Pentotren
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT PENTOTREN (PENTOTREN)
Composition:
Active substance: pentoxifylline;
1 ml of solution contains pentoxifylline 0.5 mg;
Excipients: sodium chloride; potassium chloride; calcium chloride dihydrate; magnesium chloride hexahydrate; sodium lactate solution; water for injections.
Pharmaceutical form. Infusion solution.
Main physicochemical properties: clear, colorless or slightly yellowish liquid.
Pharmacotherapeutic group. Peripheral vasodilators. Purine derivatives.
ATC code C04AD03.
Pharmacological properties.
Pharmacodynamics.
Pentoxifylline is a methylxanthine derivative. The mechanism of action of pentoxifylline is associated with inhibition of phosphodiesterase and accumulation of cAMP in vascular smooth muscle cells, blood cells, as well as in other tissues and organs. Pentoxifylline inhibits platelet and erythrocyte aggregation, increases their flexibility, reduces elevated plasma fibrinogen concentration, and enhances fibrinolysis, thereby decreasing blood viscosity and improving its rheological properties. In addition, pentoxifylline exerts a weak myotropic vasodilating effect, slightly reduces total peripheral vascular resistance, and demonstrates a positive inotropic effect. As a result of pentoxifylline administration, microcirculation and tissue oxygen supply are improved, most notably in the extremities and CNS, and to a moderate extent in the kidneys. The drug slightly dilates coronary vessels.
Pharmacokinetics.
The main pharmacologically active metabolite, 1-(5-hydroxyhexyl)-3,7-dimethylxanthine (metabolite I), is detected in blood plasma at concentrations exceeding those of the unchanged substance by two times and exists in reversible biochemical equilibrium with it. Therefore, pentoxifylline and its metabolite should be considered as an active entity. The elimination half-life of pentoxifylline is 1.6 hours.
Pentoxifylline is completely metabolized, with over 90% excreted by the kidneys as non-conjugated, water-soluble, polar metabolites. Less than 4% of the administered dose is excreted in feces. In patients with severe renal impairment, metabolite excretion is delayed. In patients with hepatic dysfunction, the elimination half-life of pentoxifylline is prolonged.
Clinical characteristics.
Indications.
Atherosclerotic encephalopathy; ischemic cerebral stroke; dyscirculatory encephalopathy; peripheral circulatory disorders caused by atherosclerosis, diabetes mellitus (including diabetic angiopathy), inflammation; tissue trophic disorders associated with venous damage or impaired microcirculation (post-thrombophlebitic syndrome, trophic ulcers, gangrene, frostbite); obliterative endarteritis; angioneuropathy (Raynaud's disease); ocular circulation disorders (acute, subacute, chronic insufficiency of blood flow in the retina and choroid); vascular-origin inner ear function disorders accompanied by hearing loss.
Contraindications.
Hypersensitivity to pentoxifylline, other methylxanthines, or any of the excipients of the medicinal product; massive bleeding (risk of exacerbating hemorrhage); extensive retinal hemorrhage, intracerebral hemorrhage (risk of exacerbating hemorrhage); if retinal hemorrhage occurs during treatment with pentoxifylline, the medicinal product should be discontinued immediately; acute phase of myocardial infarction and severe cardiac arrhythmia; gastric and/or intestinal ulcers; hemorrhagic diathesis; pregnancy or breastfeeding period.
Interaction with other medicinal products and other forms of interactions.
The blood glucose-lowering effect characteristic of insulin or oral antidiabetic agents may be enhanced. Therefore, patients receiving medication for diabetes mellitus should be closely monitored.
There is information about cases of increased anticoagulant activity in patients who concurrently received pentoxifylline and vitamin K antagonists. When initiating or changing the dosage of pentoxifylline, monitoring of anticoagulant activity in these patients is recommended.
Pentoxifylline may enhance the hypotensive effect of antihypertensive agents and other drugs that may cause a reduction in arterial blood pressure.
Concomitant use of pentoxifylline and theophylline in some patients may lead to increased theophylline blood levels. Therefore, an increased frequency and severity of theophylline-related adverse reactions may occur.
In some patients, concomitant use of the drug with ciprofloxacin may lead to increased serum concentrations of pentoxifylline. As a result, the frequency and severity of adverse reactions associated with concomitant use of the drugs may increase.
Potential additive effect with platelet aggregation inhibitors: due to an increased risk of bleeding, concomitant use of platelet aggregation inhibitors (e.g., clopidogrel, eptifibatide, tirofiban, epoprostenol, iloprost, abciximab, anagrelide, non-steroidal anti-inflammatory drugs (NSAIDs), except selective COX-2 inhibitors, acetylsalicylates (acetylsalicylic acid (ASA)/lysine acetylsalicylate (LAS)), ticlopidine, dipyridamole) with pentoxifylline should be administered with caution.
Concomitant use with cimetidine may increase plasma concentrations of pentoxifylline and metabolite I.
Pentoxifylline should not be used concomitantly with ketorolac due to an increased risk of bleeding and/or prolonged prothrombin time.
Special precautions for use
At the first signs of an anaphylactic/anaphylactoid reaction, treatment with the drug should be discontinued immediately.
When administering pentoxifylline to patients with chronic heart failure, circulatory compensation should be achieved prior to treatment.
In patients with diabetes mellitus receiving insulin or oral antidiabetic agents, high doses of the drug may potentiate the blood glucose-lowering effect of these agents. In such cases, the dose of insulin or oral antidiabetic agents should be reduced, and particularly careful monitoring of the patient is required.
Pentoxifylline may be prescribed to patients with systemic lupus erythematosus or other connective tissue disorders only after a thorough assessment of potential risks and benefits.
Since there is a risk of developing aplastic anemia during pentoxifylline therapy, regular monitoring of complete blood counts is required.
In patients with renal impairment (creatinine clearance below 30 mL/min) or severe hepatic dysfunction, elimination of pentoxifylline may be delayed. Appropriate monitoring is necessary.
Particular caution and close monitoring are required in patients:
- with cardiac arrhythmias;
- with myocardial infarction;
- with arterial hypotension;
- with severe atherosclerosis of cerebral and coronary vessels, especially in the presence of concomitant arterial hypertension and cardiac rhythm disorders (in these patients, treatment with the drug may provoke angina attacks, arrhythmias, and arterial hypertension);
- with renal impairment (creatinine clearance below 30 mL/min);
- with severe hepatic insufficiency;
- with a high predisposition to bleeding, e.g., due to anticoagulant therapy or coagulation disorders;
- with a history of gastric or duodenal ulcer, or who have recently undergone surgery (increased risk of bleeding, requiring regular monitoring of hemoglobin and hematocrit levels);
- in whom a decrease in arterial pressure poses a high risk (e.g., patients with severe ischemic heart disease or stenosis of vessels supplying blood to the brain);
- who are receiving concomitant treatment with pentoxifylline and vitamin K antagonists;
- who are receiving concomitant treatment with pentoxifylline and antidiabetic agents;
- who are receiving concomitant treatment with pentoxifylline and ciprofloxacin (see section "Interaction with other medicinal products and other forms of interaction");
- who are receiving concomitant treatment with pentoxifylline and theophylline (see section "Interaction with other medicinal products and other forms of interaction").
Important information about excipients
This medicinal product contains 6.2 mg/mL of sodium, which should be taken into account in patients on a sodium-restricted diet.
Use during pregnancy or breastfeeding
Pregnancy
Experience with the use of this medicinal product in pregnant women is insufficient. Therefore, the drug should not be administered during pregnancy.
Breastfeeding period
Pentoxifylline passes into breast milk in small amounts. Breastfeeding should be discontinued if pentoxifylline treatment is required.
Ability to influence the speed of reactions while driving or operating machinery
No effect.
Method of Administration and Dosage
Intravenous infusions are the most effective and best-tolerated forms of parenteral administration of the medicinal product. The dosage regimen is determined by the physician and depends on the severity of circulatory disorders, body weight, and treatment tolerance. Infusion should be administered only if the solution is clear.
Recommended treatment regimen for adults:
Intravenous infusion of 100–600 mg pentoxifylline once or twice daily. Administration of 100 mg pentoxifylline should last at least 60 minutes.
The infusion may be supplemented with oral pentoxifylline, provided that the total maximum daily dose (infusion and oral) does not exceed 1200 mg.
In severe conditions (especially in cases of persistent pain, gangrene, or trophic ulcers), pentoxifylline infusion may be administered continuously over 24 hours. In such cases, the dose should be calculated at 0.6 mg/kg/hour. The calculated daily dose for a patient weighing 70 kg is 1000 mg; for a patient weighing 80 kg – 1150 mg. Regardless of body weight, the maximum daily dose must not exceed 1200 mg.
The volume of the infusion solution should be individually adjusted according to concomitant diseases and the patient's condition, averaging 1–1.5 L per day.
The duration of parenteral treatment is determined by the treating physician.
After improvement of the patient's condition, continuation of treatment with the tablet form of the drug is recommended.
Method of Administration
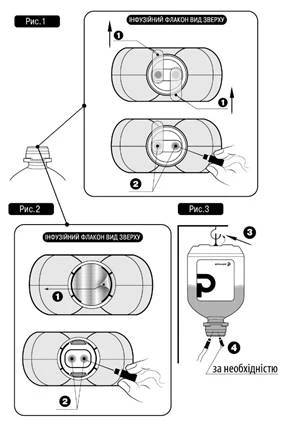
Do not insert the needle(s) into non-designated areas of the polymer bottle; use only sterile ports!
For intravenous infusion therapy, follow this procedure:
- Remove the protective plastic cap with tamper-evident seal (if present).
- Remove protective valve(s) № 1 as shown in Fig. 1 and Fig. 2 (the manufacturer may use different types and materials for protective valves).
- Remove the needle cap and insert the needle into either of the designated ports № 2 of the infusion bottle (see Fig. 1 and Fig. 2).
- The other sterile port may be used either for adding other medicinal products into the infusion bottle (№ 4, see Fig. 3), or, if necessary, for a venting needle to improve flow rate (№ 4, see Fig. 3).
- Hang the solution bottle using the special ring № 3 located at the bottom of the bottle (see Fig. 3).
Children
There is no experience with the use of this medicinal product in children.
Overdose
Symptoms: weakness, dizziness, decreased arterial blood pressure, loss of consciousness, arrhythmia, tachycardia, drowsiness or excitement, coma, hyperthermia, areflexia, tonic-clonic seizures, coffee-ground vomiting as a sign of gastrointestinal bleeding, nausea, flushing.
Treatment: symptomatic. Specific emergency measures to prevent bleeding may be required.
Adverse reactions.
The adverse reactions listed below have been observed during clinical trials and in the post-marketing period. Frequency is unknown.
Eye disorders: vision disorders, lacrimation, conjunctivitis, retinal hemorrhage, retinal detachment, scotoma.
Gastrointestinal disorders: hypersalivation, gastrointestinal disturbances, feeling of pressure in the stomach, nausea, vomiting, anorexia, flatulence, diarrhea, constipation, intestinal atony.
Hepatobiliary disorders: exacerbation of cholecystitis, cholestatic hepatitis, intrahepatic cholestasis.
Nervous system disorders: dizziness, headache, aseptic meningitis, tremor, paresthesia, seizures.
Psychiatric disorders: excitement and sleep disturbances, anxiety, hallucinations.
Cardiovascular disorders: arrhythmia, tachycardia, angina pectoris, cardialgia, fluctuations in blood pressure, chest tightness, hot flushes, bleeding, peripheral edema.
Blood and lymphatic system disorders: thrombocytopenia with thrombocytopenic purpura, aplastic anemia (partial or complete cessation of all blood cell production, pancytopenia), which may be fatal, leukopenia/neutropenia, hypofibrinogenemia.
Immune system disorders: anaphylactic reactions, anaphylactoid reactions, angioneurotic edema, bronchospasm, and anaphylactic shock.
Skin and subcutaneous tissue disorders: pruritus, rash, skin redness, urticaria, toxic epidermal necrolysis, Stevens-Johnson syndrome, increased nail fragility.
General disorders and administration site conditions: cases of hypoglycemia, increased sweating, elevated body temperature, chills.
Laboratory findings: increased transaminase levels, increased alkaline phosphatase levels.
Reporting of suspected adverse reactions.
Reporting suspected adverse reactions after a medicinal product's registration is an important procedure. It enables ongoing monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are required to report any suspected adverse reactions through the national reporting system.
Shelf life. 3 years.
The shelf life is defined as the use of the medicinal product until the last day of the month.
Storage conditions.
Store in the original packaging at a temperature not exceeding 25 °C. Do not freeze.
Keep out of reach and sight of children.
Packaging.
200 ml in bottles.
Prescription category. Prescription only.
Manufacturer. JSC "Pharmaceutical Company "Darnytsia".
Manufacturer's address and place of business.
13, Boryspilska Street, Kyiv, 02093, Ukraine.