Pregabalin-teva
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT Pregabalin-Teva
Composition:
Active ingredient: pregabalin;
1 capsule contains pregabalin 75 or 150 mg;
Excipients: mannite (E 421), pregelatinized corn starch, talc, titanium dioxide (E 171), red iron oxide (E 172) (75 mg capsules), yellow iron oxide (E 172) (75 mg and 150 mg capsules), gelatin.
Pharmaceutical form. Hard capsules.
Main physicochemical properties:
75 mg capsules – opaque capsules with a pink cap and an ivory-colored body, with a black imprint "75" on the body;
150 mg capsules – opaque ivory-colored capsules with a black imprint "150" on the body.
Pharmacotherapeutic group. Antiepileptics, other antiepileptic drugs.
ATC code: N03AX16.
Pharmacological Properties.
Pharmacodynamics.
The active substance is pregabalin, a gamma-aminobutyric acid analogue [(S)-3-(aminomethyl)-5-methylhexanoic acid].
Mechanism of action.
Pregabalin binds to the auxiliary subunit (α2-δ protein) of voltage-dependent calcium channels in the central nervous system (CNS).
Clinical efficacy and safety.
- Neuropathic pain.
The efficacy of pregabalin has been demonstrated in clinical studies for the treatment of diabetic neuropathy, postherpetic neuralgia, and spinal cord injury. The efficacy of the drug has not been studied in other types of neuropathic pain.
Pregabalin was evaluated in 10 controlled clinical trials lasting up to 13 weeks with a twice-daily dosing regimen and in trials lasting up to 8 weeks with a three-times-daily regimen. Overall, the safety and efficacy profiles for twice-daily and three-times-daily dosing regimens were similar.
In clinical trials lasting up to 12 weeks in which the drug was used for the treatment of neuropathic pain, a reduction in peripheral and central pain was observed after the first week and persisted throughout the treatment period.
In controlled clinical trials of peripheral neuropathic pain, a 50% improvement on the pain rating scale was observed in 35% of patients receiving pregabalin and in 18% of patients receiving placebo. Among patients who did not experience somnolence, such improvement was observed in 33% of patients receiving pregabalin and in 18% of patients in the placebo group. Among patients who experienced somnolence, the proportion of responders was 48% in the pregabalin group and 16% in the placebo group.
In a controlled clinical trial studying centrally mediated neuropathic pain, a 50% improvement on the pain rating scale was observed in 22% of patients receiving pregabalin and in 7% of patients receiving placebo.
- Epilepsy.
Adjunctive therapy. Pregabalin was studied in three controlled clinical trials lasting 12 weeks with twice-daily or three-times-daily dosing regimens. Overall, the safety and efficacy profiles for twice-daily and three-times-daily dosing regimens were similar.
A reduction in seizure frequency was observed as early as the first week.
Children. The efficacy and safety of pregabalin as adjunctive therapy in epilepsy have not been established in children under 12 years of age and in adolescents. Adverse reactions observed in a pharmacokinetic and tolerability study involving patients aged 3 months to 16 years (n=65) with partial seizures were similar to those in adults. Results from a 12-week placebo-controlled study involving 295 children aged 4 to 16 years and a 14-day placebo-controlled study involving 175 children aged 1 month to < 4 years, designed to evaluate the efficacy and safety of pregabalin as adjunctive therapy for partial seizures, and from two open-label safety studies of 1 year duration involving 54 and 431 children, respectively, aged 3 months to 16 years with epilepsy, indicate that adverse reactions such as pyrexia and upper respiratory tract infections occur more frequently in children than in adult patients with epilepsy (see sections "Method of administration and dosage", "Adverse reactions", and "Pharmacokinetics").
In the 12-week placebo-controlled study, children (aged 4 to 16 years) were administered pregabalin at 2.5 mg/kg/day (maximum 150 mg/day), pregabalin at 10 mg/kg/day (maximum 600 mg/day), or placebo. A reduction of at least 50% in partial seizures compared to baseline was observed in 40.6% of patients receiving pregabalin at 10 mg/kg/day (p=0.0068 vs placebo), in 29.1% of patients receiving pregabalin at 2.5 mg/kg/day (p=0.2600 vs placebo), and in 22.6% of those receiving placebo.
In the 14-day placebo-controlled study, children (aged 1 month to < 4 years) received pregabalin at 7 mg/kg/day, pregabalin at 14 mg/kg/day, or placebo. The median daily seizure frequency at baseline and at the final visit was 4.7 and 3.8, respectively, for pregabalin at 7 mg/kg/day, 5.4 and 1.4 for pregabalin at 14 mg/kg/day, and 2.9 and 2.3 for placebo. Pregabalin at 14 mg/kg/day significantly reduced the logarithmically transformed frequency of partial seizures compared to placebo (p=0.0223); pregabalin at 7 mg/kg/day did not demonstrate improvement compared to placebo.
In a 12-week placebo-controlled study of patients with primary generalized tonic-clonic (PGTC) seizures, 219 patients aged 5 to 65 years (including 66 individuals aged 5 to 16 years) received pregabalin at 5 mg/kg/day (maximum 300 mg/day), 10 mg/kg/day (maximum 600 mg/day), or placebo as adjunctive therapy. A reduction of at least 50% in the frequency of PGTC seizures was observed in 41.3%, 38.9%, and 41.7% of patients receiving pregabalin at 5 mg/kg/day, pregabalin at 10 mg/kg/day, and placebo, respectively.
Monotherapy (in patients with newly diagnosed disease). Pregabalin was studied in one controlled clinical trial lasting 56 weeks with a twice-daily dosing regimen. When pregabalin was used, equivalent efficacy compared to lamotrigine was not achieved, based on the 6-month endpoint assessment—seizure freedom. Pregabalin and lamotrigine were equally safe and well tolerated.
- Generalized anxiety disorder.
Pregabalin was studied in six controlled trials lasting 4–6 weeks, one 8-week trial involving elderly patients, and one long-term relapse prevention trial with a double-blind relapse prevention phase lasting 6 months. Reduction in symptoms of generalized anxiety disorder according to the Hamilton Anxiety Rating Scale (HAM-A) was observed as early as week 1.
In controlled clinical trials (lasting 4–8 weeks), a ≥50% improvement in the total HAM-A score from baseline to endpoint was observed in 52% of patients receiving pregabalin and in 38% of patients in the placebo group.
During controlled trials, blurred vision occurred more frequently in patients receiving pregabalin than in those receiving placebo. In most cases, this effect resolved with continued therapy. Ophthalmological examinations (including visual acuity testing, formal visual field testing, and fundus examination with dilated pupils) were performed in over 3600 patients in controlled clinical trials. Among these patients, visual acuity worsened in 6.5% of patients in the pregabalin group and in 4.8% of patients in the placebo group. Visual field changes were detected in 12.4% of patients receiving pregabalin and in 11.7% of patients in the placebo group. Fundus changes were observed in 1.7% of patients receiving pregabalin and in 2.1% of patients in the placebo group.
- Fibromyalgia.
The efficacy of pregabalin was established in one 14-week double-blind, placebo-controlled, multicenter trial (F1) and one 6-week randomized withdrawal trial (F2). These trials included patients diagnosed with fibromyalgia based on American College of Rheumatology criteria (widespread pain for at least 3 months and pain present in 11 or more of 18 specific tender points). The studies demonstrated a reduction in pain on the visual analog scale. Additional improvement was demonstrated by patient global assessment and fibromyalgia impact questionnaire.
Children. A placebo-controlled trial lasting 15 weeks was conducted in 107 children aged 12–17 years with fibromyalgia who received pregabalin at doses of 75–450 mg/day. The primary efficacy endpoint (change in overall pain intensity from baseline to week 15, measured using an 11-point rating scale) showed numerically greater improvement in patients receiving pregabalin compared to those receiving placebo, but this improvement did not achieve statistical significance. The most commonly observed adverse reactions in clinical trials were dizziness, nausea, headache, weight gain, and fatigue. The overall safety profile in adolescents was similar to that in adults with fibromyalgia.
Pharmacokinetics.
Pharmacokinetic parameters of pregabalin at steady state were similar in healthy volunteers, patients with epilepsy taking antiepileptic drugs, and patients with chronic pain.
Absorption. Pregabalin is rapidly absorbed after oral administration on an empty stomach and reaches maximum plasma concentrations within 1 hour after single and multiple doses. The calculated oral bioavailability of pregabalin is ≥ 90% and is dose-independent. Steady-state concentrations are achieved within 24–48 hours after repeated administration. The extent of pregabalin absorption is reduced when administered with food, resulting in approximately a 25–30% decrease in maximum concentration (Cmax) and prolongation of tmax to approximately 2.5 hours. However, administration of pregabalin with food did not have a clinically significant effect on the extent of absorption.
Distribution. Pregabalin crosses the blood-brain barrier in animals, as well as the placenta and into the milk of lactating animals. In humans, the apparent volume of distribution of pregabalin after oral administration is approximately 0.56 L/kg. Pregabalin does not bind to plasma proteins.
Metabolism. In humans, pregabalin undergoes minimal metabolism. After administration of a radiolabeled dose of pregabalin, approximately 98% of radioactivity is excreted in urine as unchanged pregabalin. The N-methylated derivative of pregabalin (the main metabolite detected in urine) accounted for 0.9% of the administered dose. Racemization of the S-enantiomer of pregabalin to the R-enantiomer did not occur during preclinical studies.
Elimination. Pregabalin is eliminated from systemic circulation primarily by renal excretion in unchanged form. The mean elimination half-life of pregabalin is 6.3 hours. Plasma and renal clearance of pregabalin are directly proportional to creatinine clearance. Dose adjustment is required for patients with renal impairment or undergoing hemodialysis.
Linearity/Non-linearity. The pharmacokinetics of pregabalin are linear over the entire recommended dose range. Inter-subject pharmacokinetic variability for pregabalin is low (< 20%). Pharmacokinetics of multiple doses are predictable based on single-dose data. Therefore, routine monitoring of plasma concentrations of pregabalin is not necessary.
Pharmacokinetics in specific patient groups.
Gender. There is no clinically significant effect of gender on plasma concentrations of pregabalin.
Renal impairment. Clearance of pregabalin is directly proportional to creatinine clearance. Additionally, pregabalin is effectively removed from plasma during hemodialysis (after 4 hours of hemodialysis, plasma concentrations of pregabalin decrease by approximately 50%). Since renal excretion is the main elimination pathway, dose reduction is required for patients with renal impairment, and an additional dose should be administered after hemodialysis (see section "Method of administration and dosage", table).
Hepatic impairment. Specific pharmacokinetic studies in patients with hepatic impairment have not been conducted. Since pregabalin undergoes negligible metabolism and is primarily excreted unchanged in urine, it is unlikely that hepatic impairment would affect plasma concentrations of pregabalin.
Children. The pharmacokinetics of pregabalin were evaluated in children with epilepsy (age groups: 1 to 23 months, 2 to 6 years, 7 to 11 years, and 12 to 16 years) receiving doses of 2.5, 5, 10, and 15 mg/kg/day in a pharmacokinetic and tolerability study. After oral administration of pregabalin to children under fasting conditions, the time to reach maximum plasma concentration was generally similar across all age groups, ranging from 0.5 to 2 hours after dosing. Cmax and area under the concentration-time curve (AUC) values of pregabalin increased linearly with dose in each age group. In children with body weight below 30 kg, AUC values were 30% lower, due to a 43% higher body weight-adjusted clearance in these patients compared to patients with body weight ≥ 30 kg.
The terminal elimination half-life of pregabalin averaged approximately 3–4 hours in children under 6 years of age and 4–6 hours in children aged 7 years and older.
Population pharmacokinetic analysis demonstrated that creatinine clearance was a significant covariate for oral pregabalin clearance, and body weight was a significant covariate for the apparent volume of distribution of oral pregabalin, and this relationship was similar in children and adult patients.
The pharmacokinetics of pregabalin have not been studied in patients under 3 months of age (see sections "Method of administration and dosage", "Adverse reactions", and "Pharmacodynamics").
Elderly patients. Pregabalin clearance tends to decrease with age. This reduction in oral pregabalin clearance is consistent with the age-related decline in creatinine clearance. Elderly patients with age-related renal impairment may require dose reduction of pregabalin (see section "Method of administration and dosage", Table 1).
Lactation. The pharmacokinetics of pregabalin were evaluated in 10 breastfeeding women who received 150 mg every 12 hours (daily dose 300 mg), at least 12 weeks postpartum. Breastfeeding did not affect or had minimal effect on the pharmacokinetics of pregabalin. Pregabalin was excreted into breast milk, with average steady-state concentrations approximately 76% of maternal plasma concentrations. The calculated infant dose received via breast milk (assuming average milk intake of 150 mL/kg/day) from a woman taking pregabalin at 300 mg/day or at the maximum dose of 600 mg/day is 0.31 or 0.62 mg/kg/day, respectively. These calculated doses represent approximately 7% of the maternal daily dose normalized to mg/kg.
Clinical characteristics.
Indications.
Neuropathic pain.
Pregabalin-Teva is indicated for the treatment of peripheral or central neuropathic pain in adults.
Epilepsy.
Pregabalin-Teva is indicated as adjunctive therapy in adults with partial seizures with or without secondary generalization.
Generalized anxiety disorder.
Pregabalin-Teva is indicated for the treatment of generalized anxiety disorder in adults.
Fibromyalgia.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
Interaction with other medicinal products and other forms of interaction.
Since pregabalin is predominantly excreted unchanged in urine, undergoes negligible metabolism in the human body (less than 2% of the dose is excreted in urine as metabolites), does not inhibit in vitro metabolism of other drugs, and does not bind to plasma proteins, it is unlikely that pregabalin would cause pharmacokinetic interactions or be the object of such interactions.
In vivo studies and population pharmacokinetic analysis.
Accordingly, in in vivo studies, no clinically significant pharmacokinetic interactions were observed between pregabalin and phenytoin, carbamazepine, valproic acid, lamotrigine, gabapentin, lorazepam, oxycodone, or ethanol. Population pharmacokinetic analysis showed that oral antidiabetic agents, diuretics, insulin, phenobarbital, tiagabine, and topiramate had no clinically significant effect on pregabalin clearance.
Oral contraceptives, norethisterone and/or ethinylestradiol.
Concomitant administration of pregabalin and oral contraceptives, norethisterone and/or ethinylestradiol does not affect the steady-state pharmacokinetics of either agent.
Medicinal products affecting the CNS.
Pregabalin may potentiate the effects of ethanol and lorazepam. In the post-marketing period, cases of respiratory depression, coma, and death have been reported in patients who concurrently took pregabalin with opioids and/or other CNS depressants. Pregabalin is likely to enhance the cognitive and motor impairment caused by oxycodone.
Interactions in elderly patients.
No specific pharmacodynamic interaction studies were conducted in elderly volunteers. Drug interaction studies have been conducted only in adult patients.
Special precautions for use.
Patients with diabetes mellitus.
According to current clinical practice, some diabetic patients whose body weight increased during pregabalin treatment may require adjustment of antihyperglycemic medicinal products.
Hypersensitivity reactions.
Hypersensitivity reactions, including cases of angioedema, have been reported. If symptoms of angioedema occur, such as swelling of the face, perioral area, or upper airways, pregabalin should be discontinued immediately.
Severe skin adverse reactions.
Rare cases of severe skin adverse reactions, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), have been reported during pregabalin treatment. These reactions may be life-threatening or fatal. When prescribing this medicinal product, patients should be informed about the signs and symptoms, and skin reactions should be closely monitored. If signs or symptoms suggestive of these reactions occur, pregabalin should be discontinued immediately and alternative treatment considered (if necessary).
Dizziness, somnolence, loss of consciousness, confusion, and psychiatric disturbances.
Pregabalin use has been associated with cases of dizziness and somnolence, which may increase the risk of accidental injuries (falls) in elderly patients. During the post-marketing period, cases of loss of consciousness, confusion, and psychiatric disturbances have been reported. Therefore, patients should be advised to exercise caution until they are aware of the potential effects of this medicinal product.
Visual disorders.
In controlled studies, blurred vision was reported more frequently in patients receiving pregabalin than in those receiving placebo. In most cases, these effects resolved with continued treatment. In clinical trials involving ophthalmological examinations, the incidence of decreased visual acuity and visual field changes was higher in patients receiving pregabalin compared to placebo group patients; however, the incidence of fundus changes was higher in the placebo group.
During the post-marketing period, adverse reactions affecting the visual organs, including vision loss, blurred vision, or other changes in visual acuity, have been reported, most of which were temporary. These visual symptoms may resolve or diminish after discontinuation of pregabalin.
Renal impairment.
Cases of renal failure have been reported. This effect was sometimes reversible after discontinuation of pregabalin.
Discontinuation of concomitant antiepileptic medicinal products.
There is currently insufficient data on whether concomitant antiepileptic medicinal products can be discontinued after seizure control has been achieved by adding pregabalin to therapy, to switch to pregabalin monotherapy.
Withdrawal symptoms.
Withdrawal symptoms have been observed after discontinuation of both short-term and long-term pregabalin treatment. Reported events include insomnia, headache, nausea, anxiety, diarrhea, flu-like syndrome, restlessness, depression, suicidal thoughts, pain, seizures, hyperhidrosis, and dizziness. The occurrence of withdrawal symptoms after stopping pregabalin may indicate drug dependence (see section "Adverse reactions"). This information should be communicated to the patient prior to initiating treatment. If pregabalin treatment needs to be discontinued, it is recommended to do so gradually over at least 1 week, regardless of the indication (see section "Dosage and administration").
Seizures, including status epilepticus and generalized seizures, may occur during treatment with pregabalin or shortly after discontinuation.
Regarding discontinuation of long-term pregabalin treatment, available data suggest that the frequency and severity of withdrawal symptoms may be dose-dependent.
Heart failure.
During the post-marketing period, cases of heart failure have been reported in some patients receiving pregabalin. This reaction typically occurred during pregabalin treatment for neuropathic pain in elderly patients with pre-existing cardiovascular disorders. Pregabalin should be used with caution in such patients. This condition may resolve upon discontinuation of pregabalin.
Treatment of central neuropathic pain due to spinal cord injury.
During treatment of central neuropathic pain due to spinal cord injury, the overall incidence of adverse reactions, CNS-related adverse reactions, and particularly somnolence, was higher. This may be explained by an additive effect of other medicinal products (e.g., antispastic agents) required for the treatment of this condition. This should be taken into account when prescribing pregabalin to such patients.
Suicidal thoughts and behavior.
Cases of suicidal thoughts and behavior have been reported in patients receiving antiepileptic medicinal products for various indications. A meta-analysis of data from randomized, placebo-controlled trials of antiepileptic medicinal products showed a slightly increased risk of suicidal thoughts and behavior. The mechanism underlying this risk is unknown. During the post-marketing period, cases of suicidal thoughts and behavior have been reported in patients receiving pregabalin (see section "Adverse reactions"). An epidemiological study using a self-controlled design (comparing treatment periods with non-treatment periods in individual patients) demonstrated an increased risk of new-onset suicidal behavior and fatal outcomes due to suicide in patients receiving pregabalin.
Patients (and caregivers) should seek medical help if signs of suicidal thoughts or behavior occur. Patients should be carefully monitored for signs of suicidal thoughts and behavior, and appropriate treatment should be considered. If suicidal thoughts or behavior occur, discontinuation of pregabalin therapy should be considered.
Worsening of lower gastrointestinal tract function.
During the post-marketing period, events related to worsening of lower gastrointestinal tract function (e.g., intestinal obstruction, paralytic ileus, constipation) have been reported with pregabalin use, particularly when used concomitantly with medicinal products that may cause constipation, such as opioid analgesics. When pregabalin is used concomitantly with opioids, measures to prevent constipation should be taken (especially in women and elderly patients).
Concomitant use with opioids.
Caution is recommended when prescribing pregabalin concomitantly with opioids due to the risk of CNS depression (see section "Interaction with other medicinal products and other forms of interaction"). In a case-control study of individuals using opioids, an increased risk of opioid-related mortality was observed in patients receiving pregabalin concomitantly with an opioid compared to those receiving opioids alone (adjusted odds ratio [aOR], 1.68 [95% CI, 1.19–2.36]). This increased risk was observed at low pregabalin doses (≤ 300 mg, aOR 1.52 [95% CI, 1.04–2.22]) with a trend toward increased risk at higher pregabalin doses (> 300 mg, aOR 2.55 [95% CI 1.24–5.06]).
Misuse, abuse, or dependence.
Pregabalin may cause drug dependence, which may occur during therapeutic dose use. Cases of misuse and abuse have been reported. Patients with a history of substance abuse may have a higher risk of misuse, abuse, and dependence, and therefore pregabalin should be used with caution in such patients. Before prescribing pregabalin, the risk of misuse, abuse, or dependence should be carefully assessed.
Patients receiving pregabalin should be carefully monitored for symptoms of misuse, abuse, or dependence, such as development of tolerance, exceeding the prescribed dose, and drug-seeking behavior.
Encephalopathy.
Cases of encephalopathy have been reported, occurring primarily in patients with concomitant conditions that may predispose to encephalopathy.
Respiratory depression.
Serious respiratory depression has been reported with pregabalin use. Patients with impaired respiratory function, respiratory or neurological disorders, renal impairment, concomitant use of CNS depressants, and elderly patients may be at higher risk of this serious adverse reaction. Dose adjustment may be required in such patients (see section "Dosage and administration").
Women of childbearing potential/contraception.
Use of pregabalin during the first trimester of pregnancy may cause major congenital malformations in the fetus. Pregabalin should not be used during pregnancy unless the benefit to the pregnant woman clearly outweighs the potential risk to the fetus. Women of childbearing potential should use effective contraception during treatment (see section "Pregnancy and breastfeeding").
Pregnancy and breastfeeding.
Women of childbearing potential/contraception for women and men.
Women of childbearing potential should use effective contraception during treatment (see section "Special precautions for use").
Pregnancy.
Animal studies have shown reproductive toxicity. It has been demonstrated that pregabalin crosses the placenta in rats (see section "Pharmacokinetics"). Pregabalin may cross the placenta in humans.
Major congenital malformations
Data from an observational study conducted in Scandinavian countries, involving over 2700 pregnancies, showed a higher prevalence of major congenital malformations (MCM) in the pediatric population (live or stillborn children) exposed to pregabalin during the first trimester compared to the unexposed population (5.9% vs. 4.1%).
The risk of MCM in children whose mothers used pregabalin during the first trimester of pregnancy was slightly higher compared to children not exposed in utero (adjusted prevalence ratio and 95% confidence interval: 1.14 (0.96–1.35)) and compared to children exposed to lamotrigine (1.29 (1.01–1.65)) or duloxetine (1.39 (1.07–1.82)).
Analysis of specific malformations showed a higher risk of nervous system malformations, eye malformations, orofacial clefts, urinary tract malformations, and genital organ malformations, although the number of such malformations was small and estimates imprecise.
Pregabalin should not be used during pregnancy unless clearly necessary (when the benefit to the pregnant woman clearly outweighs the potential risk to the fetus).
Breastfeeding.
A small amount of pregabalin has been detected in human breast milk. Women should be informed that breastfeeding is not recommended during pregabalin treatment.
Fertility.
There are no clinical data on the effect of pregabalin on female fertility.
In a clinical study assessing the effect of pregabalin on sperm motility, healthy male volunteers received pregabalin 600 mg/day. After 3 months of treatment with pregabalin, no effect on sperm motility was observed.
In fertility studies, negative effects on reproductive function in female rats and negative effects on reproductive function and development in male rats were demonstrated. The clinical significance of these findings is unknown.
Ability to affect reaction speed when driving or operating machinery.
Pregabalin may have a slight or moderate influence on the ability to drive and use machinery. Pregabalin may cause dizziness and somnolence and may affect the ability to drive and operate machinery. Therefore, patients should be advised to refrain from driving, operating complex machinery, and other potentially hazardous activities until it is known whether this medicinal product affects their ability to perform such activities.
Dosage and Administration
The medication should be administered at a dose of 150 to 600 mg per day, divided into 2 or 3 doses. Pregabalin may be taken independently of food intake. The medicinal product is intended exclusively for oral administration.
Neuropathic Pain.
The initial dose of pregabalin is 150 mg per day, divided into 2 or 3 doses. Depending on individual patient response and tolerability, the dose may be increased after 3–7 days to 300 mg per day and, if necessary, further increased to the maximum dose of 600 mg per day after another 7 days.
Fibromyalgia.
The recommended dose of the medication for most patients is 300–450 mg per day. Treatment should be initiated at a dose of 75 mg twice daily (150 mg/day), which may be increased, based on efficacy and tolerability, to 150 mg twice daily (300 mg/day) within one week. For patients in whom a dose of 300 mg/day is insufficiently effective, the dose may be increased to 225 mg twice daily (450 mg/day). Although a study on the use of a 600 mg/day dose exists, there is no evidence that its use provides additional benefit. Moreover, this dose was associated with poorer tolerability. Due to dose-dependent adverse reactions, doses exceeding 450 mg per day are not recommended. Since pregabalin is primarily eliminated via the kidneys, dosage adjustment is required for patients with impaired renal function.
Epilepsy.
The initial dose of pregabalin is 150 mg per day, divided into 2 or 3 doses. Depending on individual patient response and tolerability, the dose may be increased to 300 mg per day after one week. After another week, the dose may be further increased to the maximum of 600 mg per day.
Generalized Anxiety Disorders.
The daily dose ranges from 150 to 600 mg, divided into 2 or 3 doses. The need for pregabalin treatment should be regularly reassessed.
Treatment with pregabalin may be initiated at a dose of 150 mg per day. Depending on individual response and tolerability, the dose may be increased to 300 mg per day after the first week of treatment. During the following week, the dose may be increased to 450 mg per day. After another week, the dose may be further increased to the maximum of 600 mg per day.
Discontinuation of Pregabalin.
According to current clinical practice, if pregabalin needs to be discontinued, it is recommended to gradually taper the medication over at least 1 week, regardless of the indication.
Patients with Impaired Renal Function.
Pregabalin is eliminated from systemic circulation in unchanged form, primarily via the kidneys. Since pregabalin clearance is directly proportional to creatinine clearance, dosage reduction in patients with impaired renal function should be individualized according to creatinine clearance (CLcr), as indicated in the table below and calculated using the following formula:
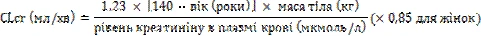
Pregabalin is effectively removed from plasma by hemodialysis (50% of the drug within 4 hours). For patients undergoing hemodialysis, the daily dose of pregabalin should be adjusted according to renal function. In addition to the daily dose, an additional dose of the medication should be administered immediately after each 4-hour hemodialysis session (see table).
| Creatinine clearance (CLcr) (mL/min) |
Total daily pregabalin dose* |
Dosing regimen |
|
| Initial dose (mg per day) |
Maximum dose (mg per day) |
||
| ≥ 60 |
150 |
600 |
2 or 3 times daily |
| ≥ 30−< 60 |
75 |
300 |
2 or 3 times daily |
| ≥ 15−< 30 |
25–50 |
150 |
1 or 2 times daily |
| < 15 |
25 |
75 |
Once daily |
| Supplemental dose after hemodialysis (mg) |
|||
| 25 |
100 |
Single dose+ |
|
* The total daily dose (mg/day) should be divided by the number of doses to obtain the number of milligrams per dose.
- Additional dose – is a single additional dose.
Patients with hepatic impairment.
Dose adjustment is not required in patients with hepatic impairment.
Geriatric patients.
A dose reduction of pregabalin may be necessary in geriatric patients due to impaired renal function.
Children.
The safety and efficacy of pregabalin in children (under 18 years of age) have not been established. The information currently available is presented in the section "Adverse reactions" as well as in the sections "Pharmacodynamics" and "Pharmacokinetics", however, based on this information, no dosage recommendations can be provided for this category of patients.
Overdose.
Following marketing of the drug, the most commonly reported adverse reactions in cases of pregabalin overdose were somnolence, confusion, agitation, and restlessness. Seizures have also been reported.
Coma has been reported rarely.
Treatment of pregabalin overdose consists of general supportive measures and, if necessary, may include hemodialysis (see section "Dosage and administration", table).
Adverse Reactions
In the clinical development program for pregabalin, more than 8900 patients received the drug, including 5600 patients in double-blind, placebo-controlled studies. The most commonly reported adverse reactions were dizziness and somnolence. Adverse reactions were generally mild to moderate in severity. In all controlled studies, the discontinuation rate due to adverse reactions was 12% among patients receiving pregabalin and 5% among those receiving placebo. The most common adverse reactions leading to discontinuation of study medication in the pregabalin group were dizziness and somnolence.
Below are listed all adverse reactions occurring more frequently than with placebo and in more than one patient. These adverse reactions are categorized by system organ class and frequency: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1000 to <1/100); rare (≥1/10,000 to <1/1000); very rare (<1/10,000); frequency not known (cannot be estimated from available data). Within each frequency grouping, adverse effects are listed in order of decreasing severity.
During treatment of central neuropathic pain due to spinal cord injury, the overall incidence of adverse reactions, CNS-related adverse reactions, and particularly somnolence, increased.
The adverse reactions listed below may be related to the underlying disease and/or concomitant use of other medicinal products.
Additional adverse reactions reported during the post-marketing period are indicated in italics in the list below.
Infections and infestations: common – nasopharyngitis.
Immune system disorders: uncommon – hypersensitivity; rare – angioedema, allergic reactions, anaphylactoid reactions.
Blood and lymphatic system disorders: uncommon – neutropenia.
Metabolism and nutrition disorders: common – increased appetite; uncommon – anorexia, hypoglycemia.
Psychiatric disorders: common – euphoric mood, confusion, irritability, disorientation, insomnia, decreased libido; uncommon – hallucinations, panic attacks, restlessness, agitation, depression, depressed mood, elevated mood, aggression, mood swings, depersonalization, word-finding difficulty, pathological dreams, increased libido, anorgasmia, apathy; rare – disinhibition, suicidal behaviour, suicidal ideation; frequency not known – drug dependence.
Nervous system disorders: very common – dizziness, somnolence, headache; common – ataxia, coordination disorder, tremor, dysarthria, amnesia, memory impairment, attention disturbance, paresthesia, hypesthesia, sedation, balance disorder, lethargy; uncommon – syncope, stupor, loss of consciousness, psychomotor hyperactivity, dyskinesia, postural dizziness, intention tremor, nystagmus, cognitive impairment, psychiatric disorder, speech disorder, hyporeflexia, hyperesthesia, burning sensation, ageusia, malaise, apathy, perioral paresthesia, myoclonus; rare – seizures, parosmia, hypokinesia, dysgraphia, hypoalgesia, dependence, cerebellar syndrome, cogwheel rigidity, coma, delirium, encephalopathy, extrapyramidal disorder, Guillain-Barré syndrome, intracranial hypertension, manic reactions, paranoid reactions, sleep disorders, parkinsonism.
Eye disorders: common – blurred vision, diplopia, conjunctivitis; uncommon – peripheral vision loss, visual disturbance, eye swelling, visual field defect, reduced visual acuity, eye pain, asthenopia, photopsia, dry eyes, increased lacrimation, eye irritation, blepharitis, accommodation disorder, subconjunctival hemorrhage, photophobia, retinal edema; rare – vision loss, keratitis, oscillopsia, altered depth perception, mydriasis, strabismus, brightness of vision, anisocoria, corneal ulcer, exophthalmos, oculomotor nerve paralysis, iritis, keratoconjunctivitis, miosis, night blindness, ophthalmoplegia, optic nerve atrophy, optic disc edema, ptosis, uveitis.
Ear and labyrinth disorders: common – vertigo; uncommon – hyperacusis.
Cardiac disorders: uncommon – tachycardia, first-degree atrioventricular block, sinus bradycardia, congestive heart failure, arterial hypotension, hypertension, flushing, hyperemia, feeling of cold in extremities; rare – QT interval prolongation, sinus tachycardia, sinus arrhythmia.
Respiratory, thoracic and mediastinal disorders: common – pharyngolaryngeal pain; uncommon – dyspnea, epistaxis, cough, nasal congestion, rhinitis, snoring, dryness of nasal mucosa; rare – pulmonary edema, throat tightness, laryngospasm, apnea, atelectasis, bronchiolitis, hiccups, pulmonary fibrosis, yawning; frequency not known – respiratory depression.
Gastrointestinal disorders: common – vomiting, nausea, constipation, diarrhea, flatulence, abdominal distension, dry mouth, gastroenteritis; uncommon – gastroesophageal reflux disease, hypersalivation, oral hypesthesia, cholecystitis, cholelithiasis, colitis, gastrointestinal hemorrhage, melena, tongue swelling, rectal bleeding; rare – ascites, pancreatitis, tongue swelling, dysphagia, aphthous stomatitis, esophageal ulcer, periodontal abscess.
Hepatobiliary disorders: uncommon – elevated liver enzymes*; rare – jaundice; very rare – liver failure, hepatitis.
Skin and subcutaneous tissue disorders: common – pressure ulcers; uncommon – papular rash, urticaria, hyperhidrosis, itching, alopecia, dry skin, eczema, hirsutism, skin ulcers, vesiculobullous rash; rare – Stevens-Johnson syndrome, cold sweat, toxic epidermal necrolysis, exfoliative dermatitis, lichenoid dermatitis, melanosis, nail disorders, petechial rash, purpura, pustular rash, skin atrophy, skin necrosis, skin and subcutaneous nodules.
Musculoskeletal and connective tissue disorders: common – muscle spasms, arthralgia, back pain, limb pain, neck spasms; uncommon – joint swelling, myalgia, muscle twitching, neck pain, muscle stiffness; rare – rhabdomyolysis.
Renal and urinary disorders: uncommon – urinary incontinence, dysuria, albuminuria, hematuria, kidney stone formation, nephritis; rare – renal failure, oliguria, urinary retention, acute renal failure, glomerulonephritis, pyelonephritis.
Reproductive system and breast disorders: common – erectile dysfunction, impotence; uncommon – sexual dysfunction, delayed ejaculation, dysmenorrhea, breast pain, leukorrhea, menorrhagia, metrorrhagia; rare – amenorrhea, breast discharge, breast enlargement, gynecomastia, cervicitis, balanitis, epididymitis.
General disorders and administration site conditions: common – peripheral edema, edema, gait disturbance, falls, feeling drunk, unusual sensations, fatigue; uncommon – generalized edema, facial swelling, chest tightness, pain, increased body temperature, thirst, chills, asthenia, general weakness, malaise, abscess, cellulitis, photosensitivity reactions; rare – granuloma, self-harm, retroperitoneal fibrosis, shock.
Investigations: common – weight increased; uncommon – increased blood creatine phosphokinase, increased blood glucose, decreased platelet count, increased blood creatinine, decreased blood potassium, weight decreased; rare – decreased white blood cell count.
* Increase in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels.
Symptoms of drug withdrawal have been observed following discontinuation of both short- and long-term pregabalin treatment. Reported reactions include: insomnia, headache, nausea, diarrhea, influenza-like syndrome, restlessness, depression, suicidal thoughts, pain, anxiety, seizures, hyperhidrosis, and dizziness. These symptoms may indicate drug dependence. Patients should be informed of this at the beginning of therapy.
Regarding withdrawal after long-term pregabalin treatment, data suggest that the frequency and severity of withdrawal symptoms may be dose-dependent (see sections "Dosage and Administration" and "Special Warnings and Precautions for Use").
Paediatric population. The safety profile of pregabalin established in five studies involving paediatric patients with partial seizures with or without secondary generalization (a 12-week efficacy and safety study in patients aged 4 to 16 years, n=295; a 14-day efficacy and safety study in patients aged 1 month to less than 4 years, n=175; a pharmacokinetic and tolerability study, n=65; and two open-label, one-year safety studies, n=54 and n=431) was similar to that observed in adult epilepsy studies. The most commonly observed adverse events in the 12-week pregabalin therapy study were somnolence, pyrexia, upper respiratory tract infections, increased appetite, weight gain, and nasopharyngitis. The most commonly observed adverse events in the 14-day pregabalin therapy study were somnolence, upper respiratory tract infections, and pyrexia (see sections "Dosage and Administration", "Pharmacodynamics", and "Pharmacokinetics").
Shelf life. 3 years.
Storage conditions. Store at temperatures not exceeding 30°C, in a place inaccessible to children.
Packaging. 14 capsules in a blister. 1, 2, or 4 blisters per carton.
Prescription status. Prescription only.
Manufacturer. PLIVA Hrvatska d.o.o.
Manufacturer's address and place of business.
Prilaz baruna Filipovića 25, 10000 Zagreb, Croatia.