Xeomin
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT KSEOMIN (XEOMIN®)
Composition:
Active substance: Clostridium Botulinum neurotoxin type A;
one vial contains 50 LD50 units or 100 LD50 units of botulinum neurotoxin Clostridium Botulinum type A (150 kDa), free from complexing proteins [botulinum neurotoxin type A, purified from cultures of Clostridium Botulinum (Hall strain)];
Excipients: sucrose, human serum albumin.
Pharmaceutical form. Powder for solution for injection.
Main physicochemical properties: white to almost white solid substance.
Pharmacotherapeutic group. Other peripherally-acting muscle relaxants. Botulinum toxin. ATC code M03AX01.
Pharmacological Properties.
Pharmacodynamics.
Botulinum neurotoxin type A blocks cholinergic transmission at neuromuscular synapses by inhibiting the release of acetylcholine. The nerve endings at neuromuscular synapses no longer respond to nerve impulses, thereby interrupting neurotransmitter secretion at the motor end plate (chemical denervation). Restoration of impulse transmission occurs through the formation of new nerve endings and new connections with motor end plates.
Mechanism of action
The mechanism by which botulinum neurotoxin type A exerts its effect on cholinergic nerve endings can be described as a four-step sequential process:
- binding: the heavy chain of botulinum neurotoxin type A binds with exclusively high selectivity and affinity to receptors located only on cholinergic nerve endings;
- internalization: invagination of the nerve ending membrane and uptake of the toxin into the nerve ending (endocytosis);
- translocation: the amino-terminal segment of the neurotoxin's heavy chain forms a pore in the vesicular membrane, the disulfide bond is cleaved, and the light chain of the neurotoxin passes through this pore into the cytosol;
- effect: after release of the light chain, it selectively cleaves the target protein (SNAP-25), which is essential for acetylcholine release.
Complete recovery of end-plate function/impulse transmission after intramuscular injection normally occurs within 3–4 months, during which time nerve endings regenerate and re-establish connections with the motor end plate.
Clinical study results
The objective of two comparative phase III studies with single doses was to demonstrate non-inferior efficacy of Xeomin compared to another product containing the traditional botulinum toxin type A complex – onabotulinumtoxinA (900 kDa): one study in patients with blepharospasm (study MRZ 60201-0003, n = 300), and another in patients with cervical dystonia (study MRZ 60201-0013, n = 463). The results indicate that Xeomin and the comparator product have equivalent efficacy and safety profiles in patients with blepharospasm and in patients with spasmodic torticollis when used at a 1:1 dose conversion ratio.
Blepharospasm
Xeomin was evaluated in a randomized, double-blind, placebo-controlled, multicenter phase 3 study involving 109 patients with blepharospasm. All patients had a clinical diagnosis of benign essential blepharospasm, a baseline score of ≥ 2 on the Jankovic Rating Scale (JRS) severity subscale, and a stable therapeutic response after prior treatment with onabotulinumtoxinA (Botox).
Patients were randomized (2:1) to receive a single dose of Xeomin (n = 75) or placebo (n = 34) at a dose similar (+/− 10 %) to the doses used during the two most recent treatment sessions with onabotulinumtoxinA prior to study entry. The maximum allowed dose in the study was 50 units per eye; the mean dose of Xeomin was 32 units per eye.
The primary efficacy endpoint was change in the JRS severity subscale score from baseline up to week 6 after injection in the intent-to-treat (ITT) population, with missing values replaced by the last observed value for that patient (last observation carried forward). The difference between the Xeomin group and the placebo group in change from baseline on the JRS severity subscale up to week 6 in the ITT population was −1.0 (95 % CI: −1.4; −0.5) points and was statistically significant (p < 0.001).
Patients who required another injection could participate in an extension of this study. Patients received up to five injections of Xeomin with a minimum interval of at least 6 weeks between two injections (total study duration: 48–69 weeks, maximum dose: 50 units per eye). Throughout the study, the median interval between injections in patients receiving NT 201 ranged between 10.14 (1st interval) and 12.00 weeks (2nd and 5th intervals).
Cervical dystonia
Xeomin was evaluated in a randomized, double-blind, placebo-controlled, multicenter phase III study involving 233 patients with cervical dystonia. All patients had a clinical diagnosis of predominantly rotational cervical dystonia and a baseline total score of ≥ 20 on the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS). Patients were randomized (1:1:1) to receive a single injection of Xeomin at a dose of 240 units (n = 81), Xeomin at a dose of 120 units (n = 78), or placebo (n = 74). The number and injection sites were determined by the investigator.
The primary efficacy variable was the change from baseline in the mean square root transformed TWSTRS score up to week 4 after injection in the ITT population, with missing values replaced by the patient's baseline value (full analysis model). Changes from baseline in the TWSTRS score up to week 4 after injection were significantly greater in the NT 201 groups compared to the placebo group (p < 0.001 for all statistical models). Furthermore, these differences were clinically meaningful, e.g., −9.0 points for the 240-unit group compared to placebo and −7.5 points for the 120-unit group compared to placebo in the full analysis model.
Patients who required another injection could participate in an extension of this study. Patients received up to five injections of Xeomin at doses of 120 or 240 units with a minimum interval of at least 6 weeks between two injections (total study duration: 48–69 weeks). Throughout the study, the median interval between injections in patients receiving NT 201 ranged between 10.00 (1st interval) and 13.14 weeks (3rd and 6th intervals).
Upper limb spasticity (in adults)
In a core (double-blind, placebo-controlled, multicenter) study involving patients with upper limb spasticity after stroke, 148 participants were randomized to receive Xeomin (n = 73) or placebo (n = 75) according to dosing recommendations for initial treatment provided in the section "Posology and method of administration". The cumulative dose after up to 6 repeated treatment cycles within the clinical study averaged 1333 units (maximum 2395 units) over a period of up to 89 weeks.
According to the predefined primary efficacy endpoint (response rate in wrist flexor muscles assessed by the Ashworth scale at week 4, with response defined as an improvement of at least 1 point on the 5-point Ashworth scale), patients receiving Xeomin (response rate: 68.5 %) were 3.97 times more likely to achieve a therapeutic response compared to those receiving placebo (response rate: 37.3 %; 95 % CI: 1.90 to 8.30; p < 0.001, ITT population).
Although fixed doses in this study were not designed to determine differences in treatment effect between women and men, a retrospective analysis showed that the response rate was higher in women (89.3 %) than in men (55.6 %), but the difference reached statistical significance only for women. However, in male patients in the Xeomin group, the Ashworth scale response rate at week 4 was consistently higher across all treated muscle groups compared to the placebo group.
In the open-label extension phase of this core study (during which flexible dosing was allowed) involving 145 patients who received up to 5 injections, the positive response rate to treatment was similar in men and women; a similar conclusion was drawn from a masked investigator data analysis study (EudraCT number 2006-003036-30), in which the efficacy and safety of Xeomin in two dilutions were evaluated in 192 patients with upper limb spasticity of various etiologies.
In another double-blind, placebo-controlled, phase III clinical study, 317 patients with upper limb spasticity after stroke (occurring at least 3 months prior) who had not previously received treatment were enrolled. During the main study phase, patients received a fixed total dose of Xeomin (400 units) administered intramuscularly into predefined primary sites (flexed elbow, flexed wrist, or clenched fist) and other affected muscle groups (n = 210). Confirmatory analysis of primary and co-primary efficacy variables at week 4 after injection demonstrated statistically significant improvements in responder status on the Ashworth scale and in changes from baseline on the Ashworth scale and the investigator's global impression of change scale.
A total of 296 patients completed treatment during the main phase and participated in the first cycle of the open-label clinical trial. Continuing the study, patients received up to three injections. Each open-label trial cycle consisted of one treatment session (administration of Xeomin at a total dose of 400 units, evenly distributed among all affected muscles) followed by a 12-week observation period. The total study duration was 48 weeks.
Treatment of shoulder muscles was investigated during an open-label phase III clinical trial involving 155 patients with clinical need for treatment of combined upper and lower limb spasticity. The study protocol allowed administration of up to 600 units of Xeomin into the upper limb.
This study demonstrated a positive relationship between increasing Xeomin doses and improvement in patient outcomes on the Ashworth scale and other efficacy variables, without compromising patient safety or Xeomin tolerability.
Glabellar frown lines (moderate to severe at maximum frown)
In efficacy studies of Xeomin for glabellar frown lines, a total of 994 patients with moderate to severe glabellar frown lines at maximum frown participated. Of these, 169 patients (≥ 18 years) received Xeomin during the main phase of a core double-blind, placebo-controlled phase III study, and 236 patients were treated during the open-label extension phase of this study. Treatment efficacy for dynamic wrinkles was defined as "none" or "mild" on a 4-point scale, assessed by the investigator at week 4 at maximum frown. The study demonstrated statistically and clinically significant efficacy of 20 units of Xeomin compared to placebo. The overall response rate was 51.5 % in the Xeomin group versus 0 % in the placebo group. During the core study, no worsening was observed in any patient receiving Xeomin. This was confirmed by a higher number of responders at day 30 based on assessment of facial dynamic wrinkles at maximum frown. Significantly more patients showed a positive treatment response among those receiving 20 units of Xeomin compared to the placebo group, as assessed by both investigators and patients.
Subgroup analysis showed that efficacy was lower in patients aged 50 years and older compared to younger patients. Of these, 113 patients were under 50 years of age and 56 were over 50 years of age. Efficacy in men was lower than in women. Thirty-three patients were male and 136 were female.
Therapeutic equivalence of Xeomin was demonstrated in a comparative study with comparator products Vistabel/Botox, which contained the botulinum toxin type A complex (onabotulinumtoxinA, 900 kDa), in two prospective, multicenter, randomized, double-blind comparative studies (n = 631) using single doses (20 and 24 units, respectively). Results showed that Xeomin and the comparator products have equivalent efficacy and safety profiles in patients with glabellar frown lines (moderate to severe) when used at a 1:1 dose conversion ratio (see section "Posology and method of administration").
Safety with long-term repeated use (20 units) for treatment of glabellar frown lines was demonstrated in a phase III study over a treatment period of up to two years with up to 8 consecutive injection cycles (MRZ 60201-0609, n = 796).
Lateral periorbital lines (moderate to severe at maximum smile) ("crow's feet")
In a phase III study, 111 patients with moderate to severe lateral periorbital lines ("crow's feet") at maximum smile were treated over one cycle with 12 units of Xeomin or placebo administered to one side (right/left eye area), comparing 3-point and 4-point injection patterns. Treatment success was defined as improvement of at least 1 point on a 4-point scale, assessed by an independent expert at week 4 using standardized digital photographs of both eyes taken at maximum smile, compared to baseline. Both 3-point and 4-point injection patterns demonstrated superiority over placebo. With the 3-point injection pattern, the success rate was 69.9 % in the Xeomin group versus 21.4 % in the placebo group, and with the 4-point pattern, it was 68.7 % versus 14.3 %, respectively. No worsening was observed in any patient treated with Xeomin. This was confirmed by a higher number of responders at day 30 based on assessment on a 4-point scale at maximum smile. Significantly more patients showed a positive treatment response among those receiving 12 units of Xeomin in one eye area compared to those receiving placebo, as assessed by both investigators and patients.
Upper facial dynamic lines
Efficacy and safety of Xeomin at doses from 54 to 64 units in combined treatment of upper facial dynamic lines (glabellar frown lines, lateral periorbital lines, and horizontal forehead lines) were evaluated in a placebo-controlled phase III study involving 156 patients. Treatment efficacy for wrinkles was assessed as "none" or "mild" at maximum frontalis muscle contraction, evaluated by the investigator using a 5-point Merz aesthetic scale. Analysis demonstrated statistically significant differences in treatment and a high response rate to Xeomin for glabellar frown lines, lateral periorbital lines, and horizontal forehead lines individually, as well as for combined treatment of all areas.
Overall, 82.9 % of patients receiving Xeomin had a positive treatment response for glabellar frown lines, whereas no patient responded to placebo. A treatment response for lateral periorbital lines was observed in 63.8 % of patients receiving Xeomin versus 2.0 % of patients receiving placebo. Overall, 71.4 % of patients receiving Xeomin had a positive treatment response for horizontal forehead lines, whereas only one patient (2.0 %) responded to placebo. When treating all three areas simultaneously, a positive response was observed in the majority of patients in the Xeomin group (54.3 %), whereas no response was observed in the placebo group (0.0 %).
Long-term safety and tolerability of 54–64 units of Xeomin were demonstrated in a prospective open-label phase III study with repeated dosing over a treatment period of more than one year with four consecutive injection cycles, involving 125 patients with moderate or severe upper facial dynamic lines.
Children
The European Medicines Agency has waived the obligation to submit results of Xeomin studies in all pediatric subpopulations for the treatment of muscle-related wrinkles, dystonia, and in children aged 0 to 24 months for the treatment of muscle spasticity (see section "Posology and method of administration" regarding use in children).
Pharmacokinetics.
General characteristics of the active substance
Classical pharmacokinetic and distribution studies of botulinum neurotoxin type A have not been conducted because the active substance is administered in very small quantities (picograms per injection) and rapidly and irreversibly binds to cholinergic nerve endings.
The native botulinum toxin is a high-molecular-weight complex that, in addition to the neurotoxin (150 kDa), contains other non-toxic proteins such as hemagglutinin and non-hemagglutinins. In contrast to traditional products containing the botulinum toxin type A complex, Xeomin contains the pure (150 kDa) neurotoxin, as it is free from complexing proteins and thus has a low content of foreign protein. The amount of foreign protein administered is considered one of the factors contributing to secondary treatment failure.
Like many other proteins, botulinum neurotoxin type A has been shown to undergo retrograde axonal transport after intramuscular injection. However, retrograde transsynaptic migration of active botulinum neurotoxin type A into the central nervous system is not observed when therapeutically relevant doses are used.
Botulinum neurotoxin type A bound to receptors undergoes endocytosis into nerve endings before reaching its target (SNAP-25), followed by intracellular degradation. Free circulating molecules of botulinum neurotoxin type A that do not bind to presynaptic receptors of cholinergic nerve endings are subject to phagocytosis and pinocytosis and degrade like any other free circulating proteins.
Distribution of the active substance in patient's body
Pharmacokinetic studies of Xeomin in humans have not been conducted for the reasons stated above.
Preclinical safety data
Preclinical data revealed no special hazard for humans based on standard safety pharmacology studies for the cardiovascular and gastrointestinal systems.
Toxicity findings from repeated-dose toxicity studies of Xeomin in animals related primarily to its pharmacodynamic effects, i.e., atony, paresis, and atrophy of injected muscles.
No signs of local intolerance were observed. Toxicological studies of Xeomin on reproductive function showed no adverse effects on fertility in male and female rabbits or direct effects on embryo/fetus or pre- and postnatal development in rats and/or rabbits. However, daily, weekly, or biweekly administration of Xeomin in embryotoxicity studies at doses causing reduced body weight in females increased the number of abortions in rabbits and slightly reduced fetal body weight in rats. Continuous systemic administration to female rabbits during (unknown) sensitive phases of organogenesis as a prerequisite for teratogenic effects was not required in these studies.
Accordingly, safety margins relative to clinical therapy were generally low in terms of high clinical doses.
Genotoxicity and carcinogenicity studies of Xeomin have not been conducted.
Clinical Characteristics.
Indications.
Xeomin is indicated for the temporary reduction of moderate to severe glabellar lines (vertical frown lines between the eyebrows) visible at maximum frowning, lateral periorbital lines (crow’s feet) visible at maximum smile, and/or horizontal forehead lines visible at maximum frontalis muscle contraction, in adult patients up to 65 years of age, when the marked appearance of these lines has a significant psychological impact on the patient; for symptomatic treatment in adults of blepharospasm, predominantly rotational-type cervical dystonia (spasmodic torticollis), and upper limb muscle spasticity.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients of the product; generalized disorders of muscular activity (e.g. myasthenia gravis, Lambert–Eaton syndrome); infection or inflammation at the intended injection site.
Interaction with other medicinal products and other forms of interaction.
Interaction studies have not been conducted.
The effect of botulinum neurotoxin may theoretically be enhanced by aminoglycoside antibiotics or other medicinal products that interfere with neuromuscular transmission, such as muscle relaxants of the tubocurarine type.
Therefore, concomitant use of Xeomin with aminoglycosides or spectinomycin requires special caution. Muscle relaxants with a peripheral mechanism of action should be used carefully, with a possible reduction in the initial dose of the muscle relaxant or by using intermediate-acting agents such as vecuronium or atracurium instead of long-acting substances.
4-Aminoquinolines may reduce the efficacy of Xeomin.
Special precautions for use
General information
Prior to administration of Xeomin, the physician should be familiar with the patient's anatomy and any anatomical changes due to previous surgical procedures.
Care should be taken to avoid injecting Xeomin into a blood vessel.
It should be noted that horizontal forehead wrinkles are not only caused by active facial expressions but may also result from loss of skin elasticity (e.g., due to aging or photo-damage). In such cases, patients may not respond to botulinum toxin products.
Xeomin should be used with caution in the following cases:
- Presence of any coagulation disorders;
- Patient use of anticoagulants or other substances that may have anticoagulant effects.
The clinical effects of botulinum neurotoxin type A may increase or decrease after repeated injections. Possible reasons for changes in clinical effects include different reconstitution methods, intervals between injections, injected muscles, and inherent variability in toxin potency resulting from biological testing or secondary non-response.
Local and distant spread of toxin effect
Adverse effects may occur due to injections of botulinum neurotoxin type A into incorrectly selected sites, potentially causing temporary paralysis of adjacent muscle groups. When used for non-cosmetic neurological indications, higher doses may cause paralysis in muscles distant from the injection site.
There have been reports of adverse effects potentially related to the spread of botulinum toxin to areas distant from the injection site (see section "Adverse reactions"). Some of these effects may be life-threatening, and fatal outcomes have been reported, sometimes associated with dysphagia, pneumonia, and/or severe exhaustion.
Excessive muscle weakness may occur in patients receiving therapeutic doses.
Patients or caregivers should be advised to seek immediate medical attention if symptoms of swallowing, speech, or breathing difficulties occur.
Dysphagia has also been reported following injections into areas unrelated to neck musculature.
Pre-existing neuromuscular disorders
There is a risk of excessive muscle weakness in patients with neuromuscular disorders. Botulinum toxin products should be administered to such patients only under specialist supervision and only when the therapeutic benefit outweighs the risk. Treatment of patients with a history of dysphagia and aspiration should be performed with particular caution.
Xeomin is not recommended for aesthetic use in patients with a history of dysphagia or aspiration.
Xeomin should be used with caution in:
- Patients with amyotrophic lateral sclerosis (ALS);
- Patients with other disorders leading to peripheral neuromuscular dysfunction;
- Patients with marked weakness or atrophy in the target muscles.
Hypersensitivity reactions
Hypersensitivity reactions have been reported with the use of botulinum neurotoxin products. In the event of serious (e.g., anaphylactic) and/or immediate hypersensitivity reactions, appropriate treatment should be initiated.
Antibody formation
Frequent use of the product increases the risk of antibody formation, which may lead to treatment failure (see section "Dosage and administration").
The possibility of antibody formation may be minimized by using the lowest effective dose and the longest clinically appropriate intervals between injections.
Indications
Benign essential blepharospasm
Injections near the muscle that elevates the upper eyelid should be avoided to reduce the risk of ptosis. Diffusion of botulinum neurotoxin type A into the inferior oblique muscle may result in diplopia. The frequency of this adverse reaction may be reduced by avoiding medial injections into the lower eyelid.
Due to the anticholinergic effects of botulinum neurotoxin type A, Xeomin should be used with caution in patients at risk of developing angle-closure glaucoma.
To prevent ectropion, injections into the lower eyelid should be avoided, and any epithelial defect should be treated intensively. This may require protective eye drops, ointments, therapeutic soft contact lenses, or eye patching or other protective measures.
Reduced blinking after injection of Xeomin into the orbicularis oculi muscle may lead to corneal exposure, persistent epithelial defects, and corneal ulceration, particularly in patients with cranial nerve disorders (e.g., facial nerve palsy). In patients with a history of eye surgery, careful assessment of corneal sensitivity is recommended.
Bruising may easily occur in the soft tissues of the eyelid. Applying gentle pressure to the injection site immediately after injection may reduce this risk.
Cervical dystonia
Xeomin should be administered cautiously near sensitive structures such as the carotid artery, lung apices, or esophagus.
Patients with akinesia or those leading a sedentary lifestyle should be informed about the importance of gradual restoration of mobility after Xeomin injection.
Patients should be informed that Xeomin injections for the treatment of cervical dystonia may lead to mild to severe dysphagia, with risk of aspiration and respiratory compromise. Medical intervention (e.g., nasogastric feeding) may be required in such cases (see also section "Adverse reactions"). Limiting the dose injected into the sternocleidomastoid muscle to 100 units reduces the likelihood of dysphagia. Patients with reduced neck muscle mass or those requiring bilateral injections into the sternocleidomastoid muscle are at higher risk. Dysphagia is believed to result from the spread of the pharmacological effect of Xeomin due to diffusion of the neurotoxin into esophageal muscles.
Upper limb spasticity
Xeomin should be administered cautiously near sensitive structures such as the carotid artery, lung apices, or esophagus.
Patients with akinesia or those leading a sedentary lifestyle should be informed about the importance of gradual restoration of mobility after Xeomin injection.
Xeomin for the treatment of focal spasticity has been studied in combination with standard medical care and is not intended as a substitute for these therapies. Xeomin is unlikely to be effective in improving range of motion in a joint affected by fixed contracture.
There have been reports of new or recurrent seizures, typically in patients predisposed to such events. A definitive causal relationship between these events and botulinum toxin injection has not been established.
Use during pregnancy or breastfeeding
Pregnancy
Adequate data on the use of botulinum neurotoxin type A in pregnant women are lacking. Animal studies have shown toxic effects on reproductive function (see also section "Preclinical safety data"). The potential risk to humans is unknown. Therefore, Xeomin should not be used during pregnancy unless clearly necessary and only if the potential benefit justifies the potential risk.
Lactation
It is unknown whether botulinum neurotoxin type A is excreted in human milk. Therefore, Xeomin should not be used during breastfeeding.
Fertility
There are no clinical data on the effect of botulinum neurotoxin type A on human fertility. In rabbits, no adverse effects on fertility in males or females were observed (see also section "Preclinical safety data").
Ability to influence reaction speed when driving or operating machinery
Xeomin has no or negligible influence on the ability to drive or operate machinery. However, patients should be advised to avoid driving and other potentially hazardous activities if they experience asthenia, muscle weakness, dizziness, visual disturbances, or eyelid ptosis.
Method of Administration and Dosage.
Due to differences in units used to define biological activity, the unit dose of Xeomin is not interchangeable with units of other botulinum toxin products.
For detailed information on clinical studies comparing Xeomin with traditional botulinum toxin type A complexes (900 kDa), see the section "Pharmacodynamics".
Xeomin may only be administered by physicians who have specialized training and experience in handling botulinum toxin type A.
The optimal dosage, frequency, and number of injection sites into muscles must be determined individually by the physician for each patient. Dose titration is required.
The recommended single doses of Xeomin must not be exceeded.
The reconstituted solution of Xeomin is administered by intramuscular injection.
After reconstitution, Xeomin should be used immediately and must be used only during a single procedure and for only one patient.
Glarellar lines (vertical frown lines between the eyebrows, visible at maximum frowning)
Dosage
After reconstitution of Xeomin (50 units/1.25 mL), the recommended injection volume of 0.1 mL (4 units) is administered into each of 5 injection sites: two injections into each corrugator supercilii muscle and one injection into the procerus muscle, corresponding to a total standard dose of 20 units. The dose may be increased by the physician up to 30 units if necessary to meet individual patient needs, with treatment intervals of at least 3 months between sessions.
Reduction in the severity of vertical frown lines (glabellar lines) generally occurs within 2–3 days, with maximum effect observed by day 30. The effect lasts up to 4 months after injection.
Method of Administration
The reconstituted Xeomin solution is administered using a fine sterile needle (e.g., 30–33G / diameter 0.20–0.30 mm / length 13 mm).
Before and during injection, the index or middle finger should be firmly pressed below the orbital rim to prevent diffusion of the solution into this area. The needle should be directed upward and medially. To reduce the risk of blepharoptosis, injections near the levator palpebrae superioris muscle and in the cranial portion of the orbicularis oculi muscle should be avoided. Injections into the corrugator supercilii muscle should be administered into the central portion of the muscle and at least 1 cm above the supraorbital ridges.
Lateral periorbital lines (crow's feet), visible at maximum smile
Dosage
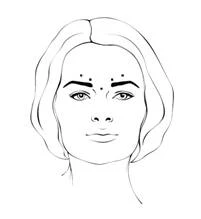
After reconstitution of Xeomin (50 units/1.25 mL), the recommended injection volume of 0.1 mL (4 units) is administered bilaterally into each of 3 injection sites. One injection of 0.1 mL is administered approximately 1 cm lateral to the bony orbital rim. The other two injections (0.1 mL each) should be administered approximately 1 cm above and below the first injection site.
The total recommended standard dose per treatment session is 12 units per side (total cumulative dose – 24 units).
Reduction of lateral periorbital lines (crow's feet) visible at maximum smile usually occurs within the first 6 days, with maximum effect observed by day 30. The effect lasts up to 4 months after injection.
There are currently no data on the efficacy and safety of more than two injections with a 4-month interval for lateral periorbital lines visible at maximum smile.
Method of Administration
The reconstituted Xeomin solution is administered using a fine sterile needle (e.g., 30–33G / diameter 0.20–0.30 mm / length 13 mm). The injection should be administered intramuscularly into the orbicularis oculi muscle directly beneath the dermis to prevent spread of Xeomin. Injections very close to the zygomaticus major muscle should be avoided to prevent lip ptosis.
For treatment of glabellar lines and crow's feet, treatment intervals must be at least 3 months. If treatment is ineffective or the effect of repeated injections diminishes, alternative treatment methods should be considered.
Horizontal forehead lines, visible at maximum frontalis muscle contraction
Dosage
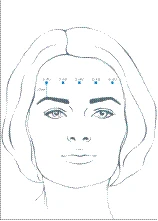
Depending on individual patient needs, the recommended total dose range is 10 to 20 units, with treatment intervals of at least 3 months between sessions. After reconstitution, a total dose of Xeomin from 10 to 20 units is administered into the frontalis muscle at 5 horizontally aligned injection sites, at least 2 cm above the bony orbital rim. Each injection site receives 2, 3, or 4 units of the drug, respectively.
Reduction in forehead lines at maximum frontalis muscle contraction typically occurs within 7 days, with maximum effect observed by day 30. The effect lasts up to 4 months after injection.
Method of Administration
The reconstituted Xeomin solution is administered using a fine sterile needle (e.g., 30–33G / diameter 0.20–0.30 mm / length 13 mm). To prevent brow ptosis, injections of Xeomin near the orbital rim should be avoided, as this may cause paralysis of the lower muscle fibers.
Treatment intervals must be at least 3 months. If treatment is ineffective or the effect of repeated injections diminishes, alternative treatment methods should be considered.
Blepharospasm
Dosage
The recommended initial dose is 1.25–2.5 units per injection site. The initial dose should not exceed 25 units per eye. The total dose should not exceed 100 units per eye per treatment session. Repeat treatment can generally be performed no more frequently than every 12 weeks. The intervals between treatment sessions should be determined based on the actual clinical needs of the individual patient.
The effect of the drug begins on average within 4 days after injection. The therapeutic effect of Xeomin typically lasts 3–4 months, although it may last significantly longer or shorter. Treatment may be repeated as needed.
If the effect of the initial dose is insufficient, the dose may be doubled during subsequent procedures. However, it has been demonstrated that no additional benefit is achieved from injecting more than 5.0 units into a single site.
Method of Administration
After reconstitution, Xeomin is administered using an appropriate sterile needle (e.g., 27–30G / diameter 0.30–0.40 mm / length 12.5 mm). Electromyographic guidance is not required during the procedure. The recommended volume per single injection is approximately 0.05–0.1 mL.
Xeomin is administered into the medial and lateral portions of the orbicularis oculi muscle of the upper eyelid and into the lateral portion of the orbicularis oculi muscle of the lower eyelid. If vision is impaired due to spasms, additional injections may be administered in the forehead area, lateral portions of the orbicularis oculi muscle, and upper facial region.
Cervical dystonia (spasmodic torticollis)
Dosage
For treatment of cervical dystonia, the dosage of Xeomin should be individually adjusted for each patient depending on head and neck posture, location of pain, muscle hypertrophy, patient body weight, and response to injections.
The recommended dose per injection site is up to 50 units, and the maximum dose for the first procedure is 200 units. The physician may prescribe doses up to 300 units during subsequent sessions, depending on the individual patient's response.
The effect of the drug usually begins within 7 days after injection. The effect of each Xeomin treatment session lasts approximately 3–4 months, although it may last significantly longer or shorter. The recommended interval between procedures is at least 10 weeks. The intervals between treatment sessions should be determined based on the actual clinical needs of the individual patient.
Method of Administration
For injections into superficial muscles, appropriate sterile needles should be used, e.g., 25–30G / diameter 0.30–0.50 mm / length 37 mm; for deep muscles, needles such as 22G / diameter 0.70 mm / length 75 mm may be used. The recommended volume per single injection site is approximately 0.1–0.5 mL.
Treatment of cervical dystonia includes injections of Xeomin into the sternocleidomastoid muscle, the levator scapulae muscle, the splenius muscles, the scalene muscles, and/or the trapezius muscles. This list is not exhaustive, as any muscle involved in controlling head position may be affected by the pathological process and require treatment. If difficulty arises in identifying individual affected muscles, electromyography or ultrasound imaging may be required. Muscle mass and degree of hypertrophy or atrophy are factors that must be considered when selecting the appropriate dose.
Injecting into multiple sites allows Xeomin to evenly cover the innervated areas of muscles prone to dystonia (especially when injecting into large muscles). The optimal number of injection sites depends on the size of the muscle to be chemically denervated.
Bilateral injections into the sternocleidomastoid muscle should not be administered, as there is an increased risk of adverse events (particularly dysphagia) if bilateral injections or doses administered into this muscle exceed 100 units.
Upper limb spasticity
Dosage
The precise dosage and number of injection sites should be individually determined for each patient depending on the size, number, and location of affected muscles, severity of spasticity, and presence of localized muscle weakness.
Recommended therapeutic doses by muscle type:
| Clinical target |
Units (range) |
Number of injection sites in one muscle |
|
| Wrist flexion |
|||
| Flexor carpi ulnaris (Flexor carpi ulnaris) |
20−100 |
1−2 |
|
| Clenched fist |
|||
| Flexor digitorum superficialis (Flexor digitorum superficialis) |
25−100 |
2 |
|
| Flexor digitorum profundus (Flexor digitorum profundus) |
25−100 |
2 |
|
| Elbow flexion |
|||
| Brachioradialis |
25−100 |
1−3 |
|
| Biceps brachii |
50−200 |
1−4 |
|
| Brachialis |
25−100 |
1−2 |
|
| Pronated forearm |
|||
| Pronator quadratus (Pronator quadratus) |
10−50 |
1 |
|
| Pronator teres (Pronator teres) |
25−75 |
1−2 |
|
| Thumb adducted to palm |
|||
| Flexor pollicis longus (Flexor pollicis longus) |
10−50 |
1 |
|
| Adductor pollicis (Adductor pollicis) |
5−30 |
1 |
|
| Flexor pollicis brevis / Opponens pollicis |
5−30 |
1 |
|
| Shoulder internal rotation/abducted shoulder/adducted shoulder |
|||
| Deltoideus, clavicular part (Deltoideus, pars clavicularis) |
20−150 |
1−3 |
|
| Latissimus dorsi (Latissimus dorsi) |
25−150 |
1−4 |
|
| Pectoralis major (Pectoralis major) |
20−200 |
1−6 |
|
| Subscapularis (Subscapularis) |
15−100 |
1−4 |
|
| Teres major (Teres major) |
20−100 |
1−2 |
|
The maximum total dose for treatment of upper limb spasticity should not exceed 500 units per treatment session, and the dose for shoulder muscles should not exceed 250 units.
The effect of the medicinal product begins, according to patient reports, 4 days after injection. The maximum effect, considered as improvement in muscle tone, is felt within 4 weeks. Overall, the treatment effect lasts 12 weeks, although it may last significantly longer or shorter. Repeated treatment can generally be performed no more frequently than every 12 weeks. The intervals between treatment procedures should be determined based on the actual clinical needs of the individual patient.
Method of administration
The reconstituted solution of Xeomin should be administered by injection using an appropriate sterile needle (e.g., a 26G/0.45 mm diameter/37 mm length needle for superficial muscles and a longer needle, e.g., 22G/0.7 mm diameter/75 mm length, for deep muscles).
If difficulties arise in identifying individual affected muscles, injections should be performed under electromyographic or ultrasound guidance. Administering injections at multiple sites allows Xeomin to evenly cover the muscle innervation areas, which is particularly important when injecting into large muscles.
Special patient groups
Clinical data from Phase III studies regarding the use of Xeomin in patients aged 65 years and older are limited. Until further studies are conducted in this age group, Xeomin is not recommended for use in patients over 65 years of age.
All indications
Treatment of glabellar lines, "crow's feet," and horizontal forehead lines
If no treatment effect is observed within one month after the first injection, the following measures should be taken:
- Analyze the reasons for lack of response to treatment, e.g., very low dose, poor injection technique, possible formation of neutralizing neurotoxin antibodies;
- Adjust the dose based on analysis of the last unsuccessful treatment attempt;
- Re-evaluate the suitability of treatment with botulinum neurotoxin type A;
- If no adverse events were observed during the first treatment cycle, an additional treatment course may be administered, provided that a minimum interval of 3 months is maintained between the initial and repeat treatment courses.
Treatment of blepharospasm, spasmodic torticollis, and upper limb spasticity
If no treatment effect is observed within one month after the first injection, the following measures should be taken:
- Reassess clinical confirmation of the neurotoxin's effect on the muscle into which the medicinal product was injected, e.g., by performing electromyography at a specialized facility;
- Analyze the reasons for lack of response to treatment, e.g., poor isolation of muscles intended for injection, very low dose, poor injection technique, fixed contracture, very weak antagonist, possible development of antibodies;
- Re-evaluate the suitability of treatment with botulinum neurotoxin type A;
- If no adverse events were observed during the first treatment cycle, an additional treatment course may be administered under the following conditions: 1) dose adjustment based on analysis of the last unsuccessful treatment attempt; 2) localization of affected muscles using electromyography; 3) adherence to the recommended minimum interval between initial and repeat treatment cycles.
Reconstitution and disposal of unused medicinal product
Prior to administration, Xeomin should be reconstituted with 9 mg/mL (0.9%) sodium chloride solution for injection. Reconstitution and dilution must be performed in accordance with good clinical practice recommendations, particularly with regard to aseptic technique.
Reconstitution of the vial contents and syringe preparation should be performed over paper towels with a plastic backing to prevent splashing. The appropriate amount of diluent is drawn into the syringe (see below). After inserting the needle vertically through the rubber stopper, the diluent is gently injected into the vial to avoid foaming. A short conical needle of size 20–27G is recommended for reconstitution. The vial should be discarded if the vacuum prevents injection of the diluent. Remove the syringe from the vial and mix Xeomin with the diluent by gently rotating and inverting, or simply inverting the vial (vigorous shaking is not permitted). If necessary, the needle used for reconstitution may remain in the vial, but the required volume of solution should be drawn using a new sterile injection syringe.
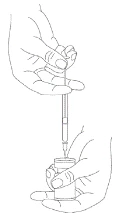
The reconstituted Xeomin medicinal product is a clear, colorless solution free of particulate matter.
Xeomin should not be used if, after reconstitution, the resulting solution is cloudy or contains visible flakes or particles.
The correct volume of diluent for the selected number of units of the medicinal product dose must be carefully determined to prevent accidental overdose. If during a single injection procedure vials of Xeomin of different volumes are used, the correct amount of diluent must be carefully determined during reconstitution to achieve a specific number of units per 0.1 mL. The amount of diluent varies depending on the selected dose of Xeomin, ranging from 50 to 100 units. Each syringe must be appropriately labeled.
For treatment of glabellar lines, "crow's feet," and horizontal forehead lines
Available Xeomin solutions with strengths of 50 and 100 units
| Administered dose (units per 0.1 ml) |
Amount of diluent (9 mg/ml (0.9%) sodium chloride for injection) |
|
| Vial contains 50 units |
Vial contains 100 units |
|
| 5 units |
1 ml |
2 ml |
| 4 units |
1.25 ml |
2.5 ml |
For the treatment of blepharospasm, cervical dystonia, and upper limb spasticity
Possible concentrations of Xeomin with 50 and 100 units of potency
| Administered dose (units per 0.1 ml) |
Volume of diluent (9 mg/ml (0.9%) sodium chloride for injection) |
|
| Vial contains 50 units |
Vial contains 100 units |
|
| 20 units |
0.25 ml |
0.5 ml |
| 10 units |
0.5 ml |
1 ml |
| 8 units |
0.625 ml |
1.25 ml |
| 5 units |
1 ml |
2 ml |
| 4 units |
1.25 ml |
2.5 ml |
| 2.5 units |
2 ml |
4 ml |
| 2 units |
2.5 ml |
5 ml |
| 1.25 units |
4 ml |
Not applicable |
From a microbiological standpoint, the medicinal product should be used immediately. If not used immediately, the duration and conditions of storage of the opened container are the responsibility of the user. Generally, storage should not exceed 24 hours at a temperature of 2 °C to 8 °C, unless reconstitution was performed under controlled and validated aseptic conditions.
Injectable solution stored for longer than 24 hours, as well as any unused injectable solution, must be discarded.
Procedure for safe disposal of vials, syringes, and used materials
Unused vials, residual reconstituted solution in vials, and/or syringes should be autoclaved. Alternatively, residual prepared Xeomin product may be inactivated by adding one of the following solutions: 70% ethanol, 50% isopropanol, 0.1% sodium dodecyl sulfate (SDS) (anionic detergent), diluted sodium hydroxide solution (0.1 N NaOH), or diluted sodium hypochlorite solution (at least 0.1% NaOCl).
After inactivation, used vials, syringes, and materials should not be emptied; they must be placed in appropriate containers and disposed of according to local requirements.
Recommendations in the event of any incident involving botulinum toxin type A
- Spilled or scattered product must be wiped up immediately: using an absorbent material soaked with one of the solutions listed above, in the case of powder, or dry absorbent material, in the case of reconstituted solution;
- Contaminated surfaces must be cleaned with an absorbent material soaked with one of the solutions described above, then dried;
- If a vial breaks, proceed as described above, carefully collect broken glass fragments, and wipe up spilled or scattered product, avoiding skin cuts;
- In case of contact with skin, wash the affected area thoroughly with large amounts of water;
- If the product enters the eyes, rinse thoroughly with large amounts of water or eye irrigation solution;
- If the product enters a wound, cut, or damaged skin area, rinse thoroughly with large amounts of water and take appropriate medical measures depending on the administered dose.
Strict adherence to instructions for handling the medicinal product and its disposal procedure is essential.
Children.
The safety and efficacy of Xeomin in children and adolescents under 17 years of age have not yet been established. Therefore, Xeomin should not be used in this patient group until further data are available.
Overdose.
Symptoms of overdose
High doses of botulinum neurotoxin type A may cause pronounced muscle paralysis with various symptoms occurring at sites distant from the injection site. Such symptoms include generalized weakness, ptosis, diplopia, difficulty breathing, difficulty speaking, paralysis of respiratory muscles, difficulty swallowing, which may lead to the development of aspiration pneumonia.
Measures to be taken in case of overdose
In the event of overdose, the patient should be under medical supervision to monitor for possible development of excessive muscle weakness or muscle paralysis. The patient may require symptomatic treatment. In case of paralysis of respiratory muscles, respiratory support is necessary.
Adverse Reactions
Undesirable effects usually occur during the first week after treatment and are transient in nature. Adverse effects may be related to the active substance, the injection procedure, or both factors.
Adverse reactions unrelated to indication
Adverse reactions associated with administration of the medicinal product
Localized pain, inflammation, paresthesia, hypoesthesia, tenderness, swelling, edema, erythema, pruritus, localized infection, hematoma, hemorrhage, and/or bruising may be related to the injection procedure.
Pain associated with needle insertion and/or anxiety may lead to parasympathetic vascular reactions, such as transient symptomatic hypotension, nausea, tinnitus, and syncope.
Adverse effects of the class of substances — botulinum toxin type A
Localized muscle weakness is one of the expected pharmacological effects of botulinum toxin.
Toxin spread
When treating with botulinum toxins for other indications, very rare cases of adverse effects related to the spread of the toxin to sites distant from the injection site have been reported, causing symptoms consistent with the effects of botulinum toxin type A (increased muscle weakness, dysphagia, and aspiration pneumonia, with fatal outcomes in some cases) (see section "Special precautions").
The adverse effects listed above cannot be completely excluded when using Xeomin.
Hypersensitivity reactions
Rare cases of serious and/or immediate hypersensitivity reactions, including anaphylaxis, serum sickness, urticaria, soft tissue swelling, and dyspnea have been reported. Some of these reactions occurred after administration of the traditional botulinum toxin type A complex — either alone or in combination with other substances known to cause such reactions.
Adverse reactions observed in clinical practice
The information below describes the frequency of adverse reactions observed in clinical practice for specific indications. Frequency is defined as follows: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (cannot be estimated from available data).
Glabellar lines (vertical frown lines between the eyebrows)
Infections and infestations
Uncommon: bronchitis, nasopharyngitis, influenza-like illness.
Psychiatric disorders
Uncommon: depression, insomnia.
Nervous system disorders
Common: headache.
Uncommon: facial paresis (brow ptosis).
Eye disorders
Uncommon: eyelid edema, eyelid ptosis, blurred vision.
Skin and subcutaneous tissue disorders
Uncommon: pruritus, skin nodules, brow ptosis.
Musculoskeletal and connective tissue disorders
Common: "Mephisto brows" (lateral elevation of the eyebrows).
Uncommon: muscle twitching, muscle spasms, facial asymmetry (brow asymmetry), sensation of heaviness.
General disorders and administration site conditions
Uncommon: injection site hematoma, injection site pain, local tenderness, weakness, fatigue, discomfort (feeling of heaviness in eyelids/eyebrows).
Vascular disorders
Uncommon: hematoma.
Latent periorbital lines visible at maximum smile ("crow's feet")
Eye disorders
Common: eyelid edema, dry eyes.
General disorders and administration site conditions
Common: injection site hematoma.
Wrinkles in the upper part of the face
Nervous system disorders
Very common: headache.
Common: hypoesthesia.
General disorders and administration site conditions
Common: injection site hematoma, injection site pain, injection site erythema, discomfort (feeling of heaviness in the forehead).
Eye disorders
Common: eyelid ptosis, dry eyes.
Skin and subcutaneous tissue disorders
Common: brow ptosis.
Musculoskeletal and connective tissue disorders
Common: facial asymmetry, "Mephisto brows" (lateral elevation of the eyebrows).
Gastrointestinal disorders
Common: nausea.
Blepharospasm
Nervous system disorders
Common: headache, facial paresis.
Eye disorders
Very common: eyelid ptosis, dry eyes.
Common: blurred vision, visual disturbance, diplopia, increased lacrimation.
Gastrointestinal disorders
Common: dry mouth, dysphagia.
Skin and subcutaneous tissue disorders
Common: rash.
General disorders and administration site conditions
Common: injection site pain, fatigue.
Musculoskeletal and connective tissue disorders
Common: muscle weakness.
Spastic torticollis
Nervous system disorders
Common: headache, presyncope, dizziness.
Uncommon: speech disorders.
Respiratory, thoracic and mediastinal disorders
Uncommon: dysphonia, dyspnea.
Gastrointestinal disorders
Very common: dysphagia.
Common: dry mouth, nausea.
Skin and subcutaneous tissue disorders
Common: hyperhidrosis.
Uncommon: rash.
Musculoskeletal and connective tissue disorders
Common: neck pain, muscle weakness, muscle pain, muscle spasms, musculoskeletal stiffness.
General disorders and administration site conditions
Common: injection site pain, asthenia.
Infections and infestations
Common: upper respiratory tract infections.
Treatment of spastic torticollis may lead to dysphagia of varying severity with a risk of aspiration, which may sometimes require medical intervention. Dysphagia may persist for 2–3 weeks after injection; however, one case of dysphagia lasting five months has been documented.
Upper limb spasticity
Nervous system disorders
Uncommon: headache, hypoesthesia.
Gastrointestinal disorders
Common: dry mouth.
Uncommon: dysphagia, nausea.
Musculoskeletal and connective tissue disorders
Uncommon: muscle weakness, limb pain, muscle pain.
General disorders and administration site conditions
Uncommon: asthenia.
Common: injection site pain, sensation of warmth.
Not known: injection site pain.
Post-marketing experience
Cases of influenza-like symptoms and hypersensitivity reactions such as swelling, edema (also distant from the injection site), erythema, pruritus, rash (localized and generalized), and dyspnea have been reported. Additionally, with a frequency of "not known", a musculoskeletal adverse reaction independent of indication — muscle atrophy — has been reported.
Reporting suspected adverse reactions
It is important to report suspected adverse reactions after the medicinal product has been authorized. This allows continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are encouraged to report any suspected adverse reactions via the national reporting system.
Shelf life
3 years.
The reconstituted solution may be stored at 2 °C to 8 °C for up to 24 hours.
Storage conditions
Store at a temperature not exceeding 25 °C. Keep out of the reach of children.
Incompatibilities
This medicinal product must not be mixed with other medicinal products except those mentioned in the section "Method of administration and dosage".
Packaging
Powder for solution for injection, 50 LD50 units or 100 LD50 units per vial. One vial per carton.
Prescription category
Prescription only.
Manufacturer
Merz Pharma GmbH & Co. KGaA.
Manufacturer's address and place of business
Ludwigstrasse 22, 64354 Reinheim, Germany.