquellos con trombocitopenia preexistente o asociada al tratamiento.
Hipersensibilidad
Se han notificado reacciones graves de hipersensibilidad en pacientes que recibieron azacitidina. En caso de reacción anafiláctica, el tratamiento con azacitidina debe interrumpirse inmediatamente y comenzar el tratamiento sintomático adecuado.
Reacciones adversas en la piel y tejido subcutáneo
La mayoría de las reacciones en la piel y tejido subcutáneo estuvieron relacionadas con el sitio de inyección. Ninguna de estas reacciones adversas condujo a la interrupción del uso de azacitidina o a la reducción de la dosis en los estudios fundamentales. La mayoría de las reacciones adversas ocurrieron durante los dos primeros ciclos de tratamiento y tendieron a disminuir en ciclos posteriores. Las reacciones adversas en el tejido subcutáneo, como erupción/irritación/prurito en el sitio de inyección, erupción cutánea, eritema y lesiones cutáneas, pueden requerir tratamiento con medicamentos concomitantes, tales como antihistamínicos, corticosteroides y medicamentos antiinflamatorios no esteroideos (AINE). Estas reacciones cutáneas deben diferenciarse de las infecciones de tejidos blandos, que a veces ocurren en el sitio de inyección. Se han notificado infecciones de tejidos blandos, incluyendo celulitis y fascitis necrotizante, que en casos raros condujeron a consecuencias fatales durante el uso poscomercialización de azacitidina. La estrategia clínica para reacciones adversas infecciosas se describe en la sección «Reacciones adversas», «Infecciones».
Reacciones adversas gastrointestinales
Las reacciones adversas gastrointestinales más comunes relacionadas con el tratamiento con azacitidina incluyeron estreñimiento, diarrea, náuseas y vómitos. Estas reacciones adversas se trataron sintomáticamente con antieméticos para controlar náuseas y vómitos; agentes antidiarreicos en caso de diarrea y laxantes y/o agentes suavizantes durante el estreñimiento.
Reacciones adversas renales
En pacientes que recibieron azacitidina se han notificado alteraciones de la función renal, desde aumento de la creatinina sérica y hematuria hasta acidosis tubular renal, insuficiencia renal y consecuencias fatales (véase la sección «Propiedades especiales de utilización»).
Reacciones adversas hepáticas
En pacientes con alta carga tumoral debido a enfermedad metastásica se han notificado insuficiencia hepática, coma hepático progresivo y resultado fatal durante el tratamiento con azacitidina (véase la sección «Propiedades especiales de utilización»).
Alteraciones cardiacas
Los datos de un estudio clínico que incluyó pacientes con antecedentes de enfermedades cardiovasculares o pulmonares mostraron un aumento de alteraciones cardiacas en pacientes con LMA de novo que recibieron tratamiento con azacitidina (véase la sección «Propiedades especiales de utilización»).
Pacientes de edad avanzada
Hay información limitada sobre la seguridad de la azacitidina en pacientes de ≥ 85 años (en el estudio AZA-AML-001 participaron 14 [5,9 %] pacientes de ≥ 85 años).
Pacientes pediátricos
En el estudio AZA-JMML-001, 28 niños (de 1 mes a < 18 años) recibieron tratamiento con azacitidina por MDS (n = 10) o leucemia mielomonocítica juvenil (LMJ) (n = 18) (véase la sección «Propiedades farmacológicas»).
En los 28 pacientes se observó al menos un evento adverso, y en 17 (60,7 %) al menos un evento relacionado con el tratamiento. Las reacciones adversas más comunes en la población pediátrica general fueron hipertermia, eventos hematológicos, incluyendo anemia, trombocitopenia y neutropenia febril, y trastornos gastrointestinales, incluyendo estreñimiento y vómitos.
En tres participantes se observaron eventos relacionados con el tratamiento que condujeron a la interrupción del medicamento (hipertermia, progresión de la enfermedad y dolor abdominal).
En el estudio AZA-AML-004, 7 niños (de 2 a 12 años) recibieron tratamiento con azacitidina por LMA durante la recaída molecular tras la primera remisión completa [CR1] (véase la sección «Propiedades farmacológicas»).
En los siete pacientes se notificó al menos un evento adverso relacionado con el tratamiento. Las reacciones adversas más frecuentes fueron neutropenia, náuseas, leucopenia, trombocitopenia, diarrea y aumento de alanina aminotransferasa (ALT). En dos pacientes ocurrió un evento relacionado con el tratamiento que condujo a la interrupción del medicamento (neutropenia febril, neutropenia).
Durante el estudio clínico en un número limitado de niños que recibieron azacitidina, no se identificaron nuevas señales de riesgo. El perfil general de seguridad fue comparable al de la población adulta.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la autorización del medicamento es de gran importancia. Permite una vigilancia continua de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar cualquier caso sospechoso de reacción adversa y la falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en: https://aisf.dec.gov.ua.
Período de validez.
3 años.
Después de la reconstitución
Cuando el medicamento Azacitidina Accord se reconstituye con agua para inyección no refrigerada, la estabilidad química y física de la solución reconstituida durante su uso se ha demostrado a 25 °C durante 60 minutos y a una temperatura de entre 2 °C y 8 °C durante 8 horas.
El período de validez de la solución reconstituida puede ampliarse si se reconstituye con agua para inyección refrigerada (entre 2 °C y 8 °C). Cuando el medicamento Azacitidina Accord se reconstituye con agua para inyección refrigerada (entre 2 °C y 8 °C), la estabilidad química y física de la solución reconstituida durante su uso se ha demostrado a una temperatura entre 2 °C y 8 °C durante 22 horas.
Desde el punto de vista microbiológico, el producto reconstituido debe usarse inmediatamente. Si no se usa inmediatamente, el usuario es responsable de la duración y condiciones de almacenamiento durante el período de uso, que no debe exceder de 8 horas a una temperatura entre 2 °C y 8 °C si se reconstituye con agua para inyección no refrigerada, y de 22 horas si se reconstituye con agua para inyección refrigerada (entre 2 °C y 8 °C).
Condiciones de conservación.
El medicamento no requiere condiciones especiales de conservación.
Mantener fuera del alcance de los niños.
Incompatibilidades.
No mezclar con otros medicamentos, excepto los indicados en la sección «Modo de administración y dosis».
Envase. 100 mg en un frasco, 1 frasco por caja.
Categoría de dispensación. Bajo receta médica.
Fabricante.
Accord Healthcare Polska Sp. z o.o. Sklad Importera/Accord Healthcare Polska Sp. z o.o. Magazyn Importera.
Dirección del fabricante y lugar de actividad.
ul. Lutomierska 50, Pabianice, 95-200, Polonia / ul. Lutomierska 50, Pabianice, 95-200, Poland.
Titular del registro. Accord Healthcare S.L.U.
Las consultas sobre calidad deficiente del medicamento, cuestiones relacionadas con la seguridad de su uso, uso inadecuado o reclamaciones se aceptan las 24 horas (24/7) al teléfono: +380993100335 o por correo electrónico: [email protected].
Dirección del titular del registro. World Trade Center, Moll de Barcelona, s/n, Edificio Este, 6ª planta, 08039 Barcelona, España / World Trade Center, Moll de Barcelona, s/n, Edifici Est 6a planta, 08039 Barcelona, Spain.