Amikacid
Ucrania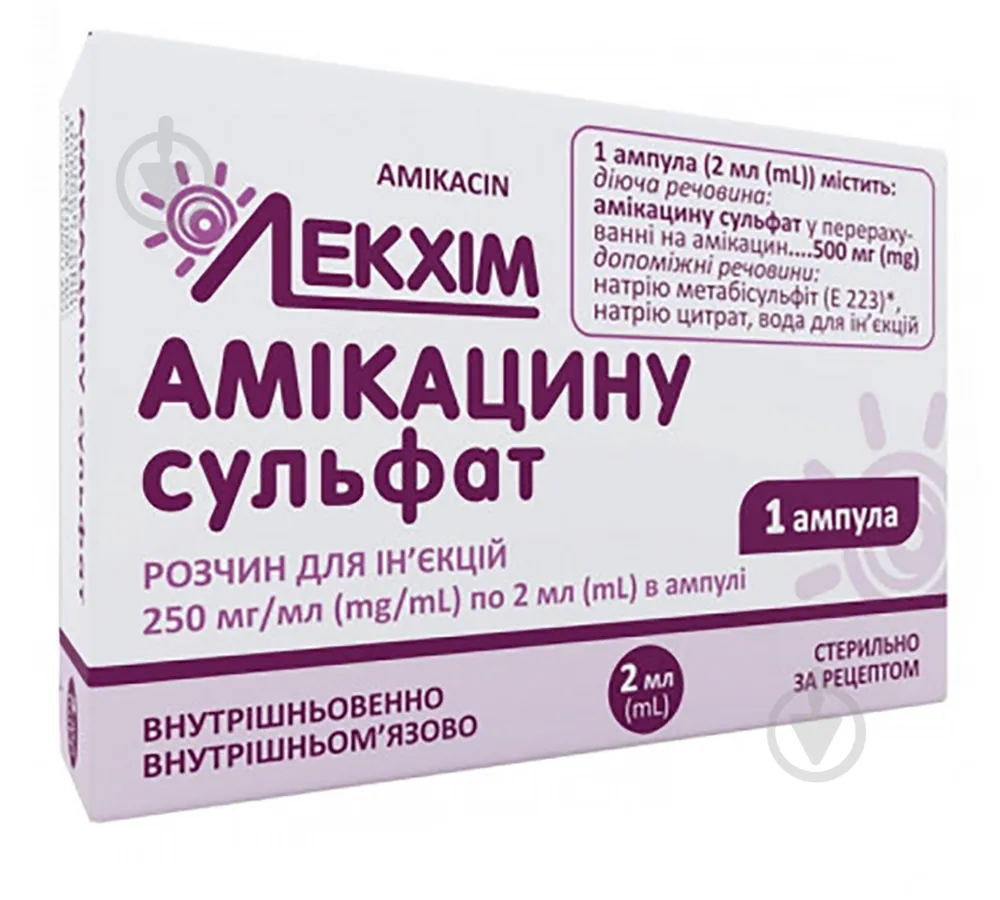
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO AMIKACID (AMIKACID)
Composición:
Principio activo: sulfato de amikacina;
1 ml de solución contiene sulfato de amikacina equivalente a 250 mg de amikacina;
Excipientes: citrato de sodio, metabisulfito de sodio (E 223), ácido sulfúrico, agua para inyección.
Forma farmacéutica. Solución inyectable.
Propiedades físico-químicas principales: solución transparente, incolora o ligeramente amarillenta.
Grupo farmacoterapéutico. Agentes antibacterianos de uso sistémico. Antibacterianos aminoglucósidos. Otros aminoglucósidos. Amikacina.
Código ATC J01G B06.
Propiedades farmacológicas.
Farmacodinámica.
La amikacina es un antibiótico semisintético del grupo de los aminoglucósidos de amplio espectro.
Mecanismo de acción
La amikacina ejerce un efecto bactericida. Al unirse a la subunidad 30S de los ribosomas de los microorganismos, interfiere la formación del complejo de ARN de transferencia y ARN mensajero, bloquea la síntesis de proteínas y además destruye las membranas citoplasmáticas de las bacterias.
Espectro de acción in vitro
La amikacina es eficaz in vitro frente a una serie de microorganismos patógenos gramnegativos y grampositivos. Concentraciones inhibitorias mínimas (CIM) (en µg/ml):
a) Microorganismos sensibles a la amikacina:
| Patógeno |
CMIn 75 % |
CMIn 100 % |
| Pseudomonas aeruginosa |
0,8 |
12,5 |
| Providencia rettgeri |
1,6 |
1,6 |
| Staphylococcus aureus |
1,6 |
3,1 |
| Klebsiella spp. |
1,6 |
6,2 |
| Enterobacter spp. |
1,6 |
6,2 |
| Providencia stuartii |
3,1 |
3,1 |
| Citrobacter spez. |
3,1 |
6,2 |
| Serratia spp. |
3,1 |
6,2 |
| Proteus vulgaris/morganii |
3,1 |
6,2 |
| Escherichia coli |
6,2 |
12,5 |
| Proteus mirabilis |
6,2 |
12,5 |
| Patógeno |
CIM 50 % |
CIM 90 % |
| Acinetobacter |
2,41 |
50,0 |
Amikacina no pierde actividad frente a enzimas que inactivan otras aminoglucósidos y puede permanecer activa frente a cepas de Proteus rettgeri, Serratia marcescens y Nocardia asteroides resistentes a kanamicina, gentamicina y tobramicina.
b) Microorganismos resistentes a la amikacina:
| Patógeno |
CIМ 75 % |
CIМ 100 % |
| Enterococcus |
25,0 |
50,0 |
| Neisseria gonorrhoeae |
25,0 |
50,0 |
Haemophilus influenzae, Streptococcus, microorganismos anaerobios – son insensibles a la amikacina.
La sensibilidad a la amikacina puede determinarse utilizando métodos estandarizados: el método de dilución en serie y el método de difusión con disco.
Los siguientes son los criterios recomendados de sensibilidad:
| Patógeno |
Diámetro (mm) de la zona de difusión en disco (disco con 30 µg de amikacina) |
(CMI [µg/ml]) |
| sensible |
≥ 17 |
≤ 16 |
| intermedio |
15-16 |
17-31 |
| resistente |
≤ 14 |
≥ 32 |
Farmacocinética.
La administración de amikacina puede realizarse bien en forma de dos o tres dosis diarias, bien como dosis única diaria. Tanto con la administración dos veces al día como con la dosis única se observa una correlación lineal progresiva entre los cambios en los parámetros cinéticos y la concentración en suero sanguíneo con respecto a la cantidad de amikacina administrada.
Absorción
Administración intramuscular: la amikacina se absorbe rápidamente tras la administración intramuscular.
Tras la inyección de 250 mg (3,7 mg/kg), 375 mg (5 mg/kg) y 500 mg (7,5 mg/kg), se determinan concentraciones en suero de 12 µg/ml, 16 µg/ml y 21 µg/ml respectivamente a la 1 hora. A las 10 horas, los niveles correspondientes en suero son de 0,3 µg/ml, 1,2 µg/ml y 2,1 µg/ml.
Administración intravenosa: cuando se administra a adultos una dosis única de 500 mg (7,5 mg/kg) mediante una infusión corta de 30 minutos, el nivel en sangre alcanza 38 µg/ml al final de la infusión, y 24 µg/ml, 18 µg/ml y 0,75 µg/ml a los 30 minutos, 1 hora y 10 horas, respectivamente, tras la administración.
Una infusión de 30 minutos con una dosis única de 1000 mg (15 mg/kg) produce un nivel en sangre de 77 µg/ml al final de la infusión, y de 47 µg/ml y 1 µg/ml a la 1 hora y a las 12 horas tras la infusión, respectivamente.
El área bajo la curva farmacocinética «concentración-tiempo» (AUC) tras la administración de 15 mg/kg de amikacina oscila entre 137 mg/l/h y 193 mg/l/h en pacientes adultos.
Distribución
El volumen de distribución en adultos es de 24 litros, lo que representa aproximadamente el 28 % del peso corporal. La unión a proteínas oscila entre el 0 % y el 11 %.
Tras la administración de la dosis recomendada, se alcanzan concentraciones terapéuticas en tejidos óseos, corazón, vesícula biliar y pulmones, así como concentraciones significativas en orina, bilis, esputo, secreción bronquial, intersticio, líquido pleural y líquido sinovial.
Como otros antibióticos aminoglucósidos, la amikacina generalmente alcanza concentraciones insuficientes y, sobre todo, impredecibles en el líquido cefalorraquídeo, incluso en presencia de inflamación meníngea.
En niños pequeños, el nivel en el líquido cefalorraquídeo representa aproximadamente el 10-20 % de la concentración en suero, y puede aumentar hasta el 50 % en caso de meningitis.
La amikacina atraviesa la barrera placentaria, y el nivel en suero fetal puede alcanzar entre el 16 % y el 50 % de la concentración sérica materna.
No se sabe si la amikacina se excreta en la leche materna. Sin embargo, se han detectado pequeñas cantidades de otros antibióticos aminoglucósidos en la leche materna.
Metabolismo
No se metaboliza.
Eliminación
Se elimina por los riñones mediante filtración glomerular.
Con función renal normal, aproximadamente el 92 % de la dosis intramuscular se excreta sin cambios en la orina durante las primeras 8 horas y aproximadamente el 98 % en 24 horas (Qo=0,02). El aclaramiento sérico es de 100 ml/min, y el aclaramiento renal es de 94 ml/min con función renal normal.
El período medio de eliminación de la amikacina en voluntarios sanos es de 2 horas. Incluso con una dosis única diaria de 15 mg/kg, el período de semivida no cambia, aunque puede aumentar hasta 3-4 horas en pacientes con patologías concomitantes y hasta 10 horas en pacientes de edad avanzada.
Farmacocinética en situaciones clínicas especiales
Pacientes con alteraciones de la función renal. En pacientes con insuficiencia renal o con velocidad de filtración glomerular reducida, la amikacina se elimina mucho más lentamente. Por tanto, debe controlarse cuidadosamente la función renal y, si es necesario, ajustarse la dosis.
No debe administrarse amikacina en forma de dosis única a pacientes con alteraciones de la función renal (aclaramiento de creatinina <50 ml/min): una dosis única de amikacina de 15 mg/kg en pacientes con función renal moderadamente o moderadamente reducida produce concentraciones plasmáticas de 82-95 µg/ml. La constante de eliminación, el período de semivida, el AUC, el aclaramiento total del plasma y la velocidad de acumulación cambian considerablemente en función de la función renal. Esto significa que en pacientes con aclaramiento de creatinina <50 ml/min, especialmente en aquellos con mayor deterioro de la función renal (aclaramiento de creatinina <25 ml/min), los cambios en los parámetros farmacocinéticos son muy pronunciados, por lo que la administración de amikacina conlleva un aumento del tiempo de permanencia de la sustancia en el organismo. Este problema no depende de la dosis, y si se reduce la dosis, se pierde el efecto beneficioso del alto nivel en sangre.
Pacientes de edad avanzada. En pacientes de edad avanzada (aclaramiento medio de creatinina de 64 ml/min), el nivel en sangre tras una infusión de 30 minutos de 15 mg/kg fue de 55 µg/ml, que disminuyó a 5,4 µg/ml a las 12 horas. El AUC tras la administración de 15 mg/kg de amikacina aumenta hasta 255 mg/l/h en pacientes de edad avanzada y posiblemente incluso más en pacientes muy ancianos.
Datos preclínicos
Existen estudios a corto plazo en animales sobre carcinogenicidad. No se han realizado estudios de mutagenicidad. La amikacina no afectó la fertilidad en animales tras su administración a ratas en una dosis 10 veces superior a la dosis diaria humana. Sin embargo, con el uso de aminoglucósidos se ha observado un efecto fetotóxico y/o teratogénico.
Características clínicas.
Indicaciones.
Infecciones causadas por cepas de microorganismos sensibles a la amikacina, resistentes a otros aminoglucósidos.
Debe seguirse las recomendaciones oficiales sobre el uso adecuado de los medicamentos antibacterianos.
Contraindicaciones.
- Insuficiencia renal;
- neuritis del nervio auditivo;
- hipersensibilidad a la amikacina o a cualquier otro antibiótico del grupo de los aminoglucósidos y sus derivados;
- hipersensibilidad a cualquiera de los excipientes del medicamento;
- miastenia grave;
- alteración de la función del aparato vestibular;
- azotemia (nitrógeno residual superior a 150 mg %);
- tratamiento previo con fármacos oto- o nefrotóxicos.
Interacción con otros medicamentos y otros tipos de interacciones.
Farmacéuticamente incompatible con penicilinas, heparina, cefalosporinas, capreomicina, anfotericina B, hidroclorotiazida, eritromicina, nitrofurantoína, vitaminas del grupo B y C, cloruro de potasio.
Debe evitarse la administración simultánea o sucesiva de amikacina con otros fármacos neurotóxicos, ototóxicos o nefrotóxicos, especialmente bacitracina, cisplatino, anfotericina B, ciclosporina, tacrolimus, cefaloridina, paromomicina, viomicina, polimixina B, colistina, vancomicina u otros aminoglucósidos, debido al potencial de efectos aditivos.
La amikacina muestra sinergismo en combinación con carbenicilina, penicilina bencilada y cefalosporinas (en pacientes con insuficiencia renal crónica grave, la administración concomitante con antibióticos beta-lactámicos puede provocar una disminución de la eficacia de los aminoglucósidos).
El riesgo de efecto nefrotóxico aumenta con la administración concomitante de amikacina junto con penicilina, cefalosporinas, sulfonamidas, vancomicina, metoxiflurano, enflurano, antiinflamatorios no esteroideos (AINE), agentes de contraste radiológico, ciclosporina, cisplatino, anfotericina B, cefalotina, polimixina y diuréticos (especialmente con furosemida).
Se han notificado casos de mayor nefrotóxicidad tras la administración parenteral simultánea de antibióticos aminoglucósidos y cefalosporinas. La administración concomitante de cefalosporinas puede provocar un aumento del nivel sérico de creatinina.
El riesgo de ototoxicidad aumenta cuando se administra amikacina junto con diuréticos de acción rápida (furosemida y ácido etacrínico), especialmente si el diurético se administra por vía intravenosa. Los diuréticos pueden aumentar la toxicidad de los aminoglucósidos al modificar la concentración del antibiótico en el plasma sanguíneo y en los tejidos. La administración concomitante con furosemida y ácido etacrínico, que también tienen efecto ototóxico, puede provocar sordera irreversible.
El ácido nalidíxico, la polimixina B, el cisplatino y la vancomicina aumentan el riesgo de ototoxicidad y nefrototoxicidad.
El indometacina, el fenilbutazona y otros AINE que alteran el flujo sanguíneo renal pueden ralentizar la velocidad de eliminación de la amikacina. Si se administra amikacina simultáneamente con indometacina por vía intravenosa en recién nacidos prematuros, se produce un aumento de la concentración plasmática del fármaco y un riesgo de toxicidad. La administración parenteral de indometacina aumenta el riesgo de efectos tóxicos de los aminoglucósidos (aumento de la semivida T1/2 y disminución del aclaramiento).
Existe un riesgo aumentado de hipocalcemia con la administración concomitante de aminoglucósidos y bifosfonatos.
Existe un riesgo aumentado de nefrototoxicidad y, posiblemente, de ototoxicidad, cuando los aminoglucósidos se administran junto con compuestos de platino.
El riesgo de depresión respiratoria aumenta con la administración concomitante de amikacina junto con éter etílico y bloqueadores de la transmisión neuromuscular.
La amikacina potencia el efecto miorrelajante de los fármacos tipo curare.
No se recomienda administrar amikacina a pacientes bajo los efectos de anestésicos, miorrelajantes o a pacientes que reciben transfusiones de grandes volúmenes de productos sanguíneos con conservantes citratos, debido al riesgo de bloqueo neuromuscular y de parálisis respiratoria.
La amikacina disminuye la eficacia de los medicamentos utilizados en el tratamiento de la miastenia.
La administración simultánea de tiamina (vitamina B1) con amikacina puede provocar la destrucción de la tiamina por el componente reactivo de sulfito presente en la amikacina.
Las combinaciones de antibióticos amikacina+ceftazidima y amikacina+cefoperazona muestran el mayor efecto aditivo y sinérgico frente a Pseudomonas aeruginosa.
En caso de tratamiento con múltiples antibióticos, no debe mezclarse la amikacina en la misma jeringa o frasco con otros medicamentos antibacterianos.
Características de aplicación.
Antes de la administración del medicamento, se debe determinar la sensibilidad de los microorganismos aislados.
No se debe utilizar amikacina en pacientes con hipersensibilidad a otros aminoglucósidos debido al riesgo de reacción alérgica cruzada.
Debe tenerse precaución al administrar el medicamento a pacientes con insuficiencia renal preexistente o con alteraciones auditivas o vestibulares previas. En caso de pruebas audiométricas insatisfactorias, la dosis del fármaco debe reducirse o interrumpirse el tratamiento.
Se recomienda que los pacientes con infecciones inflamatorias del tracto urinario consuman abundantes líquidos.
Los pacientes que reciben aminoglucósidos por vía parenteral deben estar bajo estricta vigilancia clínica debido al riesgo potencial de ototoxicidad y nefrotoxicidad asociado con su uso. No se ha establecido la seguridad del tratamiento prolongado (más de 14 días). Se deben observar precauciones adecuadas respecto a la dosificación y una hidratación suficiente.
Durante el tratamiento, se debe controlar semanalmente, al menos una vez por semana, la función renal, el nervio auditivo y el aparato vestibular.
Si se prevé que la terapia dure 7 días o más en pacientes con alteración de la función renal, o 10 días o más en pacientes sin alteración renal, se debe realizar una audiometría antes del inicio del tratamiento y repetirla durante la terapia. El tratamiento con amikacina debe interrumpirse ante quejas de acúfenos, sensación subjetiva de pérdida auditiva o si las audiometrías posteriores muestran una pérdida significativa de la respuesta en frecuencias altas.
Neuro-/ototoxicidad
El efecto tóxico principal del fármaco tras la administración parenteral es su acción sobre el VIII par craneal, que inicialmente se manifiesta como sordera en el rango de frecuencias altas.
La neurotoxicidad, que se presenta como afectación vestibular y/o ototoxicidad bilateral, puede ocurrir en pacientes que reciben aminoglucósidos. El riesgo de complicaciones ototóxicas es considerablemente mayor en pacientes con alteración de la función renal, así como en aquellos que reciben dosis elevadas del medicamento o terapia prolongada durante 5-7 días. La pérdida auditiva de alta frecuencia suele aparecer primero y solo puede detectarse mediante pruebas audiométricas. Puede presentarse vértigo, lo que indica daño vestibular.
Otros signos de neurotoxicidad pueden incluir entumecimiento, hormigueo cutáneo, fasciculaciones musculares y convulsiones. El riesgo de ototoxicidad por el uso de aminoglucósidos aumenta con niveles persistentemente altos de concentraciones séricas pico o elevadas. Los pacientes que desarrollan lesión vestibular o coclear pueden no presentar síntomas negativos iniciales que adviertan del daño tóxico del VIII par craneal. Pueden presentarse sordera bilateral completa o parcial irreversible o vértigo tras la finalización del tratamiento. La ototoxicidad inducida por aminoglucósidos suele ser irreversible.
El uso de amikacina en pacientes con antecedentes de reacción alérgica a aminoglucósidos o en aquellos con lesión subclínica renal o del VIII par craneal provocada por el uso previo de fármacos nefrotóxicos u ototóxicos, como estreptomicina, dihidroestreptomicina, gentamicina, tobramicina, kanamicina, becamicina, neomicina, polimixinas B, colistina, cefaloridina o viomicina, debe considerarse con precaución, ya que la toxicidad puede ser aditiva. El uso de amikacina en estos pacientes solo debe considerarse bajo prescripción médica cuando el beneficio esperado supere el riesgo potencial.
La administración concomitante de sulfato de amikacina y diuréticos de acción rápida, como derivados del ácido etacrínico, furosemida o manitol (especialmente si el diurético se administra por vía intravenosa), puede provocar sordera irreversible.
Los pacientes con mutación en el ADN mitocondrial (especialmente el reemplazo del nucleótido 1555 A por G en el gen del ARNr 12S) tienen un riesgo aumentado de ototoxicidad, incluso si los niveles séricos de aminoglucósidos durante el tratamiento se mantienen dentro del rango recomendado. Para estos pacientes, se debe considerar la posibilidad de tratamientos alternativos.
En pacientes con antecedentes familiares de mutaciones en el ADN mitocondrial o con sordera inducida por aminoglucósidos, se debe considerar la posibilidad de utilizar tratamientos alternativos o realizar pruebas genéticas antes de la administración del medicamento.
Bloqueo neuromuscular
Se han notificado casos de bloqueo neuromuscular y parálisis respiratoria tras la administración parenteral, instilación local (como en irrigaciones ortopédicas o abdominales o en el tratamiento local de empiema) e incluso tras la administración oral de aminoglucósidos. Debe considerarse la posibilidad de parálisis respiratoria cuando los aminoglucósidos se administran por cualquier vía, especialmente en pacientes que reciben anestésicos, bloqueadores neuromusculares o transfusiones masivas de sangre anticoagulada con citrato. En caso de presentarse bloqueo neuromuscular, las sales de calcio pueden revertir este efecto, aunque puede ser necesaria ventilación artificial. El bloqueo neuromuscular y la parálisis muscular se han demostrado en animales de laboratorio que recibieron dosis altas de amikacina.
No se debe administrar amikacina a pacientes con miastenia grave. El medicamento debe usarse con precaución en pacientes con miastenia, enfermedad de Parkinson, botulismo (los aminoglucósidos pueden alterar la transmisión neuromuscular, provocando un mayor debilitamiento de la musculatura esquelética), deshidratación, insuficiencia renal, recién nacidos (especialmente prematuros) y pacientes de edad avanzada.
Nefrotoxicidad
Los aminoglucósidos son potencialmente nefrotóxicos. La probabilidad de desarrollar nefrotoxicidad es mayor en pacientes con alteración de la función renal, así como con el uso del fármaco en dosis altas o durante períodos prolongados (estos pacientes requieren control diario de la función renal).
Antes de iniciar el tratamiento, se debe corregir el equilibrio hidroelectrolítico en los pacientes. Durante el tratamiento con sulfato de amikacina, los pacientes deben ingerir una cantidad adecuada de líquidos, determinar frecuentemente la concentración de creatinina en plasma y, si es necesario, ajustar el esquema de dosificación. En pacientes con alteración de la función renal, la dosis diaria debe reducirse y/o prolongarse el intervalo entre dosis según la concentración sérica de creatinina, para prevenir la acumulación del fármaco en sangre y minimizar el riesgo de ototoxicidad. Se debe evaluar regularmente la actividad funcional renal. Es necesario realizar análisis de orina antes y durante el tratamiento.
La administración de amikacina puede alterar los siguientes parámetros de laboratorio: alaninaminotransferasa sérica, aspartato aminotransferasa, bilirrubina, lactato deshidrogenasa, fosfatasa alcalina, nitrógeno ureico en sangre, creatinina, iones de calcio, magnesio, potasio, sodio.
Si aparecen signos de irritación renal (presencia de cilindros urinarios, albuminuria, microhematuria, leucocituria, disminución del aclaramiento de creatinina, disminución de la densidad específica de la orina, aumento del nitrógeno ureico en sangre, creatinina sérica u oliguria), se debe aumentar la hidratación y reducir la dosis. Estos síntomas suelen desaparecer tras finalizar el tratamiento. Es especialmente importante el monitoreo de la función renal en pacientes de edad avanzada durante el tratamiento con aminoglucósidos. En pacientes ancianos, se debe reducir la dosis de amikacina debido a la disminución de la función renal y a la posible reducción de la masa corporal, que puede no detectarse con pruebas de cribado convencionales, como el nitrógeno ureico en sangre o la creatinina sérica.
Si aparecen signos de ototoxicidad (por ejemplo, vértigo, zumbidos o acúfenos en los oídos o pérdida auditiva) o nefrotoxicidad (por ejemplo, disminución del aclaramiento de creatinina, oliguria), se debe interrumpir o reducir la dosis de amikacina. Si aparecen signos de azotemia o empeoramiento de la oliguria, el tratamiento debe suspenderse.
Debe evitarse la administración simultánea y/o secuencial de otros medicamentos sistémicos, orales o tópicos que sean neurotóxicos o nefrotóxicos, especialmente bacitracina, cisplatino, anfotericina B, cefaloridina, paromomicina, viomicina, polimixina B, colistina, vancomicina u otros aminoglucósidos. Otros factores que pueden aumentar el riesgo de toxicidad incluyen la edad avanzada y la deshidratación.
Otro
No se debe administrar simultáneamente dos aminoglucósidos ni sustituir un fármaco por otro si el primer aminoglucósido se ha utilizado durante 7-10 días. Un nuevo curso de tratamiento no debe iniciarse antes de 4-6 semanas.
En ausencia de mejoría clínica, debe considerarse la posibilidad de resistencia microbiana. En tales casos, se debe suspender el tratamiento y comenzar una terapia adecuada.
Los aminoglucósidos se absorben rápida y casi completamente tras la aplicación local, excepto en la vejiga urinaria, debido a procedimientos quirúrgicos. Se han notificado casos de sordera irreversible, insuficiencia renal y muerte por bloqueo neuromuscular tras la aplicación del fármaco aminoglucósido en áreas quirúrgicas, tanto pequeñas como grandes.
Los aminoglucósidos deben usarse con precaución en recién nacidos prematuros y a término debido a la inmadurez renal de estos pacientes y, por consiguiente, al prolongado período de semivida de eliminación de estos fármacos en el suero.
Se han notificado casos de infarto macular, que puede provocar pérdida total de la visión, tras la administración intravítrea (inyección en el ojo) de amikacina.
El medicamento contiene metabisulfito sódico, que puede provocar reacciones de tipo alérgico, incluyendo síntomas de anafilaxia y ataques de asma de diversa gravedad, especialmente en pacientes con asma bronquial.
Al administrarse simultáneamente con cefalosporinas, se recomienda inyectar ambos fármacos en sitios diferentes (en caso de administración intramuscular) con un intervalo de al menos 1 hora.
2 ml del medicamento contienen menos de 1 mmol (23 mg) de sodio, es decir, es prácticamente libre de sodio.
4 ml del medicamento contienen menos de 1,3 mmol (29,86 mg) de sodio, por lo que debe tenerse precaución al administrar el fármaco a pacientes que siguen una dieta controlada en sodio.
Uso durante el embarazo o la lactancia.
Debido a que la amikacina atraviesa la placenta y puede ejercer efectos ototóxicos y nefrotóxicos sobre el feto, el medicamento está contraindicado durante el embarazo.
Dado que la amikacina penetra en la leche materna en bajas concentraciones y puede afectar la microflora intestinal del lactante, durante el tratamiento con amikacina debe suspenderse la lactancia.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
El medicamento generalmente no afecta la velocidad de reacción, pero debe considerarse la posibilidad de efectos adversos del sistema nervioso central y de los órganos sensoriales, como somnolencia, alteración de la transmisión neuromuscular, descoordinación motora y vértigo.
Vía de administración y dosis.
Amikacina debe administrarse por vía intramuscular o intravenosa. La dosis habitual para niños a partir de 12 años y adultos es de 5 mg/kg de peso corporal cada 8 horas, o 7,5 mg/kg de peso corporal cada 12 horas. La dosis máxima para adultos es de 15 mg/kg de peso corporal al día. En casos graves y en infecciones provocadas por Pseudomonas, la dosis diaria debe dividirse en 3 administraciones. La dosis diaria máxima es de 1,5 g. La dosis total del tratamiento no debe superar los 15 g. La duración del tratamiento suele ser de 3 a 7 días con administración intravenosa y de 7 a 10 días con administración intramuscular.
En recién nacidos prematuros, la dosis inicial recomendada es de 10 mg/kg de peso corporal, seguida de 7,5 mg/kg de peso corporal cada 18-24 horas durante 7-10 días. En recién nacidos a término y niños menores de 12 años, la dosis inicial es de 10 mg/kg de peso corporal, seguida de 7,5 mg/kg de peso corporal cada 12 horas durante 7-10 días.
En pacientes con alteraciones de la función renal, la dosis diaria debe reducirse y/o aumentarse los intervalos entre administraciones. La dosis debe ajustarse según los niveles de creatinina en plasma y el peso corporal del paciente. El intervalo entre administraciones del antibiótico se calcula multiplicando el valor del nivel de creatinina en plasma por 9; por ejemplo: si el nivel de creatinina es de 2 mg, el medicamento debe administrarse cada 18 horas.
La administración intravenosa de amikacina en adultos y niños debe realizarse mediante infusión, utilizando un volumen de líquido suficiente para una perfusión gota a gota durante 60-90 minutos (a una velocidad de 50 gotas por minuto), y en recién nacidos durante 1-2 horas. La concentración del solución de amikacina por vía intravenosa no debe exceder los 5 mg/ml. La inyección intravenosa de amikacina debe realizarse muy lentamente (durante 2-7 minutos). La solución para administración parenteral debe prepararse inmediatamente antes de su uso y emplearse inmediatamente después de su preparación.
Para la administración intravenosa pueden utilizarse los siguientes vehículos: solución isotónica de cloruro de sodio al 0,9 %, solución de glucosa al 5 %, solución de Ringer con lactato para inyección que contiene glucosa (5 %).
Niños.
Se recomienda utilizar este medicamento con precaución en el tratamiento de recién nacidos prematuros y a término, ya que debido al desarrollo incompleto del sistema excretor, la eliminación de los aminoglucósidos puede prolongarse, provocando efectos de toxicidad.
Sobredosificación.
Pueden aparecer efectos ototóxicos y nefrotóxicos del medicamento, así como signos de bloqueo neuromuscular: zumbidos en los oídos, pérdida auditiva, trastornos auditivos, erupciones cutáneas, cefalea, mareo, fiebre, escalofríos, parestesias, disminución de la función renal (hasta insuficiencia renal), depresión o parálisis respiratoria, y reacciones tóxicas (ataxia, alteraciones urinarias, sed, pérdida de apetito, náuseas, vómitos).
Tratamiento: para revertir el bloqueo de la transmisión neuromuscular y sus consecuencias: hemodiálisis o diálisis peritoneal; agentes anticolinesterásicos, sales de calcio, ventilación mecánica, y otro tratamiento sintomático y de soporte. El nivel de amikacina debe reducirse mediante hemofiltración arteriovenosa continua.
Reacciones adversas.
La lista de reacciones adversas se presenta por clases de órganos y sistemas, según el término preferente de MedDRA y la frecuencia, utilizando las siguientes categorías: muy frecuentes (≥1/10), frecuentes (≥1/100, <1/10), poco frecuentes (≥1/1000, <1/100), raras (≥1/10000, <1/1000), muy raras (<1/10000) y frecuencia desconocida (no puede determinarse la frecuencia a partir de los datos disponibles).
Infecciones e infestaciones: poco frecuentes – superinfecciones o colonización por bacterias resistentes o levaduras.
Trastornos del sistema hematopoyético: raros – eosinofilia, anemia, leucopenia, granulocitopenia, trombocitopenia.
Trastornos del aparato gastrointestinal: raros – náuseas, vómitos, diarrea.
Trastornos del sistema endocrino y del metabolismo: poco frecuentes – hipomagnesemia.
Trastornos de la piel y del tejido subcutáneo: erupciones cutáneas, prurito, urticaria, hiperemia cutánea, edema de angioedema de Quincke.
Trastornos del sistema inmunitario: frecuencia desconocida – shock anafiláctico, reacciones de hipersensibilidad, reacciones anafilactoides, reacciones anafilácticas.
Trastornos del sistema cardiovascular: raros – hipotensión, taquicardia, miocarditis.
Trastornos del sistema nervioso: cefalea, somnolencia, efecto neurotóxico (fasciculaciones musculares, sensación de entumecimiento, hormigueo, crisis epilépticas), posible disminución de la audición, trastornos vestibulares, mareo, parestesias, temblor, convulsiones, encefalopatía; en casos aislados – alteración de la conducción neuromuscular, posible desarrollo de bloqueo neuromuscular (parálisis muscular, depresión respiratoria).
Trastornos de los órganos de los sentidos: ototoxicidad (hipoacusia, pérdida de audición, zumbidos en los oídos, trastornos vestibulares laberínticos, sordera parcialmente reversible o irreversible), efecto tóxico sobre el aparato vestibular (descoordinación de movimientos, mareo, náuseas, vómitos).
Trastornos oculares: ceguera, infarto de retina*.
Trastornos del sistema hepatobiliar: alteración de la función hepática (aumento de la actividad de las transaminasas hepáticas, hiperbilirrubinemia), hepatomegalia, necrosis hepática.
Trastornos renales y urinarios: alteración de la función renal (oliguria, proteinuria, microhematuria, albuminuria, cilindruria, hiperazotemia, hematuria, aumento de los niveles de creatinina); raros – necrosis aguda, nefritis intersticial, insuficiencia renal aguda, nefropatía tóxica, azotemia, eritrocitos modificados en la orina, presencia de leucocitos en la orina.
Trastornos del aparato respiratorio y del mediastino: apnea, broncoespasmo.
Trastornos del sistema musculoesquelético y del tejido conectivo: artralgia, calambres musculares.
Trastornos generales y en el sitio de administración: frecuencia desconocida – dolor en el sitio de inyección y tromboflebitis, esplenomegalia; raros – hipertermia.
*La amikacina no está indicada para su uso intravítreo. Se han descrito casos de ceguera e infarto de retina tras la administración intravítrea (inyección en el ojo) de amikacina.
El metabisulfito sódico, componente del medicamento, puede provocar reacciones de hipersensibilidad graves y broncoespasmo.
Todos los aminoglucósidos pueden causar ototoxicidad, nefrotoxicidad y bloqueo neuromuscular. Estos efectos tóxicos son más frecuentes en pacientes con insuficiencia renal, en aquellos que reciben otros medicamentos ototóxicos o nefrotóxicos, y en pacientes que reciben tratamiento durante períodos más prolongados y/o con dosis superiores a las recomendadas.
La alteración de la función renal suele ser reversible tras la interrupción del tratamiento.
El efecto tóxico sobre el VIII par craneal puede provocar pérdida auditiva y alteración de la coordinación. La amikacina afecta principalmente la función auditiva. El daño coclear incluye sordera de alta frecuencia, que puede detectarse mediante pruebas audiométricas y que precede a las manifestaciones clínicas de pérdida auditiva (ver sección «Precauciones de uso»).
Notificación de reacciones adversas sospechadas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite continuar con el seguimiento de la relación beneficio-riesgo del medicamento. Los profesionales de la salud deben informar sobre cualquier reacción adversa sospechosa.
Período de validez. 2 años.
Condiciones de conservación. Conservar en el envase original a una temperatura no superior a 25 °C. No congelar. Mantener fuera del alcance de los niños.
Incompatibilidades.
La solución de sulfato de amikacina no debe mezclarse directamente con otros aminoglucósidos, penicilinas, heparina, cefalosporinas, capreomicina, anfotericina B, tiopental, hidroclorotiazida, eritromicina, nitrofurantoína, vitaminas del grupo B y C, ni cloruro potásico. Si se requiere la administración de dos medicamentos, deben administrarse por separado y de forma secuencial. En caso de uso combinado de varios antibióticos, la amikacina no debe mezclarse en la misma jeringa o frasco con otros agentes antibacterianos.
Envase.
2 ml o 4 ml en frascos; 1 frasco por caja de cartón.
2 ml o 4 ml en frascos; 5 frascos en envase blíster; 2 envases blíster por caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. Sociedad con responsabilidad limitada «Novofarm-Biosintez».
Dirección del fabricante y lugar de actividad.
Ucrania, 11700, región de Zhytomyr, distrito de Zvyagel, ciudad de Zvyagel, calle Zhytomyrska, 38.