Haemate P
Italia
Contenido
- Folleto informativo: información para el usuario
- HAEMATE P 500 UI / 1200 UI
- 1. Qué es HAEMATE P y para qué se utiliza
- 2. Qué debe saber antes de usar HAEMATE P
- 3. Cómo utilizar HAEMATE P
- 4. Posibles efectos adversos
- La siguiente información está destinada exclusivamente a médicos o profesionales sanitarios
Folleto informativo: información para el usuario
HAEMATE P 500 UI / 1200 UI
polvo y disolvente para disolución inyectable o para perfusión
HAEMATE P 1000 UI / 2400 UI
polvo y disolvente para disolución inyectable o para perfusión
Factor VIII de la coagulación humana
Factor de von Willebrand humano
Lea todo el prospecto detenidamente antes de empezar a usar este medicamento, porque contiene información importante para usted.
- Conserve este prospecto. Puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico, farmacéutico o enfermero.
- Este medicamento se le ha recetado exclusivamente a usted. No se lo dé a otras personas, aunque sus síntomas sean iguales a los suyos, ya que podría resultar peligroso.
- Si experimenta algún efecto adverso, incluidos los no mencionados en este prospecto, informe a su médico, farmacéutico o enfermero. Véase la sección 4.
Contenido de este prospecto:
- Qué es HAEMATE P y para qué se utiliza
- Qué debe saber antes de recibir HAEMATE P
- Cómo usar HAEMATE P
- Posibles efectos adversos
- Cómo conservar HAEMATE P
- Contenido del envase y demás información
1. Qué es HAEMATE P y para qué se utiliza
HAEMATE P pertenece a un grupo de medicamentos denominados hemostáticos o antihemorrágicos, utilizados para detener los sangrados. Este medicamento contiene como principios activos el factor VIII de la coagulación humana y el factor de von Willebrand humano, obtenidos a partir de la fracción líquida de la sangre (plasma). Estos dos factores participan en el proceso de coagulación sanguínea.
HAEMATE P está indicado para la prevención y el tratamiento de hemorragias debidas a:
- déficit congénito del factor VIII en sangre (hemofilia A), carencia adquirida de este factor o producción de sustancias que interfieren con la acción del factor VIII (anticuerpos anti-factor VIII o inhibidores);
- niveles reducidos del factor de von Willebrand en sangre (enfermedad de von Willebrand), en el contexto de intervenciones quirúrgicas cuando el tratamiento exclusivo con desmopresina no es eficaz o está contraindicado.
Haemate P contiene tanto FVIII como VWF. Si padece hemofilia A, su médico le recetará Haemate P indicando el número de unidades de FVIII que necesita. Si padece enfermedad de von Willebrand, su médico le recetará Haemate P indicando el número de unidades de VWF que necesita.
2. Qué debe saber antes de usar HAEMATE P
No utilice HAEMATE P
- si es alérgico al factor VIII de la coagulación humana, al factor von Willebrand humano o a alguno de los demás componentes de este medicamento (indicados en la sección 6).
Advertencias y precauciones
Trazaibilidad
Se recomienda encarecidamente que, cada vez que se administre Haemate P, se registren el nombre y el número de lote del medicamento, con el fin de mantener un seguimiento del lote utilizado.
Consulte a su médico, farmacéutico o enfermero antes de usar HAEMATE P.
La formación de inhibidores (anticuerpos) es una complicación conocida que puede ocurrir durante el tratamiento con todos los medicamentos que contienen factor VIII. Los inhibidores, especialmente a niveles elevados, impiden que el tratamiento actúe correctamente, y usted o su hijo serán sometidos a un control estrecho para detectar la aparición de estos inhibidores. Si HAEMATE P no controla la hemorragia suya o de su hijo, informe inmediatamente a su médico.
Además, durante el tratamiento con HAEMATE P, su médico realizará controles periódicos para verificar si su organismo ha producido sustancias que interfieran con la acción del factor von Willebrand (anticuerpos neutralizantes o inhibidores), especialmente si padece una forma grave de deficiencia del factor von Willebrand (enfermedad de von Willebrand tipo 3).
En ambos casos, si presenta niveles elevados de estas sustancias inhibitoras, el tratamiento con HAEMATE P podría no ser eficaz y se requerirían terapias alternativas.
Si padece deficiencia del factor von Willebrand (enfermedad de von Willebrand), este medicamento puede provocar la formación de coágulos sanguíneos en las venas (episodios trombóticos, embolia pulmonar), especialmente si:
- debe someterse o ha sido sometido recientemente a una intervención quirúrgica (período perioperatorio, especialmente en ausencia de profilaxis tromboembólica);
- se ha levantado demasiado pronto tras permanecer inmovilizado en cama (mobilización precoz);
- es obeso;
- se le han administrado dosis elevadas de este medicamento;
- padece cáncer;
- presenta un aumento excesivo de los niveles de factor VIII en sangre, especialmente si utiliza este medicamento durante mucho tiempo. Su médico le mantendrá bajo control estrecho para detectar oportunamente los primeros signos de formación de coágulos sanguíneos en las venas (ver la sección “Posibles efectos adversos”).
Como con otros medicamentos obtenidos del plasma sanguíneo humano (de origen plasmático) administrados por inyección en vena (vía endovenosa), pueden producirse reacciones alérgicas (hipersensibilidad) que se manifiestan con los siguientes síntomas:
- irritación de la piel (urticaria), que puede extenderse a todo el cuerpo (urticaria generalizada);
- sensación de opresión en el pecho (torácica);
- dificultad para respirar (disnea);
- disminución de la presión arterial (hipotensión);
- reacción alérgica grave (anafilaxia);
- shock. Si presenta estos síntomas, interrumpa inmediatamente el tratamiento y consulte a su médico, quien adoptará una terapia adecuada.
Si sabe que padece enfermedades cardíacas o que tiene riesgo de padecerlas, informe a su médico o farmacéutico.
Si es portador de un dispositivo de acceso venoso central (DAVC), su médico deberá considerar el riesgo de complicaciones relacionadas con el DAVC, entre las cuales: infecciones locales, bacterias en sangre (bacteriemia) y formación de coágulos en el vaso sanguíneo (trombosis) donde se ha insertado el catéter.
Seguridad vírica
Para los medicamentos derivados de sangre o plasma humano se adoptan ciertas medidas de seguridad para prevenir la transmisión de infecciones al paciente. Estas medidas incluyen:
- la selección cuidadosa de los donantes;
- el análisis de cada donación y del pool de plasma (conjunto de varias donaciones) para detectar la posible presencia de virus/infecciones;
- la inclusión de procesos de fabricación capaces de inactivar o eliminar virus.
A pesar de estas medidas, al administrar medicamentos derivados de sangre o plasma humano, el riesgo de transmisión de infecciones no puede excluirse por completo.
Esto también se aplica a virus u otros agentes infecciosos emergentes o desconocidos.
Las medidas adoptadas se consideran eficaces frente a virus con envoltura lipídica, como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y el virus de la hepatitis C (VHC), y frente a algunos virus sin envoltura, como el virus de la hepatitis A (VHA). Sin embargo, estas medidas tienen una eficacia limitada frente al parvovirus B19, que puede causar infecciones graves, especialmente si está embarazada, si tiene problemas en el sistema inmunitario (pacientes con inmunodeficiencia) o si padece algún tipo de anemia (por ejemplo, anemia falciforme o anemia hemolítica).
Si se le administran regular o repetidamente medicamentos derivados del plasma sanguíneo, su médico puede recomendarle una vacunación adecuada contra la hepatitis A y B.
Niños y adolescentes
Las advertencias y precauciones indicadas son aplicables tanto a adultos como a niños.
Otros medicamentos y HAEMATE P
Informe a su médico, farmacéutico o enfermero si está utilizando, ha utilizado recientemente o podría utilizar cualquier otro medicamento.
No se conocen interacciones entre HAEMATE P y otros medicamentos.
Embarazo, lactancia y fertilidad
Si está embarazada, cree que podría estarlo, planea quedarse embarazada o está en periodo de lactancia, consulte a su médico, farmacéutico o enfermero antes de usar este medicamento.
No se han realizado estudios de reproducción en animales con HAEMATE P.
Debido a la rareza de la hemofilia A en la mujer, no existen experiencias disponibles sobre el uso del factor VIII durante el embarazo y la lactancia.
No existen estudios clínicos sobre el tratamiento sustitutivo con VWF durante el embarazo o la lactancia.
Por tanto, el FVIII y el VWF deben utilizarse durante el embarazo y la lactancia solo en casos de absoluta necesidad y bajo el control directo del médico.
Conducción y uso de máquinas
No se han observado efectos sobre la capacidad para conducir ni para utilizar maquinaria.
HAEMATE P contiene sodio
Haemate P 500 U.I. / 1200 U.I. contiene 35 mg de sodio (componente principal de la sal de cocina) por vial, equivalente al 1,8 % de la ingesta diaria máxima recomendada de sodio para un adulto.
Haemate P 1000 U.I. / 2400 U.I. contiene 70 mg de sodio (componente principal de la sal de cocina) por vial, equivalente al 3,5 % de la ingesta diaria máxima recomendada de sodio para un adulto.
3. Cómo utilizar HAEMATE P
Utilice este medicamento siguiendo siempre exactamente las instrucciones de su médico o farmacéutico. Si tiene dudas, consulte al médico, al farmacéutico o al enfermero.
El tratamiento con este medicamento debe realizarse bajo estricta supervisión médica por un especialista en el tratamiento de la hemofilia.
El médico determinará la dosis adecuada para usted y la duración del tratamiento en función de sus niveles sanguíneos de factor VIII y de factor von Willebrand, del lugar y la gravedad del sangrado, así como de su estado de salud general.
Después de la administración del medicamento, el médico lo mantendrá bajo observación para controlar la posible aparición de reacciones alérgicas (ver el apartado "Advertencias y precauciones").
Si utiliza este medicamento durante mucho tiempo, puede producirse un aumento excesivo de los niveles de factor VIII (ver el apartado "Advertencias y precauciones"). Por lo tanto, tras 24-48 horas de tratamiento, el médico evaluará la necesidad de reducir la dosis o de aumentar el intervalo entre las administraciones.
Uso en niños
Para el tratamiento de la hemofilia A, no existen datos disponibles sobre el uso de este medicamento en niños.
Para el tratamiento de la enfermedad de von Willebrand, la dosis será determinada por el médico en función del peso corporal, siguiendo las mismas indicaciones que para adultos.
Si utiliza más HAEMATE P del que debe
No se conocen las consecuencias de una sobredosis con este producto.
Si cree que se le ha administrado una cantidad excesiva de este medicamento, informe inmediatamente a su médico o acuda al centro hospitalario más cercano.
Sin embargo, si ha tomado más HAEMATE P del debido, informe inmediatamente a su médico o al farmacéutico.
Debe saber que si se le administran dosis elevadas de este medicamento, puede producirse la formación de coágulos sanguíneos en las venas (riesgo trombótico).
4. Posibles efectos adversos
Como todos los medicamentos, este medicamento puede causar efectos adversos, aunque no todas las personas los
presentan.
En niños no tratados previamente con medicamentos que contienen factor VIII, la formación de anticuerpos inhibidores
(ver sección 2) puede ser muy frecuente (más de 1 paciente de cada 10); sin embargo, en pacientes que han
recibido tratamiento previo con factor VIII (más de 150 días de tratamiento), el riesgo es poco frecuente
(menos de 1 paciente de cada 100). Si esto ocurre, el medicamento podría dejar de actuar correctamente y usted o su hijo podrían presentar una hemorragia persistente. Si esto sucede, debe contactar inmediatamente con su médico.
Asimismo, pueden presentarse los siguientes efectos adversos:
Muy raros (pueden afectar hasta 1 de cada 10.000 personas):
- producción de sustancias que interfieren con la acción del factor de von Willebrand, especialmente si padece una forma grave de enfermedad de von Willebrand (tipo 3). En tal caso, debe ser estrechamente vigilado para detectar la formación de estos inhibidores;
- fiebre;
- reacciones alérgicas (hipersensibilidad), que incluyen:
- hinchazón del rostro, labios, boca, lengua o garganta debido a acumulación de líquidos, que puede provocar dificultad para tragar y respirar (angioedema);
- sensación de ardor o picazón en el lugar de la inyección;
- escalofríos;
- aumento del flujo sanguíneo en la cara y el cuello con sensación de calor (rubor);
- irritación de la piel (urticaria), incluso generalizada (urticaria generalizada);
- dolor de cabeza (cefalea);
- descenso de la presión arterial (hipotensión);
- letargo, sensación de cansancio;
- náuseas;
- aumento de la frecuencia cardíaca (taquicardia);
- sensación de opresión en el pecho (torácica);
- hormigueo;
- vómitos;
- dificultad para respirar (disnea);
- reacción alérgica grave (anafilaxia grave), incluido shock en casos aislados;
- formación de coágulos en la sangre (trombosis), en venas (trombosis venosa profunda) o en órganos como los pulmones (embolia pulmonar), especialmente si tiene factores de riesgo conocidos o si padece enfermedad de von Willebrand y recibe productos que contienen el complejo de FVIII con VWF, con concentraciones plasmáticas elevadas de FVIII:C (ver la sección “Advertencias y precauciones”).
Frecuencia no conocida (cuya frecuencia no puede determinarse a partir de los datos disponibles):
- aumento del volumen sanguíneo circulante (hipervolemia). En tal caso, su médico lo vigilará estrechamente para detectar los primeros síntomas de este aumento, especialmente si recibe dosis altas o repetidas con frecuencia, si su organismo ha producido sustancias que neutralizan el efecto del medicamento (los llamados inhibidores) y si va a ser sometido o ya ha sido sometido a una intervención quirúrgica;
- destrucción de glóbulos rojos (hemólisis), especialmente si su grupo sanguíneo es A, B o AB. En tal caso, su médico controlará sus análisis de sangre;
- transmisión de virus y otros microorganismos que pueden causar enfermedades (patógenos) (ver la sección “Advertencias y precauciones”).
Comunicación de los efectos adversos
Si experimenta algún efecto adverso, incluidos aquellos no mencionados en este prospecto, consulte a su
médico o farmacéutico.
Asimismo, puede comunicar directamente los efectos adversos a través del sistema nacional de notificación en la dirección: www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Al comunicar los efectos adversos, usted puede contribuir a proporcionar más información sobre la seguridad de este
medicamento.
5. Cómo conservar HAEMATE P
Mantenga este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el envase tras “Cad.”. La fecha de
caducidad se refiere al último día de ese mes.
Conservar a una temperatura no superior a 25°C y en su envase original para proteger el medicamento de la
luz.
No congele. Si la solución reconstituida no se administra inmediatamente, debe utilizarse dentro de las 3 horas.
No tire medicamentos por los desagües ni a la basura doméstica. Consulte con su farmacéutico cómo eliminar los
medicamentos que ya no utiliza. Esto ayudará a proteger el medio ambiente.
6. Contenido del envase y otra información
Qué contiene HAEMATE P
HAEMATE P 500 UI / 1200 UI polvo y disolvente para solución inyectable o para perfusión
- Los principios activos son factor VIII (FVIII:C) de plasma humano y factor de von Willebrand (VWF:RCo).
- Un frasco de polvo contiene 500 UI de factor VIII y 1200 UI de factor de von Willebrand. Tras la reconstitución con 10 ml de agua para preparaciones inyectables, HAEMATE P 500 contiene: o aproximadamente 50 UI por ml (500 UI/10 ml) de factor VIII; o aproximadamente 120 UI por ml (1200 UI/10 ml) de factor de von Willebrand.
- Los demás componentes son: o frasco de polvo: albúmina humana, ácido aminoacético, cloruro sódico, citrato sódico, hidróxido sódico o ácido clorhídrico (en pequeñas cantidades para la corrección del pH); o frasco de disolvente: agua para preparaciones inyectables.
HAEMATE P 1000 UI / 2400 UI polvo y disolvente para solución inyectable o para perfusión
- Los principios activos son factor VIII (FVIII:C) de plasma humano y factor de von Willebrand (VWF:RCo). Un frasco de polvo contiene 1000 UI de factor VIII y 2400 UI de factor de von Willebrand. Tras la reconstitución con 15 ml de agua para preparaciones inyectables, HAEMATE P 1000 contiene: o aproximadamente 66,6 UI por ml (1000 UI/15 ml) de factor VIII; o aproximadamente 160 UI por ml (2400 UI/15 ml) de factor de von Willebrand.
- Los demás componentes son: o frasco de polvo: albúmina humana, ácido aminoacético, cloruro sódico, citrato sódico, hidróxido sódico o ácido clorhídrico (en pequeñas cantidades para la corrección del pH); o frasco de disolvente: agua para preparaciones inyectables.
Descripción del aspecto de HAEMATE P y contenido del envase
Haemate P se presenta como un polvo de color blanco o amarillo pálido o como un sólido frágil, y se suministra con un frasco de agua para preparaciones inyectables como disolvente.
Tras la filtración o extracción, la solución del producto reconstituido debe ser transparente o ligeramente opalescente, y no debe contener partículas visibles ni cambios de color.
HAEMATE P 500 UI / 1200 UI polvo y disolvente para solución inyectable o para perfusión
Caja que contiene: 1 frasco de polvo, 1 frasco de agua para preparaciones inyectables de 10 ml, 1 sistema de transferencia con filtro 20/20 - Mix2Vial, conjunto de administración (envase interior): 1 jeringa desechable de 10 ml sin aguja, 1 conjunto para perfusión, 2 torundas impregnadas de alcohol, 1 vendaje no estéril.
HAEMATE P 1000 UI / 2400 UI polvo y disolvente para solución inyectable o para perfusión
Caja que contiene: 1 frasco de polvo, 1 frasco de agua para preparaciones inyectables de 15 ml, 1 sistema de transferencia con filtro 20/20 - Mix2Vial, conjunto de administración (envase interior): 1 jeringa desechable de 20 ml sin aguja, 1 conjunto para perfusión, 2 torundas impregnadas de alcohol, 1 vendaje no estéril.
Titular de la autorización de comercialización y fabricante
CSL Behring GmbH – Emil-von-Behring-Str. 76 – D-35041 Marburg – Alemania
Representante en Italia
CSL Behring S.p.A. – Viale del Ghisallo, 20 - 20151 Milán – Italia
La siguiente información está destinada exclusivamente a médicos o profesionales sanitarios
Advertencias especiales y precauciones adecuadas de uso
Hemofilia A
La formación de anticuerpos neutralizantes (inhibidores) frente al factor VIII constituye una complicación conocida en el tratamiento de pacientes con hemofilia A. Dichos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (UB) por ml de plasma mediante el ensayo modificado. El riesgo de desarrollar inhibidores está relacionado con la gravedad de la enfermedad y con el tiempo de exposición al factor VIII, siendo mayor durante los primeros 20 días de exposición. Rara vez pueden desarrollarse inhibidores después de los primeros 100 días de exposición.
Se han observado casos de aparición de inhibidores recurrentes (de bajo título) tras el cambio de un producto basado en FVIII a otro, en pacientes previamente tratados con más de 100 días de exposición y con antecedentes de desarrollo previo de inhibidores. Por ello, se recomienda monitorizar cuidadosamente a todos los pacientes para detectar la reaparición de inhibidores tras cualquier cambio de un producto a otro.
La relevancia clínica del desarrollo de inhibidores dependerá del título del inhibidor: los inhibidores de bajo título, ya sean temporales o que permanezcan constantemente en niveles bajos, tendrán menor impacto en el riesgo de respuesta clínica insuficiente en comparación con los inhibidores de alto título.
En general, todos los pacientes tratados con productos basados en factor VIII de la coagulación deben ser monitorizados cuidadosamente para detectar el desarrollo de inhibidores mediante observaciones clínicas adecuadas y pruebas de laboratorio. Si no se alcanzan los niveles plasmáticos esperados de actividad del factor VIII, o si la hemorragia no se controla con una dosis adecuada, debe realizarse un análisis para determinar si están presentes inhibidores del factor VIII. En pacientes con niveles elevados de inhibidor, la terapia con factor VIII podría no ser eficaz, y deben considerarse otras opciones terapéuticas. La gestión de estos pacientes debe estar a cargo de médicos con experiencia en el tratamiento de la hemofilia y de los inhibidores del factor VIII.
Enfermedad de von Willebrand
Existe el riesgo de que ocurran episodios trombóticos, incluyendo embolia pulmonar, especialmente en pacientes con factores de riesgo clínicos o de laboratorio conocidos (por ejemplo, durante el período perioperatorio, particularmente en ausencia de profilaxis trombótica o de movilización precoz, en casos de obesidad, sobredosificación de HAEMATE P o en pacientes con cáncer). Por lo tanto, los pacientes en riesgo deben ser monitorizados para detectar los primeros signos de trombosis. Si procede, debe instaurarse un régimen de profilaxis contra la tromboembolia venosa, de acuerdo con las recomendaciones vigentes.
Al utilizar productos que contienen factor VIII y VWF, el médico tratante debe tener presente que un tratamiento prolongado puede provocar un aumento excesivo del nivel de FVIII:C. Los pacientes que reciben productos que contienen FVIII:C y VWF:RCo deben ser monitorizados cuidadosamente para evitar un aumento excesivo de los niveles plasmáticos de FVIII:C, con el consiguiente aumento del riesgo de eventos trombóticos.
Los pacientes con enfermedad de von Willebrand, especialmente del tipo 3, pueden desarrollar anticuerpos neutralizantes del VWF (inhibidores). Si no se alcanzan los niveles esperados de actividad de VWF:RCo en plasma, o si la dosis administrada no logra controlar eficazmente la hemorragia, será conveniente realizar una prueba adecuada para confirmar la posible presencia de inhibidores del VWF. En pacientes con título elevado de inhibidores, la terapia puede resultar ineficaz, y será conveniente considerar otras opciones terapéuticas.
Posología
Hemofilia A
Monitorización del tratamiento
Durante el tratamiento, se recomienda una determinación adecuada de los niveles de factor VIII para establecer la dosis a administrar y la frecuencia con que deben repetirse las infusiones. Algunos pacientes pueden presentar variabilidad en su respuesta al factor VIII, alcanzando diferentes niveles de recuperación in vivo y mostrando diferentes vidas medias. Los cálculos de dosis basados en el peso corporal pueden requerir ajustes en pacientes con bajo peso o sobrepeso. En particular, durante intervenciones quirúrgicas mayores, es indispensable un control preciso de la terapia sustitutiva mediante el seguimiento de los parámetros de coagulación (actividad plasmática del factor VIII).
Los pacientes deben ser monitorizados para detectar el desarrollo de inhibidores del factor VIII. Véase también el apartado 2.
La posología y la duración de la terapia sustitutiva dependen de la gravedad del déficit de factor VIII, de la localización y magnitud de la hemorragia, así como de las condiciones clínicas del paciente.
Es importante calcular la dosis utilizando el número de UI de FVIII:C especificado.
El número de unidades de factor VIII a administrar se expresa en Unidades Internacionales (U.I.), referidas al estándar actual de la OMS (WHO) para productos concentrados de factor VIII.
La actividad del factor VIII en plasma se expresa en porcentaje (relativo al plasma humano normal) o preferiblemente en U.I. (de acuerdo con el Estándar Internacional para el factor VIII en plasma).
Una unidad internacional de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
El cálculo de la dosis necesaria de factor VIII se basa en el dato empírico de que 1 U.I. de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en plasma aproximadamente en un 2% de la actividad normal (2 U.I./dl). La dosis necesaria se determina utilizando la siguiente fórmula:
Unidades requeridas = peso corporal [kg] × aumento deseado de factor VIII [% o U.I./dl] × 0,5.
La dosis y la frecuencia de administración deben basarse siempre en la eficacia clínica obtenida en cada caso individual.
En los siguientes episodios hemorrágicos, la actividad del factor VIII no debe descender por debajo del nivel de actividad plasmática indicado (en % del normal o en U.I./dl). La siguiente tabla puede utilizarse como referencia para la dosificación en caso de eventos hemorrágicos o intervenciones quirúrgicas:
| Gravedad de la hemorragia / Tipo de intervención quirúrgica | Nivel requerido de Factor VIII (% o U.I./dl) | Frecuencia de las dosis (horas) / Duración del tratamiento (días) |
| Hemorragia | ||
| Hemoartrosis en fase inicial, hemorragias intramusculares o de la cavidad oral. | 20 – 40 | Repetir la infusión cada 12-24 horas durante al menos 1 día hasta la resolución del episodio hemorrágico o cicatrización, según lo indique la desaparición del dolor. |
| Hemoartrosis más extensas, hemorragias intramusculares o hematomas. | 30 – 60 | Repetir la infusión cada 12-24 horas durante 3-4 días o más hasta la resolución del dolor y de la discapacidad aguda. |
| Hemorragias con riesgo vital. | 60 - 100 | Repetir la infusión cada 8-24 horas hasta la resolución del evento. |
| Cirugía | ||
| Cirugía menor, incluyendo extracciones dentales. | 30 – 60 | Cada 24 horas, durante al menos 1 día, hasta alcanzar la curación. |
| Cirugía mayor | 80 – 100 (pre y postoperatorio) | Repetir la infusión cada 8-24 horas hasta alcanzar una cicatrización adecuada; posteriormente continuar el tratamiento durante al menos 7 días para mantener una actividad de Factor VIII entre el 30-60% (U.I./dl). |
Para la profilaxis a largo plazo de hemorragias en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de Factor VIII por kg de peso corporal, con intervalos de 2-3 días. En algunos casos, especialmente en pacientes más jóvenes, pueden ser necesarios intervalos más cortos o dosis más elevadas.
Si no se alcanzan los niveles plasmáticos esperados de actividad del Factor VIII o si la hemorragia no se controla con una dosis adecuada, los pacientes deben ser monitorizados para detectar la posible aparición de un inhibidor del Factor VIII. En pacientes con niveles elevados de inhibidor, el tratamiento con Factor VIII puede no ser eficaz, por lo que deben considerarse otras medidas terapéuticas. En estos casos, además, el tratamiento debe realizarse bajo la responsabilidad de médicos expertos en el tratamiento de la hemofilia.
Población pediátrica
No hay datos clínicos disponibles en niños.
Enfermedad de von Willebrand
Es importante calcular la dosis utilizando el número de UI de VWF:RCo indicado.
La administración de 1 UI/kg de VWF:RCo determina, en general, un aumento del título de VWF:RCo circulante igual a 0,02 UI/ml (2%).
Se deben alcanzar niveles de VWF:RCo > 0,6 UI/ml (60%) y de FVIII:C > 0,4 UI/ml (40%).
Normalmente, para lograr la hemostasia se recomienda la administración de 40-80 UI/kg de Factor von Willebrand (VWF:RCo) y de 20-40 UI de FVIII:C/kg de peso corporal.
Puede ser necesaria la administración de una dosis inicial de 80 UI/kg de Factor von Willebrand, especialmente en pacientes con enfermedad de von Willebrand tipo 3: en este caso, de hecho, el mantenimiento de niveles adecuados puede requerir el uso de dosis más elevadas en comparación con otros tipos de enfermedad de von Willebrand.
Prevención del evento hemorrágico en caso de intervención quirúrgica o episodio traumático grave: para prevenir un sangrado excesivo durante o después de una intervención quirúrgica, la administración debe realizarse 1-2 horas antes de dicha intervención.
Posteriormente, deben administrarse dosis adecuadas cada 12-24 horas.
La dosis a administrar y la duración del tratamiento dependen de la situación clínica individual, del tipo y gravedad de la hemorragia y de los niveles de VWF:RCo y de FVIII:C.
Cuando se utilicen preparados que contienen Factor VIII y Factor von Willebrand, el médico debe tener presente que un tratamiento prolongado puede provocar un aumento excesivo del título de FVIII:C. Para evitar un aumento excesivo de FVIII:C, tras 24-48 horas de tratamiento, debe considerarse la conveniencia de reducir la dosis y/o aumentar el intervalo entre administraciones, o bien utilizar productos basados en VWF con un bajo contenido de FVIII.
Población pediátrica
En niños, la dosis está relacionada con el peso corporal y, en general, se siguen las mismas pautas indicadas para adultos. La frecuencia de administración debe adaptarse siempre para garantizar la eficacia clínica en cada caso individual.
Instrucciones para el uso, manipulación y eliminación
Los productos no utilizados y los residuos derivados de este medicamento deben eliminarse de conformidad con los requisitos legales locales.
Instrucciones generales:
La solución debe presentarse clara o ligeramente opalescente. Tras la filtración/la extracción (véase más adelante), el producto reconstituido debe inspeccionarse visualmente antes de la administración para comprobar la ausencia de partículas o decoloraciones. Aunque se sigan cuidadosamente las instrucciones siguientes, no es raro que puedan quedar floculos o partículas. El filtro Mix2Vial, incluido en el envase, eliminará dichas partículas. Tras la filtración, no utilizar soluciones turbias ni que contengan floculos o partículas. La filtración no influye en el cálculo final de la dosis.
La reconstitución y la extracción deben realizarse en condiciones asépticas.
Reconstitución:
Llevar el disolvente a temperatura ambiente. Asegurarse de haber retirado las tapas abatibles de los frascos y de que los tapones hayan sido desinfectados con una solución antiséptica, y esperar a que esta se haya secado antes de abrir el envase de Mix2Vial.
1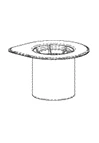 |
|
2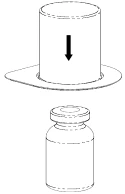 |
|
3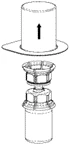 |
|
4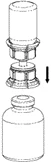 |
|
5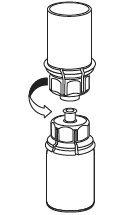 |
|
6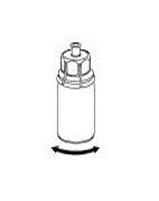 |
|
7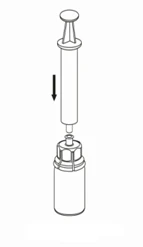 |
|
Extracción y administración:
8 |
|
9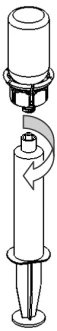 |
|
Para la inyección de HAEMATE P se recomienda el uso de jeringas desechables de plástico, ya que las superficies de vidrio de las jeringas tienden a bloquearse con este tipo de soluciones.
Modo de administración
Antes de la administración, la preparación reconstituida debe llevarse a temperatura ambiente o corporal. Inyectar lentamente por vía endovenosa, a una velocidad cómoda para el paciente. Tener cuidado de que no entre sangre en la jeringa que contiene el producto. Una vez que el producto se ha transferido a la jeringa, debe utilizarse inmediatamente.
En caso de que sea necesario administrar dosis más elevadas de Factor VIII, se puede proceder mediante infusión, transfiriendo el producto reconstituido a un sistema de infusión adecuado.
La velocidad de inyección o infusión no debe superar los 4 ml/minuto. Mantener al paciente bajo observación para detectar cualquier reacción inmediata. Si se produce alguna reacción relacionada con la administración de HAEMATE, reducir la velocidad de infusión o interrumpir la administración según el estado clínico del paciente.
Efectos no deseados
Es conveniente mantener una vigilancia estrecha en todos los pacientes con el fin de observar la aparición de los primeros signos de hipervolemia.
Además, los pacientes con grupos sanguíneos A, B y AB deben ser monitorizados para detectar posibles signos de hemólisis intravascular y/o reducción del valor del hematocrito.
Los pacientes que reciben productos que contienen FVIII:C y VWF:RCo deben ser cuidadosamente monitorizados para evitar un aumento excesivo de los niveles plasmáticos de FVIII:C, con el consiguiente incremento del riesgo de eventos trombóticos, incluyendo la embolia pulmonar.
Incompatibilidades
Este medicamento no debe mezclarse con otros medicamentos, disolventes ni diluyentes, excepto con aquellos mencionados en la lista de excipientes.
Caducidad
3 años a 25°C
Después de la reconstitución, el producto ha demostrado estabilidad físico-química durante 3 horas a temperatura ambiente (máximo +25°C).
Desde el punto de vista microbiológico, y dado que HAEMATE P no contiene conservantes, el producto reconstituido debe utilizarse inmediatamente. Si no se administra inmediatamente, el tiempo de conservación durante su uso y las condiciones previas a su administración son responsabilidad del usuario. En ningún caso se deben superar las 3 horas de conservación a temperatura ambiente.
Una vez que el producto ha sido aspirado en la jeringa, debe utilizarse inmediatamente.
Para más información, consultar el Resumen de las Características del Producto.