Lemtrada
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU LEMTRADA (LEMTRADA®)
Skład:
substancja czynna: alemtuzumab;
1,2 ml koncentratu zawiera 12 mg alemtuzumabu;
1 fiolka zawiera 12 mg alemtuzumabu;
substancje pomocnicze: fosforan sodu (E 339); chlorek potasu (E 508); fosforan dwusodowy dihydrazyn; kwas fosforowy dwuchlorek potasu (E 340); polysorbat 80 (E 433); chlorek sodu; woda do wstrzykiwań.
Postać leku. Koncentrat do sporządzenia roztworu do infuzji.
Główne właściwości fizykochemiczne: przezroczysty koncentrat o barwie od bezbarwnej do lekko żółtej, pH 7,0–7,4.
Grupa farmakoterapeutyczna. Immunosupresyjne. Selektiwne immunosupresyjne. Alemtuzumab. Kod ATC L04A A34.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania. Alemtuzumab jest humanizowanym monoklonalnym przeciwciałem, otrzymanym metodą rekombinowanego DNA, skierowanym przeciwko glikoproteinie CD52 znajdującej się na powierzchni komórek i mającej masę cząsteczkową 21–28 kD. Alemtuzumab jest przeciwciałem klasy IgG1 kappa z ludzkimi regionami stałymi i zmiennymi oraz regionami determinującymi komplementarność pochodzącymi od monoklonalnego przeciwciała mysiego (produkowanego w organizmie szczura). Przybliżona masa cząsteczkowa przeciwciała wynosi 150 kD.
Alemtuzumab wiąże się z antygenem CD52 znajdującym się na powierzchni komórek, który występuje w wysokich stężeniach na limfocytach T (CD3+) i B (CD19+) oraz w niższych stężeniach – na komórkach NK, monocytach i makrofagach. CD52 występuje w niewielkich stężeniach lub wcale na neutrofilach, komórkach plazmatycznych i komórkach macierzystych szpiku kostnego. Działanie alemtuzumabu realizuje się poprzez zależny od przeciwciał cytolizys komórek oraz lizys pośrednictwem dopełniacza po wiązaniu się z limfocytami T i B na powierzchni komórek.
Mechanizm, dzięki któremu lek Lemtrada zapewnia swoje efekty terapeutyczne w stwardnieniu rozsianym (SR), nie został jeszcze jednoznacznie wyjaśniony. Badania wskazują jednak na obecność efektów immunomodulujących poprzez eliminację i repopulację limfocytów, w tym:
- zmiany w liczbie, proporcjach i właściwościach niektórych podgrup limfocytów po zastosowaniu leku;
- zwiększenie liczby regulacyjnych podgrup komórek T;
- zwiększenie liczby limfocytów T i B pamięci;
- przejściowy wpływ na komponenty odporności wrodzonej (tj. neutrofile, makrofagi, komórki NK).
Redukcja liczby krążących limfocytów B i T pod wpływem leku Lemtrada oraz ich późniejsza repopulacja mogą zmniejszać potencjał do nawrotów choroby, co ostatecznie spowalnia jej postęp.
Efekty farmakodynamiczne. Lek Lemtrada obniża poziomy krążących limfocytów T i B po każdym cyklu leczenia, przy czym najniższe poziomy tych komórek odnotowano po 1 miesiącu od zakończenia cyklu leczenia (odpowiadało to najwcześniejszemu punktowi czasowemu oceny po leczeniu w badaniach fazy III). W miarę upływu czasu następuje repopulacja limfocytów, przy czym przywrócenie poziomów komórek B zwykle zachodzi w ciągu 6 miesięcy. Poziomy limfocytów CD3+ i CD4+ zwiększają się wolniej, ale zazwyczaj nie wracają do wartości wyjściowych po 12 miesiącach od leczenia. U około 40 % pacjentów całkowita liczba limfocytów osiągała dolną granicę normy (DGN) po 6 miesiącach od każdego cyklu leczenia, a u około 80 % pacjentów całkowita liczba limfocytów osiągała DGN po 12 miesiącach od każdego cyklu leczenia.
Neutrofile, monocyty, eozynofile, bazofile i naturalne komórki zabójcze podlegają jedynie przejściowym zmianom pod wpływem leku Lemtrada.
Skuteczność i bezpieczeństwo kliniczne. Bezpieczeństwo i skuteczność alemtuzumabu w SR oceniano w trzech randomizowanych, ślepych dla oceniającego badaniach klinicznych z użyciem aktywnego środka porównawczego oraz jednym niekontrolowanym, ślepym dla oceniającego, rozszerzonym badaniu z udziałem pacjentów z nawrotno-remitującym stwardnieniem rozsianym (NRSR).
W tabeli 1 przedstawiono informacje dotyczące projektu badań/danych demograficznych pacjentów (badania 1, 2, 3 i 4).
Tabela 1
| Nazwa badania |
Badanie 1 CAMMS323 |
Badanie 2 CAMMS32400507 |
Badanie 3 CAMMS223 |
| Projekt badania |
Kontrolowane, randomizowane, ślepe dla oceniającego |
Kontrolowane, randomizowane, ślepe dla oceniającego i ślepe pod względem dawki |
Kontrolowane, randomizowane, ślepe dla oceniającego |
| Anamneza choroby |
Pacjenci z aktywnym SM, zdefiniowanym jako co najmniej 2 zaostrzenia w ciągu ostatnich 2 lat |
Pacjenci z aktywnym SM, zdefiniowanym jako co najmniej 2 zaostrzenia w ciągu ostatnich 2 lat lub obecność jednego lub więcej ognisk uszkodzenia z akumulacją kontrastu |
|
| Czas trwania |
2 lata |
3 lata‡ |
|
| Populacja uczestników badania |
Pacjenci, którzy wcześniej nie byli leczeni |
Pacjenci, u których stwierdzono niewystarczającą odpowiedź na wcześniejszą terapię* |
Pacjenci, którzy wcześniej nie byli leczeni |
| Charakterystyki wyjściowe |
|||
| Średni wiek (lata) |
33 |
35 |
32 |
| Średnia/mediana czasu trwania choroby |
2,0/1,6 roku |
4,5/3,8 roku |
1,5/1,3 roku |
| Średni czas wcześniejszej terapii SM (z wykorzystaniem ≥1 leku) |
Nie stosowano |
36 miesięcy |
Nie stosowano |
| % pacjentów, którzy wcześniej otrzymali ≥2 kursy leczenia SM |
Nie dotyczy |
28 % |
Nie dotyczy |
| Średnia liczba punktów w skali EDSS (Rozszerzona Skala Niepełnosprawności) na poziomie wyjściowym |
2,0 |
2,7 |
1,9 |
| Badanie 4 |
|||
| Nazwa badania |
CAMMS03409 |
||
| Projekt badania |
Niekontrolowane, ślepe dla oceniającego, badanie rozszerzone |
||
| Populacja uczestników badania |
Pacjenci, którzy brali udział w badaniach CAMMS223, CAMMS323 lub CAMMS32400507 (patrz powyższe dane wyjściowe) |
||
| Czas trwania fazy rozszerzonej |
4 lata |
||
* Określano jako pacjentów, u których wystąpił co najmniej jeden nawrót podczas leczenia interferonem beta lub acetatem glatirameru po podaniu tej terapii przez co najmniej 6 miesięcy.
‡ Pierwotny punkt końcowy badania oceniano po 3 latach. Dodatkowe obserwacje dostarczyły danych dla okresu o medianie trwania 4,8 roku (z maksymalnym okresem obserwacji 6,7 roku).
W tabeli 2 przedstawiono wyniki uzyskane w badaniach 1 i 2.
Tabela 2
| Nazwa badania |
Badanie 1 CAMMS323 (CARE-MS I) |
Badanie 2 CAMMS32400507 (CARE-MS II) |
||
| Punkty końcowe kliniczne |
Lemtrada 12 mg (N = 376) |
IFN-beta-1a s.c. (N = 187) |
Lemtrada 12 mg (N = 426) |
IFN-beta-1a s.c. (N = 202) |
| Częstość nawrotów1 Roczna częstość nawrotów (RCN) (95 % CI) |
0,18 (0,13, 0,23) |
0,39 (0,29, 0,53) |
0,26 (0,21, 0,33) |
0,52 (0,41, 0,66) |
| Stosunek częstości (95 % CI) |
0,45 (0,32, 0,63) 54,9 (p < 0,0001) |
0,51 (0,39, 0,65) 49,4 (p < 0,0001) |
||
| Inwalidzacja1 (potwierdzone pogorszenie niepełnosprawności [PPN])2 |
8,0 % (5,7, 11,2) |
11,1 % (7,3, 16,7) |
12,7 % (9,9, 16,3) |
21,1 % (15,9, 27,7) |
| Stosunek ryzyka (95 % CI) |
0,70 (0,40, 1,23) (p = 0,22) |
0,58 (0,38, 0,87) (p = 0,0084) |
||
| Pacjenci, którzy nie mieli nawrotów przez 2 lata (95 % CI) |
77,6 % (72,9, 81,6) (p < 0,0001) |
58,7 % (51,1, 65,5) |
65,4 % (60,6, 69,7) (p < 0,0001) |
46,7 % (39,5, 53,5) |
| Zmiana liczby punktów w skali EDSS w porównaniu z poziomem wyjściowym po 2 latach3 (95 % CI) |
-0,14 (-0,25, -0,02) (p = 0,42) |
-0,14 (-0,29, 0,01) |
-0,17 (-0,29, -0,05) (p < 0,0001) |
0,24 (0,07, 0,41) |
| Punkty końcowe MRI (0−2 lata) |
||||
| Mediana zmiany w % objętości ognisk uszkodzeń na obrazach MRI ważonych T2 |
-9,3 (-19,6, -0,2) (p = 0,31) |
-6,5 (-20,7, 2,5) |
-1,3 (p = 0,14) |
-1,2 |
| Liczba pacjentów z nowymi ogniskami uszkodzeń lub z zwiększeniem ognisk uszkodzeń na obrazach ważonych T2 po 2 latach |
48,5 % (p = 0,035) |
57,6 % |
46,2 % (p < 0,0001) |
67,9 % |
| Liczba pacjentów z ogniskami uszkodzeń, które gromadzą gadolin, po 2 latach |
15,4 % (p = 0,001) |
27,0 % |
18,5 % (p < 0,0001) |
34,2 % |
| Liczba pacjentów z nowymi ogniskami uszkodzeń, hipointensywnymi na obrazach ważonych T1, po 2 latach |
24,0 % (p = 0,055) |
31,4 % |
19,9 % (p < 0,0001) |
38,0 % |
| Mediana zmiany w % tkanki parenchymatycznej mózgu |
-0,867 (p < 0,0001) |
-1,488 |
-0,615 (p = 0,012) |
-0,810 |
1 Składnikami pierwotnego punktu końcowego były CRR i PPI. Badanie uznawano za udane, jeśli osiągnięto co najmniej jeden z tych dwóch składników pierwotnego punktu końcowego.
2 PPI definiowano jako zwiększenie wyniku co najmniej o 1,0 punktu w skali rozszerzonej stopnia niepełnosprawności (EDSS) w porównaniu z wartością wyjściową skali EDSS (dla pacjentów z wartością wyjściową skali EDSS na poziomie 0 uwzględniano zwiększenie o 1,5 punktu), które utrzymywało się przez 6 miesięcy.
3 Oceny dokonano z wykorzystaniem mieszanego modelu dla powtarzanych pomiarów.
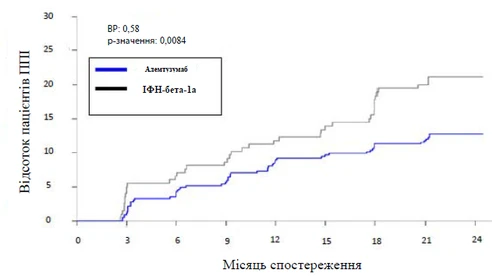
Rys. Czas do potwierdzonego pogorszenia niepełnosprawności (przez okres 6 miesięcy) w badaniu 2
Ciężkość nawrotu
W badaniu wpływu leczenia na częstość nawrotów dodatkowe analizy przeprowadzone w ramach badania 1 (CAMMS323) wykazały, że stosowanie leku Lemtrada w dawce 12 mg/dobę prowadziło do istotnej statystycznie mniejszej liczby pacjentów z ciężkimi nawrotami (zmniejszenie o 61 %, p = 0,0056) oraz do istotnej statystycznie mniejszej liczby nawrotów wymagających stosowania leków steroidowych (zmniejszenie o 58 %, p < 0,0001) w porównaniu z grupą stosującą IFNB-1a.
Dodatkowe analizy przeprowadzone w ramach badania 2 (CAMMS32400507) wykazały, że stosowanie leku Lemtrada w dawce 12 mg/dobę prowadziło do istotnej statystycznie mniejszej liczby pacjentów z ciężkimi nawrotami (zmniejszenie o 48 %, p = 0,0121) oraz do istotnej statystycznie mniejszej liczby nawrotów wymagających stosowania leków steroidowych (zmniejszenie o 56 %, p < 0,0001) lub hospitalizacji (zmniejszenie o 55 %, p = 0,0045) w porównaniu z grupą stosującą IFNB-1a.
Potwierdzona poprawa stopnia niepełnosprawności (PPN)
Czas do wystąpienia PPN określano jako zmniejszenie wyniku co najmniej o 2 punkty w skali EDSS w porównaniu z wartością wyjściową skali EDSS, które utrzymywało się przez co najmniej 6 miesięcy. PPN jest wskaźnikiem trwałej poprawy stopnia niepełnosprawności. W badaniu 2 PPN osiągnęło 29 % pacjentów otrzymujących lek Lemtrada, podczas gdy w grupie stosującej podskórnie IFNB-1a tej końcowej punktu osiągnęło jedynie 13 % pacjentów. Różnica ta była istotna statystycznie (p = 0,0002).
W badaniu 3 (badanie fazy II CAMMS223) oceniano bezpieczeństwo i skuteczność leku Lemtrada u pacjentów z RRWS przez 3 lata. Na czas włączenia do badania u pacjentów wynik skali EDSS wynosił 0–3,0 punktu, mieli oni co najmniej 2 epizody kliniczne WS w ciągu ostatnich 2 lat oraz ≥ 1 ognisko uszkodzenia gromadzące gadolin. Pacjenci wcześniej nie otrzymywali żadnej terapii z powodu WS. Pacjenci otrzymywali lek Lemtrada w dawce 12 mg/dobę (N = 108) lub 24 mg/dobę (N = 108), podawany raz dziennie przez 5 dni w miesiącu 0 oraz przez 3 dni w miesiącu 12, lub podskórne wstrzykiwania IFNB-1a w dawce 44 μg (N = 107), podawane trzy razy w tygodniu przez 3 lata. 46 pacjentów otrzymało trzeci cykl leczenia lekiem Lemtrada w dawce 12 mg/dobę lub 24 mg/dobę przez 3 dni w miesiącu 24.
Po 3 latach leczenia lekiem Lemtrada zaobserwowano zmniejszenie ryzyka PPI utrzymującego się przez 6 miesięcy o 76 % (stosunek ryzyka 0,24 [95 % CI: 0,110, 0,545], p < 0,0006) oraz zmniejszenie CRR o 67 % (stosunek częstości 0,33 [95 % CI: 0,196, 0,552], p < 0,0001) w porównaniu z odpowiednimi wartościami przy stosowaniu podskórnych wstrzykiwań IFNB-1a. Stosowanie leku Lemtrada w dawce 12 mg/dobę prowadziło do istotnie niższych wyników w skali EDSS (poprawa w porównaniu z wartością wyjściową) przez 2 lata obserwacji w porównaniu z IFNB-1a (p < 0,0001).
W podgrupie pacjentów z RRWS z 2 lub więcej nawrotami w ciągu poprzedniego roku oraz co najmniej 1 ogniskiem uszkodzenia gromadzącym gadolin w trybie ważonym T1 na poziomie wyjściowym częstość roczna nawrotów wynosiła 0,26 (95 % CI: 0,20, 0,34) w grupie leczenia lekiem Lemtrada (n = 205) i 0,51 (95 % CI: 0,40, 0,64) w grupie leczenia IFNB-1a (n = 102) (p < 0,0001). Analiza ta uwzględniała dane wyłącznie z badań fazy III (CAMMS324 i CAMMS323) z powodu różnic w algorytmach odczytu danych MRI między badaniami fazy II a badaniami fazy III. Wyniki te uzyskano w analizie post hoc i należy je interpretować ostrożnie.
Dane długoterminowe dotyczące skuteczności
Badanie 4 było badaniem fazy III, wieloośrodkowym, otwartym, ślepym dla oceniającego, rozszerzonym badaniem oceny skuteczności i bezpieczeństwa, które objęło pacjentów z RRWS, którzy brali udział w badaniach 1, 2 lub 3 (wcześniejsze badania fazy II i III), w celu określenia długoterminowej skuteczności i bezpieczeństwa stosowania leku Lemtrada. Badanie to zapewniło uzyskanie danych dotyczących skuteczności i bezpieczeństwa leczenia w okresie, którego mediana trwała 6 lat, począwszy od momentu włączenia do badań 1 i 2. Pacjenci w tym rozszerzonym badaniu (badanie 4) mogli w razie potrzeby otrzymać dodatkowy(-e) cykl(-e) leczenia lekiem Lemtrada w przypadku udokumentowanego nawrotu aktywności choroby, zdefiniowanego jako wystąpienie ≥ 1 nawrotu WS i/lub pojawienie się ≥ 2 nowych lub zwiększenia się ≥ 2 istniejących ognisk uszkodzenia w mózgu lub rdzeniu kręgowym w badaniu rezonansu magnetycznego (MRI). Dodatkowy(-e) cykl(-e) leczenia lekiem Lemtrada podawano w dawce 12 mg/dobę przez 3 kolejne dni (łączna dawka – 36 mg) nie wcześniej niż 12 miesięcy po poprzednim cyklu leczenia.
91,8 % pacjentów, którzy otrzymywali lek Lemtrada w dawce 12 mg w badaniach 1 i 2, zostało włączone do badania 4. 82,7 % tych pacjentów ukończyło badanie. Około połowa (51,2 %) pacjentów, którzy początkowo otrzymywali lek Lemtrada w dawce 12 mg/dobę w badaniach 1 lub 2 i którzy zostali włączeni do badania 4, otrzymała jedynie te początkowe 2 cykle leczenia lekiem Lemtrada i nie otrzymała żadnych innych leków modyfikujących przebieg choroby przez 6 lat obserwacji.
46,6 % pacjentów, którzy początkowo otrzymywali lek Lemtrada w dawce 12 mg/dobę w badaniach 1 lub 2, otrzymało dodatkowe cykle z powodu udokumentowanych dowodów aktywacji przebiegu WS (wystąpienie nawrotu i/lub zmiany w MRI) i zgodnie z decyzją lekarza prowadzącego o ponownym cyklu leczenia. Żadna z cech na początku badania nie pozwalała na zidentyfikowanie pacjentów, którzy później otrzymali jeden lub więcej dodatkowych cykli leczenia.
Przez 6 lat po początkowym leczeniu lekiem Lemtrada pacjenci, którzy byli nadal obserwowani, wykazywali podobne częstości nawrotów WS, powstawanie ognisk uszkodzenia w mózgu w badaniu MRI oraz utratę tkanki mózgu, co było zgodne z efektami leczenia lekiem Lemtrada podczas badań 1 i 2, a także głównie stabilne lub lepsze wyniki stopnia niepełnosprawności. Biorąc pod uwagę okres obserwacji w badaniu 4, pacjenci, którzy początkowo otrzymywali lek Lemtrada w badaniach 1 i 2, mieli odpowiednio CRR 0,17 i 0,23, PPI zaobserwowano u 22,3 % i 29,7 %, a PPN osiągnęli 32,7 % i 42,5 %. Co roku w ramach badania 4 pacjenci z obu głównych badań nadal wykazywali niskie ryzyko powstawania nowych ognisk uszkodzenia w obrazach ważonych T2 (od 27,4 % do 33,2 %) lub ognisk uszkodzenia gromadzących gadolin (od 9,4 % do 13,5 %), a mediana rocznej zmiany procentowej objętości mózgu w tkance parenchymatycznej mieściła się w zakresie od 0,19 % do –0,09 %.
U pacjentów, którzy otrzymali jeden lub dwa dodatkowe cykle leczenia lekiem Lemtrada, zaobserwowano poprawę częstości nawrotów, aktywności choroby w badaniu MRI oraz średniej liczby punktów w stopniu niepełnosprawności po pierwszym lub drugim powtórnym cyklu leczenia lekiem Lemtrada (cykle 3 i 4) w porównaniu z wynikami w poprzednim roku. U tych pacjentów CRR zmniejszyło się z 0,79 w roku poprzedzającym cykl 3 do 0,18 w roku po tym cyklu, a średnia liczba punktów w ocenie skali EDSS zmniejszyła się z 2,89 do 2,69. Procent pacjentów z powstawaniem nowych lub zwiększeniem istniejących ognisk uszkodzenia w obrazach ważonych T2 zmniejszył się z 50,8 % w roku poprzedzającym cykl 3 do 35,9 % w roku po tym cyklu, a dla ognisk uszkodzenia gromadzących gadolin zmniejszenie to wyniosło z 32,2 % do 11,9 %. Podobne poprawy w CRR, średniej liczbie punktów w ocenie skali EDSS oraz ogniskach uszkodzenia w obrazach ważonych T2 i ogniskach uszkodzenia gromadzących gadolin obserwowano również po cyklu 4 w porównaniu z poprzednim rokiem. Te poprawy utrzymywały się dalej, jednak nie można wyciągnąć jednoznacznych wniosków dotyczących długoterminowej skuteczności leczenia (np. po 3 i 4 latach po dodatkowych cyklach leczenia), ponieważ wielu pacjentów zakończyło udział w badaniu przed osiągnięciem tych punktów czasowych.
Korzyści i ryzyka 5 i więcej cykli leczenia nie są obecnie ustalone.
Immunogenność
Tak jak inne leki białkowe, ten lek jest potencjalnie immunogenny. Dane wskazują na istnienie pewnego odsetka pacjentów, u których uzyskano pozytywne wyniki testów na obecność przeciwciał przeciwko alemtuzumabowi przy użyciu testu immunoenzymatycznego (ELISA) z późniejszym potwierdzeniem metodą konkurencyjnego wiązania. Próbki, dla których uzyskano pozytywne wyniki, dodatkowo oceniano pod kątem obecności hamowania in vitro metodą cytometrii przepływowej. W klinicznych badaniach WS pobierano próbki surowicy krwi pacjentów po 1, 3 i 12 miesiącach po każdym cyklu leczenia w celu określenia obecności przeciwciał przeciwko alemtuzumabowi. U około 85 % pacjentów, którzy otrzymywali lek Lemtrada, podczas badania uzyskano pozytywne wyniki dotyczące obecności przeciwciał przeciwko alemtuzumabowi, przy czym u ≥ 90 % tych pacjentów uzyskano również pozytywne wyniki testu na obecność przeciwciał hamujących wiązanie alemtuzumabu in vitro. U pacjentów, u których wytwarzane były przeciwciała przeciwko alemtuzumabowi, miało to miejsce w ciągu 15 miesięcy po pierwszym podaniu leku. W trakcie 2 cykli leczenia nie zaobserwowano żadnej zależności między obecnością przeciwciał przeciwko alemtuzumabowi lub przeciwciał hamujących przeciwko alemtuzumabowi a zmniejszeniem skuteczności leku, zmianami w jego farmakodynamice lub wystąpieniem niepożądanych reakcji, w tym związanych z infuzjami. Wysokie miana przeciwciał przeciwko alemtuzumabowi, obserwowane u niektórych pacjentów, były związane z niepełnym eliminowaniem limfocytów po trzecim lub czwartym cyklu leczenia, jednak nie zaobserwowano wyraźnego wpływu przeciwciał przeciwko alemtuzumabowi na kliniczną skuteczność lub profil bezpieczeństwa leku Lemtrada.
Dane dotyczące częstości powstawania przeciwciał w dużej mierze zależą od czułości i specyficzności testu. Ponadto na uzyskane dane dotyczące częstości pozytywnych wyników testu na obecność przeciwciał (w tym przeciwciał hamujących) mogą wpływać różne czynniki, takie jak metodologia testu, szczegóły postępowania z próbkami, czas pobierania próbek, współistniejące leki oraz choroba podstawowa. W związku z tym porównywanie częstości powstawania przeciwciał przeciwko lekowi Lemtrada z częstością powstawania przeciwciał przeciwko innym lekom może być niepoprawne.
Pacjenci w wieku dziecięcym
Europejski Urząd ds. Leków zwolnił producenta z obowiązku przedstawienia wyników badań zastosowania alemtuzumabu u dzieci w wieku od urodzenia do mniej niż 10 lat w leczeniu stwardnienia rozsianego (informacje dotyczące stosowania leku u dzieci zawarte są w sekcji „Sposób stosowania i dawki”).
Europejski Urząd ds. Leków udzielił producentowi odroczenia obowiązku przedstawienia wyników badań zastosowania leku Lemtrada w jednej lub kilku podgrupach populacji pediatrycznej przy RRWS (informacje dotyczące stosowania leku u dzieci zawarte są w sekcji „Sposób stosowania i dawki”).
Farmakokinetyka. Właściwości farmakokinetyczne alemtuzumabu oceniano u 216 pacjentów z RRWS, którzy otrzymywali lek w postaci wlewów dożylnych w dawce 12 mg/dobę lub 24 mg/dobę przez 5 kolejnych dni, a następnie przez 3 kolejne dni po 12 miesiącach od początku pierwszego cyklu leczenia. Stężenia leku w surowicy krwi wzrastały z każdą kolejną dawką w trakcie cyklu leczenia, przy czym najwyższe stężenia odnotowano po ostatniej infuzji w cyklu leczenia. Stosowanie leku w dawce 12 mg/dobę prowadziło do średniego Cmax 3014 ng/ml w dniu 5 pierwszego cyklu leczenia i 2276 ng/ml w dniu 3 drugiego cyklu leczenia. Faza alfa okresu półtrwania wynosiła około 4–5 dni i była porównywalna w różnych cyklach leczenia, co prowadziło do osiągnięcia niskich lub nieokreślonych stężeń w surowicy krwi około 30 dni po każdym cyklu leczenia.
Alemtuzumab jest białkiem, oczekiwanym szlakiem metabolizmu którego jest rozkład do małych peptydów i pojedynczych aminokwasów przez szeroki zakres enzymów proteolitycznych. Klasycznych badań biotransformacji nie przeprowadzano.
Ze względu na dostępne dane nie można wyciągnąć pewnych wniosków dotyczących wpływu rasy i płci pacjenta na farmakokinetykę alemtuzumabu. Farmakokinetykę alemtuzumabu w RRWS nie badano u pacjentów w wieku powyżej 55 lat.
Dane doksyliczne dotyczące bezpieczeństwa
Kancerogeneza i mutageneza
Nie przeprowadzono żadnych badań potencjału kancerogennego lub mutagennego alemtuzumabu.
Plodność i rozrodczość
Wewnętrzne podanie alemtuzumabu w dawkach do 10 mg/kg/dobę przez 5 kolejnych dni (zwiększenie AUC o 7,1 razy w porównaniu z zastosowaniem zalecanej dawki dobowej leku) nie powodowało żadnego wpływu na płodność i funkcje rozrodcze u samców transgenicznych myszy huCD52. Liczba normalnych plemników istotnie zmniejszyła się (< 10 %) w porównaniu z kontrolnymi zwierzętami, a odsetek plemników zniekształconych (z odłączonymi lub bez głów) istotnie wzrósł (do 3 %). Jednak te zmiany nie wpływały na płodność i dlatego nie uznano ich za niekorzystne.
U samic myszy, które otrzymywały alemtuzumab dożylne w dawkach do 10 mg/kg/dobę (zwiększenie AUC o 4,7 razy w porównaniu z zastosowaniem zalecanej dawki dobowej leku) przez 5 kolejnych dni przed skojarzeniem z samcami myszy typu dzikiego, średnia liczba ciał żółtych i miejsc implantacji na mysz istotnie zmniejszyła się w porównaniu z zwierzętami otrzymującymi placebo. U ciężarnych myszy, którym podawano lek w dawce 10 mg/kg/dobę, zaobserwowano zmniejszenie przyrostu masy ciała w czasie ciąży w porównaniu z kontrolnymi zwierzętami otrzymującymi placebo.
W badaniu toksyczności rozrodczej u ciężarnych myszy, które otrzymywały alemtuzumab dożylne w dawkach do 10 mg/kg/dobę (zwiększenie AUC o 2,4 razy w porównaniu z zastosowaniem zalecanej dawki 12 mg/dobę) przez 5 kolejnych dni w czasie ciąży, zaobserwowano istotne zwiększenie liczby samic, u których wszystkie zarodki/płody zginęły lub uległy resorpcji, wraz z jednoczesnym zmniejszeniem liczby samic z żywymi płodami. Przy stosowaniu leku w dawkach do 10 mg/kg/dobę nie zaobserwowano żadnych wad rozwojowych ani odchyleń od normy w tkankach zewnętrznych, miękkich ani kostnych.
W czasie ciąży i po porodzie potomstwa zaobserwowano przenikanie leku przez łożysko i potencjalną aktywność farmakologiczną alemtuzumabu. W badaniach na myszach u potomstwa, które było narażone na alemtuzumab w okresie rozwoju wewnątrzmacicznego przy jego stosowaniu w dawkach 3 mg/kg/dobę przez 5 kolejnych dni (zwiększenie AUC o 0,6 razy w porównaniu z zastosowaniem zalecanej dawki 12 mg/dobę), zaobserwowano zmiany liczby limfocytów. Przy stosowaniu alemtuzumabu w dawkach do 10 mg/kg/dobę nie zaobserwowano żadnych zaburzeń rozwoju poznawczego, fizycznego i płciowego u potomstwa, które było narażone na alemtuzumab w okresie karmienia piersią.
Charakterystyka kliniczna.
Wskazania.
Lek Lemtrada wskazany jest jako lek modyfikujący przebieg choroby w monoterapii dorosłych z wysoce aktywnym przeływającym się stwardnieniem rozsianym (PSR), którzy należą do następujących grup:
- pacjenci, u których choroba pozostaje wysoce aktywna pomimo pełnego i odpowiedniego leczenia przynajmniej jednym lekiem modyfikującym przebieg choroby, lub
- pacjenci z szybko postępującym ciężkim przeływającym się stwardnieniem rozsianym, które charakteryzuje się 2 lub więcej niepełnosprawnymi napadami w ciągu jednego roku, z co najmniej jednym ogniskiem uszkodzenia gromadzącym gadolin, w badaniu MRI mózgu lub istotnym zwiększeniem całkowitej objętości ognisk uszkodzenia w trybie T2 w porównaniu z danymi ostatnio wykonanego MRI.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych wymienionych w składzie leku.
Zakażenie wirusem niedoboru odporności człowieka (HIV).
Ciężka aktywna infekcja (do pełnego wyzdrowienia pacjenta).
Niekontrolowana nadciśnienie tętnicze.
Wywiad rozwarstwienia ścian tętnic głowy i szyi.
Wywiad udaru mózgu.
Wywiad dławicy piersiowej lub zawału mięśnia sercowego.
Znana koagulopatia lub jednoczesne stosowanie terapii przeciwzakrzepowej lub antykoagulacyjnej.
Inne współistniejące choroby autoimmunologiczne (poza SR).
Środki ostrożności.
Zawartość fiolki należy sprawdzić przed podaniem pod kątem obecności zanieczyszczeń i zmiany koloru. Nie wolno stosować leku w przypadku obecności zanieczyszczeń lub nieprawidłowego zabarwienia.
Nie wstrząsać fiolki przed użyciem.
Do wstrzykiwania dożylnej należy pobrać 1,2 ml leku Lemtrada z fiolki do strzykawki, stosując technikę bezpieczną pod względem zakażeń. Lek należy wprowadzić do 100 ml roztworu chlorku sodu o stężeniu 9 mg/ml (0,9 %) do wlewu lub roztworu glukozy (5 %) do wlewu. Nie wolno rozpuszczać tego leku innymi rozpuszczalnikami. Aby wymieszać roztwór, należy ostrożnie odwracać worek.
Należy zachować ostrożność w celu zapewnienia sterylności przygotowanego roztworu. Rozcieńczony lek zaleca się natychmiast zastosować. Każda fiolka leku przeznaczona jest wyłącznie do jednorazowego użytku.
Wszelkie pozostałości nieużywanego leku lub odpady należy zniszczyć zgodnie z lokalnymi przepisami.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie przeprowadzono żadnych formalnych badań interakcji leków podczas stosowania leku Lemtrada w zalecanych dawkach pacjentom z SR. W jednym kontrolowanym badaniu klinicznym SR pacjenci, którzy niedawno otrzymywali interferon beta i octan glatyrameru, musieli przerwać leczenie tymi lekami 28 dni przed rozpoczęciem leczenia lekiem Lemtrada.
Szczególne wskazania dotyczące stosowania.
Nie zaleca się stosowania leku Lemtrada u pacjentów z nieczynną chorobą oraz u pacjentów, u których choroba jest stabilna na obecnej terapii.
Pacjentom leczonym lekiem Lemtrada należy dostarczyć Ulotkę dla pacjenta, Kartę informacyjną dla pacjenta oraz Instrukcję dla pacjenta. Przed rozpoczęciem leczenia pacjentom należy podać informacje o ryzykach i korzyściach wynikających ze stosowania leku oraz o konieczności przeprowadzania badań kontrolnych od początku leczenia i do zakończenia co najmniej 48-miesięcznego okresu po ostatniej infuzji drugiego cyklu leczenia lekiem Lemtrada. W przypadku przepisania dodatkowego cyklu leczenia okres obserwacji w celu oceny bezpieczeństwa powinien być wydłużony do zakończenia co najmniej 48-miesięcznego okresu po ostatniej infuzji leku.
Śledzenie
W celu zapewnienia lepszego śledzenia leków biologicznych nazwa handlowa i numer serii podanego leku powinny być wyraźnie wskazane w dokumentacji medycznej pacjenta.
Reakcje autoimmunologiczne
Leczenie tym lekiem może prowadzić do powstawania autoprzeciwciał i zwiększonego ryzyka wystąpienia stanów autoimmunologicznych, które mogą być poważne i zagrażające życiu. Zgłaszane choroby autoimmunologiczne obejmują zaburzenia tarczycy, immunologiczną zespół małopłytkowy (ITP), nefropatie (np. chorobę z przeciwciałami przeciwko błonie podstawnej kłębuszków nerkowych), autoimmunologiczne zapalenie wątroby, nabyte hemofilii A, zespół hemolityczno-mocznicowy (TTP), gruźlicę oraz autoimmunologiczne zapalenie mózgu. W okresie po rejestracji po leczeniu lekiem Lemtrada obserwowano przypadki wielu zaburzeń autoimmunologicznych. U pacjentów, u których wystąpiły reakcje autoimmunologiczne, należy przeprowadzić ocenę pod kątem innych stanów autoimmunologicznych (patrz sekcja „Przeciwwskazania”). Pacjenci i lekarze powinni być poinformowani o możliwym późnym wystąpieniu zaburzeń autoimmunologicznych po 48-miesięcznym okresie monitorowania.
Nabyte hemofilii A
W trakcie badań klinicznych oraz w okresie po rejestracji stosowania tego leku zgłaszano przypadki nabytej hemofilii A (z powstawaniem przeciwciał przeciwko czynnikowi krzepnięcia krwi VIII). Zazwyczaj u pacjentów obserwowano samoistne siniaki podskórne i duże siniaki, choć mogą również występować krwawienia do moczu, nosa, przewodu pokarmowego lub inne rodzaje krwawień. U wszystkich pacjentów z takimi objawami należy wykonać badanie krzepnięcia krwi, w tym oznaczenie aktywowanego częściowego czasu tromboplastynowego (APTT). W przypadku przedłużonego APTT pacjenta należy skierować na konsultację do hematologa. Pacjentów należy poinformować o objawach i objawach nabytej hemofilii A oraz poinformować o konieczności natychmiastowego zwracania się o pomoc medyczną w przypadku pojawienia się któregokolwiek z tych objawów.
Zespół hemolityczno-mocznicowy (TTP)
Zgłaszano rozwój TTP u pacjentów, którzy otrzymywali lek Lemtrada w okresie po rejestracji, w tym przypadki śmiertelne. TTP to poważny stan, który wymaga pilnej oceny i natychmiastowego leczenia i może się rozwinąć kilka miesięcy po ostatniej infuzji leku Lemtrada. TTP może charakteryzować się małopłytkowością, mikroangiopatyczną anemią hemolityczną, objawami neurologicznymi, podwyższoną temperaturą ciała i niewydolnością nerek.
Autoimmunologiczne zapalenie mózgu
U pacjentów, którzy otrzymywali leczenie lekiem Lemtrada, zgłaszano przypadki autoimmunologicznego zapalenia mózgu. Autoimmunologiczne zapalenie mózgu charakteryzuje się podostrym rozwojem (z szybkim postępem w ciągu kilku miesięcy) zaburzeń pamięci, zmian stanu psychicznego lub objawów psychicznych, zazwyczaj w połączeniu z pojawieniem się nowych ogniskowych objawów neurologicznych i drgawek. Pacjentom z podejrzeniem autoimmunologicznego zapalenia mózgu należy wykonać wizualizację neurologiczną (MRI), elektroencefalografię (EEG), punkcję lędźwiową oraz badania serologiczne odpowiednich biomarkerów (np. autoprzeciwciał przeciwko antygenom tkanki nerwowej) w celu potwierdzenia diagnozy i wykluczenia alternatywnej etiologii.
Immunologiczny zespół małopłytkowy (ITP)
Ciężkie przypadki ITP obserwowano u 12 (1 %) pacjentów, którzy otrzymywali leczenie w trakcie kontrolowanych badań klinicznych RR (co odpowiada częstotliwości rocznej 4,7 przypadku/1000 pacjentów-roku). W trakcie okresu dalszej obserwacji, którego mediana wynosiła 6,1 roku (przy maksymalnym czasie obserwacji 12 lat), zaobserwowano dodatkowe 12 ciężkich przypadków ITP (łączna częstotliwość roczna – 2,8 przypadków/1000 pacjentów-roku). U jednego pacjenta rozwinęła się ITP, która pozostawała nierozpoznana, dopóki nie wprowadzono wymogu miesięcznej kontroli parametrów krwi, i ten pacjent zmarł z powodu krwawienia do mózgu. W 79,5 % przypadków rozwój ITP obserwowano w ciągu 4 lat od pierwszego zastosowania leku. Jednak w niektórych przypadkach ITP pojawiała się kilka lat później. Do objawów ITP mogą należeć (zaproponowana lista nie jest wyczerpująca): zwiększona skłonność do powstawania siniaków, plamica, samoistne krwawienia na skórze i błonach śluzowych (np. krwawienie z nosa, kaszel krwisty), nasilenie krwawień miesięcznych (w porównaniu do zwykłych) lub nieregularność krwawień miesięcznych. Kaszel krwisty może również wskazywać na chorobę z przeciwciałami przeciwko błonie podstawnej kłębuszków nerkowych (patrz niżej), dlatego należy przeprowadzić odpowiednią diagnostykę różnicową. Należy przypomnieć pacjentom o konieczności monitorowania objawów, które mogą u nich wystąpić, oraz o natychmiastowym zwracaniu się o pomoc medyczną w przypadku jakichkolwiek powodów do niepokoju.
Przed rozpoczęciem leczenia lekiem należy wykonać rozwinięty morfologiczny badanie krwi z liczeniem formuły leukocytów i powtarzać je co miesiąc w trakcie leczenia i do zakończenia co najmniej 48-miesięcznego okresu po ostatniej infuzji leku. Po tym czasie badania należy wykonywać z uwzględnieniem danych klinicznych, które mogą wskazywać na rozwój ITP. W przypadku podejrzenia ITP należy natychmiast wykonać rozwinięty morfologiczny badanie krwi.
W przypadku potwierdzenia rozwoju ITP wskazane jest natychmiastowe odpowiednie leczenie, w tym natychmiastowe skierowanie pacjenta do specjalisty. Wyniki badań klinicznych RR wykazały, że przestrzeganie wymogów dotyczących monitorowania parametrów krwi i edukacji pacjentów dotyczącej pojawiania się i objawów ITP zapewnia wczesne wykrycie i leczenie ITP, przy czym większość przypadków odpowiada na leczenie farmakologiczne pierwszej linii.
Nefropatie
Nefropatie, w tym choroby z przeciwciałami przeciwko błonie podstawnej kłębuszków nerkowych (anty-BMK), obserwowano u 6 (0,4 %) pacjentów w trakcie badań klinicznych RR przy stosowaniu leku w okresie dalszej obserwacji, której mediana wynosiła 6,1 roku (przy maksymalnym czasie obserwacji 12 lat), i zazwyczaj rozwijały się w ciągu 39 miesięcy po ostatnim zastosowaniu leku Lemtrada. W trakcie badań klinicznych zarejestrowano 2 przypadki choroby z anty-BMK. Oba te przypadki były poważne, wykryto je we wczesnym okresie dzięki klinicznemu i laboratoryjnemu monitorowaniu i po leczeniu zakończyły się pomyślnie.
Objawy kliniczne nefropatii mogą obejmować podwyższenie stężenia kreatyniny w surowicy, krwawienie do moczu i/lub białkomocz. Chociaż w badaniach klinicznych takich przypadków nie obserwowano, przy chorobie z anty-BMK może wystąpić krwawienie pęcherzykowe, które manifestuje się kaszlem krwistym. Kaszel krwisty może również wskazywać na ITP lub nabyte hemofilii A (patrz wyżej), dlatego należy przeprowadzić odpowiednią diagnostykę różnicową. Należy przypomnieć pacjentom o konieczności monitorowania objawów, które mogą u nich wystąpić, oraz o natychmiastowym zwracaniu się o pomoc medyczną w przypadku jakichkolwiek powodów do niepokoju. Choroba z anty-BMK może prowadzić do niewydolności nerek, która będzie wymagać dializy i/lub przeszczepienia nerki, jeśli nie zastosuje się natychmiastowych zabiegów terapeutycznych, i może być zagrażająca życiu, jeśli pozostanie nieleczona.
Przed rozpoczęciem leczenia lekiem należy oznaczyć stężenia kreatyniny w surowicy i powtarzać to badanie co miesiąc w trakcie leczenia i do zakończenia co najmniej 48-miesięcznego okresu po ostatniej infuzji leku. Przed rozpoczęciem leczenia lekiem należy wykonać ogólne badanie moczu z mikroskopią osadu moczu i powtarzać je co miesiąc w trakcie leczenia i do zakończenia co najmniej 48-miesięcznego okresu po ostatniej infuzji leku. Wykrycie klinicznie istotnych zmian stężenia kreatyniny w surowicy w porównaniu z poziomami wyjściowymi, krwawienia do moczu nieznanego pochodzenia i/lub białkomoczu wymaga natychmiastowego badania pod kątem obecności nefropatii, w tym natychmiastowego skierowania pacjenta do specjalisty. Wczesne wykrycie i leczenie nefropatii może zmniejszyć ryzyko niekorzystnych skutków klinicznych. Po tym czasie badania należy wykonywać z uwzględnieniem danych klinicznych, które mogą wskazywać na rozwój nefropatii.
Zaburzenia tarczycy
Zaburzenia endokrynologiczne tarczycy, w tym autoimmunologiczne zaburzenia tarczycy, obserwowano u 36,8 % pacjentów, którzy otrzymywali lek Lemtrada w dawce 12 mg w trakcie badań klinicznych RR, gdzie mediana czasu trwania okresu obserwacji wynosiła 6,1 roku (z maksymalnym czasem obserwacji 12 lat) po pierwszym zastosowaniu leku Lemtrada. Częstotliwość niepożądanych zdarzeń ze strony tarczycy była wyższa u pacjentów, u których stwierdzono zaburzenia tarczycy w wywiadzie, zarówno w grupie stosowania leku Lemtrada, jak i w grupie stosowania interferonu beta 1a (IFNB-1a). Zarejestrowane autoimmunologiczne zaburzenia tarczycy obejmowały nadczynność i niedoczynność tarczycy. Większość przypadków była łagodna lub umiarkowana. Ciężkie niepożądane zdarzenia endokrynologiczne rozwinęły się u 4,4 % pacjentów, przy czym takie zjawiska jak choroba Basedowa (znana również jako choroba Gravesa), nadczynność tarczycy, niedoczynność tarczycy, autoimmunologiczne zapalenie tarczycy i wole obserwowano u więcej niż jednego pacjenta. Większość niepożądanych zdarzeń ze strony tarczycy leczono za pomocą standardowych zabiegów farmakologicznych, jednak niektórzy pacjenci wymagali leczenia chirurgicznego. U pacjentów, u których w okresie po rejestracji rozwinęło się autoimmunologiczne zapalenie tarczycy potwierdzone biopsją, wcześniej zarejestrowano autoimmunologiczne zaburzenia tarczycy.
Przed rozpoczęciem leczenia lekiem należy wykonać badania oceniające funkcję tarczycy, takie jak oznaczenie stężenia hormonu tyreotropowego, i powtarzać je co 3 miesiące w trakcie leczenia i do zakończenia 48-miesięcznego okresu po ostatniej infuzji leku. Po tym czasie badania należy wykonywać z uwzględnieniem danych klinicznych, które mogą wskazywać na rozwój zaburzeń funkcji tarczycy, lub w przypadku ciąży.
Choroby tarczycy stanowią szczególny ryzyko dla kobiet w ciąży (patrz sekcja „Stosowanie w okresie ciąży lub karmienia piersią”).
W badaniach klinicznych niepożądane zdarzenia ze strony tarczycy występowały u 74 % pacjentów z dodatnimi wynikami oznaczenia przeciwciał przeciwko peroksydazie tarczycy (TPO) na poziomie wyjściowym w porównaniu z 38 % pacjentów z ujemnymi wynikami oznaczenia tych przeciwciał na poziomie wyjściowym. Zdecydowana większość (około 80 %) pacjentów, u których po leczeniu występowały niepożądane zdarzenia ze strony tarczycy, miała ujemne wyniki oznaczenia przeciwciał przeciwko TPO na poziomie wyjściowym. W związku z tym niepożądane reakcje ze strony tarczycy mogą wystąpić u pacjentów niezależnie od statusu obecności przeciwciał przeciwko TPO przed leczeniem lekiem, dlatego należy okresowo wykonywać wszystkie odpowiednie badania, jak opisano powyżej.
Cytopenie
W trakcie badań klinicznych RR obserwowano rzadkie przypadki podejrzanych autoimmunologicznych cytopenii, takich jak neutropenia, anemia hemolityczna i pancytopenia. Należy wykonywać rozwinięte badania krwi (patrz wyżej w podsekcji o ITP) w celu monitorowania cytopenii, w tym neutropenii. W przypadku potwierdzenia rozwoju cytopenii wskazane jest natychmiastowe odpowiednie leczenie, w tym skierowanie pacjenta do specjalisty.
Autoimmunologiczne zapalenie wątroby i uszkodzenia wątroby
U pacjentów, którzy otrzymywali lek Lemtrada, obserwowano przypadki autoimmunologicznego zapalenia wątroby (w tym śmiertelne lub wymagające przeszczepienia wątroby) oraz uszkodzenia wątroby związane z infekcjami (patrz sekcja „Przeciwwskazania”). Należy ocenić funkcję wątroby przed rozpoczęciem terapii lekiem oraz co miesiąc do zakończenia co najmniej 48-miesięcznego okresu po ostatniej infuzji leku. Pacjentów należy poinformować o ryzyku autoimmunologicznego zapalenia wątroby, uszkodzenia wątroby i o odpowiednich objawach.
Zespół hemofagocytarny (HLH)
W trakcie stosowania po rejestracji leku u pacjentów, którzy otrzymywali lek Lemtrada, obserwowano przypadki HLH (w tym śmiertelne). HLH jest stanem zagrażającym życiu, patologicznej aktywacji immunologicznej, charakteryzującym się objawami i objawami nadmiernej reakcji zapalnej. HLH przejawia się podwyższoną temperaturą ciała, powiększeniem wątroby i cytopenią. Jest on powiązany z wysokim poziomem śmiertelności w przypadku braku wczesnej diagnostyki i leczenia. Obserwowano, że objawy pojawiają się w ciągu od kilku miesięcy do czterech lat po rozpoczęciu leczenia. Pacjentów należy poinformować o objawach HLH i o czasie ich pojawienia się. U pacjentów, u których wystąpiły wczesne objawy patologicznej aktywacji immunologicznej, należy natychmiast przeprowadzić ocenę stanu i rozważyć prawdopodobną diagnozę HLH.
Reakcje związane z infuzją (RIA)
W badaniach klinicznych reakcje związane z infuzją (RIA) definiowano jako każde niepożądane zdarzenie, które występuje podczas lub w ciągu 24 godzin po infuzji leku Lemtrada. Większość tych zjawisk może być spowodowana uwalnianiem cytokin podczas infuzji. U większości pacjentów, którzy otrzymywali lek Lemtrada w trakcie badań klinicznych RR, obserwowano RIA łagodnego lub umiarkowanego stopnia ciężkości podczas lub w ciągu 24 godzin po podaniu leku Lemtrada w dawce 12 mg. Częstotliwość RIA w trakcie pierwszego cyklu leczenia lekiem była wyższa w porównaniu z kolejnymi cyklami. Biorąc pod uwagę wszystkie dostępne dane dotyczące dalszej obserwacji, w tym pacjentów, którzy otrzymywali dodatkowe cykle leczenia lekiem, najczęściej występującymi RIA były ból głowy, wysypka, piressja, nudności, pokrzywka, swędzenie, bezsenność, dreszcze, zaczerwienienie, zwiększona zmęczalność, duszność, dysgezja, uczucie dyskomfortu w klatce piersiowej, ogólna wysypka, tachykardia, bradykardia, dyspepsja, zawroty głowy i ból. Ciężkie reakcje występowały u 3 % pacjentów i obejmowały ból głowy, piressję, pokrzywkę, tachykardię, migotanie przedsionków, nudności, uczucie dyskomfortu w klatce piersiowej i hipotensję tętniczą. Objawy kliniczne anafilaksji mogą być podobne do objawów klinicznych związanych z reakcjami infuzyjnymi, ale mają tendencję do bycia cięższych lub potencjalnie zagrażających życiu. Reakcje spowodowane anafilaksją rejestrowano rzadko w porównaniu z reakcjami związanymi z infuzją.
W celu złagodzenia skutków reakcji infuzyjnych u pacjentów zaleca się przeprowadzenie premedykacji (patrz sekcja „Sposób stosowania i dawki”).
Większość pacjentów w kontrolowanych badaniach klinicznych otrzymywała leki przeciwhistaminowe i/lub leki przeciwgorączkowe przed co najmniej jedną infuzją leku Lemtrada. RIA mogą występować u pacjentów pomimo premedykacji. Zaleca się obserwację stanu pacjentów pod kątem wystąpienia reakcji infuzyjnych podczas i przez co najmniej 2 godziny po infuzji leku Lemtrada. W razie potrzeby może być wskazane dłuższe obserwowanie (hospitalizacja). W przypadku wystąpienia ciężkich reakcji infuzyjnych infuzję dożylną należy natychmiast przerwać. Powinny być dostępne środki do leczenia anafilaksji i innych poważnych reakcji (patrz niżej).
Choroba Still’a dorosłych (HSD)
W trakcie stosowania po rejestracji u pacjentów, którzy otrzymywali leczenie lekiem Lemtrada, zgłaszano rozwój choroby Still’a dorosłych (HSD). HSD jest rzadką chorobą zapalną, która wymaga natychmiastowej oceny i leczenia. Pacjenci z HSD mogą mieć kombinację następujących objawów: gorączka, artretyzm, wysypka i leukocytoza przy braku infekcji, nowotworów złośliwych i innych chorób reumatycznych. Należy rozważyć możliwość przerwania lub odstawienia leczenia lekiem Lemtrada, jeśli nie stwierdzono alternatywnej etiologii wystąpienia tych objawów lub objawów.
Inne poważne reakcje, związane czasowo z infuzją leku Lemtrada
W trakcie stosowania po rejestracji leku obserwowano przypadki rzadkich, poważnych, czasem śmiertelnych i nieprzewidywalnych niepożądanych zjawisk ze strony różnych układów narządów. W większości przypadków czas do początku takiej reakcji wynosił od 1 do 3 dni po infuzji leku Lemtrada. Reakcje występowały po każdej dawce leku, a także po drugim cyklu terapii. Pacjentów należy poinformować o objawach i objawach tych uszkodzeń i o czasie ich pojawienia się. Pacjentom należy zalecić natychmiastowe zwracanie się o pomoc medyczną w przypadku wystąpienia któregokolwiek z tych objawów. Pacjenci powinni być poinformowani o możliwym późnym pojawieniu się takich zjawisk.
Udar mózgu krwotoczny
Kilku pacjentów z udarem mózgu krwotocznym miało poniżej 50 lat, nie miało w wywiadzie nadciśnienia tętniczego, zaburzeń krwawienia i nie przyjmowało leków przeciwzakrzepowych ani inhibitorów płytek. U niektórych pacjentów przed rozwojem krwawienia obserwowano podwyższenie ciśnienia tętniczego w porównaniu z jego poziomem wyjściowym.
Ischemia mięśnia sercowego i zawał mięśnia sercowego
Kilku pacjentów z udokumentowanymi przypadkami tych zaburzeń miało poniżej 40 lat i nie miało czynników ryzyka wystąpienia choroby niedokrwiennej serca. Zauważono, że u niektórych z tych pacjentów podczas infuzji leku czasowo zmieniały się w porównaniu z normalnymi wartościami ciśnienie tętnicze i/lub częstość skurczów serca.
Odwarstwienie ścian tętnic głowy i szyi
Przypadki odwarstwienia ścian tętnic głowy i szyi, w tym wielokrotnych odwarstwień, obserwowano zarówno w pierwszych dniach po wykonaniu infuzji leku Lemtrada, jak i w późniejszym okresie w ciągu pierwszego miesiąca po infuzji.
Krwawienie pęcherzykowe
Zarejestrowano przypadki tego zaburzenia, powiązanego czasowo ze stosowaniem leku, bez związku z chorobą anty-BMK (zespołem Goodpasture’a).
Małopłytkowość
Zarejestrowane przypadki małopłytkowości występowały w pierwszych dniach po infuzji leku (w przeciwieństwie do ITP). Małopłytkowość często ustępowała spontanicznie i była stosunkowo łagodna, choć w wielu przypadkach ciężkość i skutki kliniczne były nieznane.
Zapalenie osierdzia
Zgłaszano rzadkie przypadki zapalenia osierdzia, wypotu osierdziowego i innych zdarzeń osierdziowych zarówno w ramach ostrej reakcji infuzyjnej, jak i później.
Zapalenie płuc
Zgłaszano przypadki zapalenia płuc u pacjentów, którzy otrzymywali infuzje leku Lemtrada. Większość takich przypadków występowała w ciągu pierwszego miesiąca po leczeniu lekiem Lemtrada. Pacjentów należy poinformować o konieczności zgłaszania objawów zapalenia płuc, do których mogą należeć duszność, kaszel, świsty, ból w klatce piersiowej lub uczucie ściskania w klatce piersiowej oraz kaszel krwisty.
Instrukcje dotyczące infuzji, mające na celu zmniejszenie ryzyka wystąpienia poważnych reakcji, powiązanych czasowo z infuzją leku Lemtrada
- Oceny, które należy wykonać przed przeprowadzeniem infuzji:
- Należy wykonać ocenę wyjściowych parametrów EKG i podstawowych wskaźników czynności życiowych, w tym częstości skurczów serca i ciśnienia tętniczego.
- Należy wykonać badania laboratoryjne (rozwinięty morfologiczny badanie krwi z liczeniem formuły leukocytów, oznaczenie stężenia transaminaz w surowicy, oznaczenie stężenia kreatyniny w surowicy, badanie oceniające funkcję tarczycy i ogólne badanie moczu z mikroskopią osadu moczu).
- Podczas infuzji:
- Należy przeprowadzać ciągły/częsty (co najmniej co godzinę) monitoring częstości skurczów serca, ciśnienia tętniczego i ogólnego stanu klinicznego pacjentów.
- Infuzję należy przerwać:
- Należy przeprowadzać ciągły/częsty (co najmniej co godzinę) monitoring częstości skurczów serca, ciśnienia tętniczego i ogólnego stanu klinicznego pacjentów.
- W przypadku wystąpienia ciężkiego niepożądanego zdarzenia.
- Jeśli u pacjenta występują objawy kliniczne wskazujące na rozwój poważnego niepożądanego zdarzenia związanego z infuzją (ischemia mięśnia sercowego, udar mózgu krwotoczny, odwarstwienie ścian tętnic głowy i szyi lub krwawienie pęcherzykowe).
- Po infuzji:
- Zaleca się obserwację stanu pacjenta pod kątem wystąpienia reakcji infuzyjnych przez co najmniej 2 godziny po infuzji leku Lemtrada. U pacjentów, u których występują objawy kliniczne wskazujące na rozwój poważnego niepożądanego zdarzenia związanego z infuzją (ischemia mięśnia sercowego, udar mózgu krwotoczny, odwarstwienie ścian tętnic głowy i szyi lub krwawienie pęcherzykowe), należy prowadzić dokładne obserwowanie do całkowitego ustąpienia takich objawów. W razie potrzeby może być wskazane dłuższe obserwowanie (hospitalizacja). Należy poinformować pacjentów o możliwym późniejszym wystąpieniu reakcji związanych z infuzją i poinstruować ich o konieczności zgłoszenia swoich objawów lekarzowi i zwrócenia się o odpowiednią pomoc medyczną.
Stężenia płytek należy oznaczać natychmiast po infuzji w dniu 3 i 5 pierwszego cyklu infuzyjnego, a także natychmiast po infuzji w dniu 3 każdego z kolejnych cykli. W przypadku klinicznie istotnej małopłytkowości należy obserwować stan pacjenta do ustąpienia tego stanu. Należy rozważyć możliwość skierowania pacjenta na leczenie do hematologa.
Infekcje
W kontrolowanych badaniach klinicznych RR trwających do 2 lat infekcje występowały u 71 % pacjentów, którzy otrzymywali lek Lemtrada w dawce 12 mg, w porównaniu z 53 % pacjentów, którzy otrzymywali podskórne wstrzyknięcia interferonu beta 1a [IFNB-1a] (44 mcg 3 razy w tygodniu), i były one głównie łagodne lub umiarkowane. Do infekcji, które występowały częściej u pacjentów otrzymujących lek Lemtrada niż u pacjentów otrzymujących IFNB-1a, należą zapalenie nosa i gardła, infekcja dróg moczowych, infekcja górnych dróg oddechowych, zapalenie zatok, opryszcz usta, grypa i zapalenie oskrzeli. Ciężkie infekcje w kontrolowanych badaniach klinicznych RR występowały u 2,7 % pacjentów, którzy otrzymywali lek Lemtrada, w porównaniu z 1 % pacjentów, którzy otrzymywali IFNB-1a. Ciężkie infekcje w grupie przyjmowania leku Lemtrada obejmowały zapalenie wyrostka robaczkowego, zapalenie żołądka i jelit, zapalenie płuc, opryszcz pospolity i infekcję zębów. Infekcje miały zazwyczaj typowy czas trwania i ustępowały po standardowym leczeniu farmakologicznym.
Łączna częstotliwość roczna infekcji wynosiła 0,99 przy medianie czasu trwania okresu obserwacji 6,1 roku (z maksymalnym czasem obserwacji 12 lat) po pierwszym zastosowaniu leku Lemtrada w porównaniu z 1,27 w kontrolowanych badaniach klinicznych.
W badaniach klinicznych ciężkie infekcje spowodowane wirusem ospy wietrznej, w tym pierwotna ospa wietrzna i reaktywacja wirusa ospy wietrznej, u pacjentów otrzymujących lek Lemtrada w dawce 12 mg występowały częściej (0,4 %) niż u pacjentów otrzymujących IFNB-1a (0 %). Ponadto u pacjentów otrzymujących lek Lemtrada w dawce 12 mg rejestrowano przypadki (2 %) infekcji szyjki macicy spowodowane wirusem ludzkiego brodawczaka (HPV), w tym dysplazję szyjki macicy i brodawki anogenitalne. Kobietom zaleca się coroczne badanie przesiewowe na HPV.
Infekcje cytomegalowirusowe (CMV), w tym przypadki reaktywacji CMV, zarejestrowano u pacjentów otrzymujących lek Lemtrada. Większość przypadków obserwowano w ciągu 2 miesięcy po przyjęciu alemtuzumabu. Przed rozpoczęciem terapii można ocenić stan immunologiczny (serologiczny) zgodnie z lokalnymi zaleceniami.
U pacjentów otrzymujących lek Lemtrada obserwowano przypadki zakażeń, w tym reaktywację wirusa Epsteina-Barr (EBV), ciężkie i czasem śmiertelne przypadki zapalenia wątroby spowodowanego EBV.
W kontrolowanych badaniach klinicznych zgłaszano przypadki gruźlicy u pacjentów otrzymujących lek Lemtrada lub IFNB-1a. Przypadki aktywnej i utajonej (utajonej) gruźlicy, w tym kilka przypadków gruźlicy rozsianej, obserwowano u 0,3 % pacjentów, którzy otrzymywali lek Lemtrada, częściej – w regionach endemicznych pod względem tej choroby. Przed rozpoczęciem stosowania leku u wszystkich pacjentów należy sprawdzić obecność aktywnej lub nieaktywnej (utajonej) infekcji gruźlicy zgodnie z lokalnymi wytycznymi.
U pacjentów otrzymujących lek Lemtrada rejestrowano przypadki listeriozy/listeriozy opon mózgowych, zazwyczaj w ciągu 1 miesiąca po infuzji leku Lemtrada. W celu zmniejszenia ryzyka infekcji pacjentom otrzymującym lek Lemtrada należy unikać spożycia mięsa, które nie zostało poddane obróbce termicznej lub której obróbka była niewystarczająca, miękkich serów i nieprzegotowanych produktów mlecznych przez 2 tygodnie przed, podczas i przez co najmniej 1 miesiąc po infuzji leku Lemtrada.
W kontrolowanych badaniach klinicznych RR powierzchowne infekcje grzybicze, szczególnie kandydoza jamy ustnej i pochwy, u pacjentów otrzymujących lek Lemtrada występowały częściej (12 %) niż u pacjentów otrzymujących IFNB-1a (3 %).
U pacjentów z ciężkimi aktywnymi infekcjami należy odłożyć rozpoczęcie terapii lekiem Lemtrada do czasu wyzdrowienia. Pacjentów otrzymujących lek Lemtrada należy poinstruować o konieczności zgłaszania lekarzowi objawów infekcji.
Należy rozpocząć profilaktyczne stosowanie doustnego leku przeciwherpesowego od pierwszego dnia leczenia lekiem Lemtrada i kontynuować jego przyjmowanie przez co najmniej 1 miesiąc po każdym cyklu leczenia. W trakcie badań klinicznych pacjentom przepisywano acyklowir w dawce 200 mg dwa razy dziennie lub lek równoważny.
Leku Lemtrada nie stosowano do leczenia RR jednocześnie z lekami przeciwnowotworowymi lub immunosupresyjnymi ani po ich stosowaniu. Tak jak w przypadku stosowania innych immunomodulatorów, należy wziąć pod uwagę potencjalny wpływ kombinacji tych leków na układ odpornościowy pacjenta przy rozważaniu możliwości przepisania leku Lemtrada. Jednoczesne stosowanie leku Lemtrada z jakimikolwiek z tych leków może zwiększać ryzyko immunosupresji.
Obecnie brak danych dotyczących związku między stosowaniem leku Lemtrada a reaktywacją wirusa zapalenia wątroby B (HBV) lub wirusa zapalenia wątroby C (HCV), ponieważ pacjentów z objawami aktywnej lub przewlekłej infekcji wywołanej tymi wirusami wykluczono z badań klinicznych. Należy rozważyć możliwość przeprowadzenia badania przesiewowego u pacjentów z wysokim ryzykiem infekcji HBV i/lub HCV przed rozpoczęciem stosowania leku Lemtrada, a w przypadku przepisania leku Lemtrada pacjentom, którzy zostali zidentyfikowani jako nosiciele HBV i/lub HCV, należy zachować ostrożność, ponieważ ci pacjenci mogą mieć zwiększone ryzyko rozwoju nieodwracalnego uszkodzenia wątroby związanego z potencjalną reaktywacją wirusa z powodu już istniejącego stanu zakażenia wirusem.
Postępujące wieloogniskowe leukoenkefalopatia (PML)
Zgłaszano rzadkie przypadki PML (w tym śmiertelne) u pacjentów z RR po leczeniu alemtuzumabem. Stan pacjentów otrzymujących alemtuzumab należy kontrolować pod kątem objawów, które mogą wskazywać na PML. Czynnikiem ryzyka o szczególnym znaczeniu jest poprzednia terapia immunosupresyjna, w szczególności inne metody leczenia RR z znanymi czynnikami ryzyka wystąpienia PML.
Przed wystąpieniem objawów klinicznych lub objawów obecność PML można podejrzewać na podstawie rezultatów MRI. Przed początkową i ponowną terapią alemtuzumabem należy wykonać skanowanie MRI i ocenić jego wyniki pod kątem obecności objawów PML. W razie potrzeby należy wykonać dalsze badania, w tym analizę płynu mózgowo-rdzeniowego pod kątem obecności DNA wirusa JC, i powtarzane badania neurologiczne. Lekarz powinien szczególnie podkreślić objawy wskazujące na PML, na które pacjent może nie zwrócić uwagi (np. objawy poznawcze, neurologiczne lub psychiczne). Pacjentom należy również zalecić poinformowanie członków rodziny lub opiekunów o swoim leczeniu, ponieważ mogą oni zauważyć takie objawy, o których pacjent nie wie. U wszystkich pacjentów z RR, którzy stosują alemtuzumab i mają objawy neurologiczne i/lub objawy ogniskowych uszkodzeń mózgu w MRI, należy rozważyć PML jako diagnozę różnicową.
W przypadku postawienia diagnozy PML nie można rozpoczynać ani wznawiać leczenia alemtuzumabem.
Ostre niekalkulacyjne zapalenie pęcherzyka żółciowego
Lek Lemtrada może zwiększać ryzyko rozwoju ostrego niekalkulacyjnego (bez powstawania kamieni) zapalenia pęcherzyka żółciowego. W kontrolowanych badaniach klinicznych u 0,2 % pacjentów z RR, którzy otrzymywali lek Lemtrada, rozwinęło się ostre niekalkulacyjne zapalenie pęcherzyka żółciowego, w porównaniu z 0 % pacjentów, którzy otrzymywali IFNB-1a. W trakcie stosowania po rejestracji leku zgłaszano dodatkowe przypadki ostrego niekalkulacyjnego zapalenia pęcherzyka żółciowego u pacjentów otrzymujących lek Lemtrada. Czas do pojawienia się objawów wahał się od mniej niż 24 godzin do 2 miesięcy po infuzji leku Lemtrada. Większość pacjentów otrzymywała zachowawcze leczenie antybiotykami i wyzdrowiała bez konieczności leczenia chirurgicznego, choć u niektórych pacjentów wykonano cholecystektomię. Do objawów ostrego niekalkulacyjnego zapalenia pęcherzyka żółciowego należą ból brzucha, bolesność przy badaniu palpacyjnym brzucha, podwyższenie temperatury ciała, nudności i wymioty. Ostre niekalkulacyjne zapalenie pęcherzyka żółciowego jest stanem, który może być powiązany z wysokim odsetkiem chorób i śmiertelności w przypadku braku wczesnej diagnostyki i leczenia. W przypadku podejrzenia ostrego niekalkulacyjnego zapalenia pęcherzyka żółciowego należy natychmiast ocenić stan pacjenta i przepisać odpowiednie leczenie.
Choroby nowotworowe
Tak jak w przypadku stosowania innych immunomodulatorów, należy zachować ostrożność przy rozpoczęciu leczenia lekiem Lemtrada u pacjentów z istniejącymi i/lub aktualnymi chorobami nowotworowymi. Obecnie nie wiadomo, czy stosowanie leku Lemtrada wiąże się z zwiększonego ryzyka rozwoju chorób nowotworowych tarczycy, ponieważ reakcje autoimmunologiczne tarczycy same w sobie mogą być czynnikiem ryzyka rozwoju chorób nowotworowych tarczycy.
Antykoncepcja
U myszy podczas ciąży i po urodzeniu potomstwa obserwowano przenikanie leku Lemtrada przez łożysko i potencjalną aktywność farmakologiczną. Kobiety w wieku rozrodczym powinny stosować skuteczne środki antykoncepcyjne podczas leczenia i przez 4 miesiące po cyklu leczenia lekiem Lemtrada (patrz sekcja „Stosowanie w okresie ciąży lub karmienia piersią”).
Szczepienia
Zaleca się, aby pacjenci ukończyli wszystkie procedury szczepień, wymagane zgodnie z lokalnymi wytycznymi, nie później niż 6 tygodni przed rozpoczęciem terapii lekiem Lemtrada. Zdolność organizmu do odpowiedzi immunologicznej na jakiekolwiek szczepienia po leczeniu lekiem Lemtrada nie została obecnie zbadana.
Bezpieczeństwo szczepień żywymi wirusowymi szczepionkami po cyklu leczenia lekiem Lemtrada nie zostało formalnie zbadane w kontrolowanych badaniach klinicznych u pacjentów z RR, dlatego takie szczepienia nie powinny być przepisywane pacjentom z RR, którzy niedawno otrzymali cykl leczenia lekiem Lemtrada.
Badania na obecność przeciwciał przeciwko wirusowi ospy wietrznej / szczepienie przeciwko wirusowi ospy wietrznej. Tak jak w przypadku stosowania jakiegokolwiek immunomodulującego leku, przed rozpoczęciem cyklu leczenia lekiem Lemtrada u pacjentów, którzy nie chorowali na ospę wietrzną i nie byli szczepieni przeciwko wirusowi ospy wietrznej (VZV), należy wykonać badanie oznaczające przeciwciała przeciwko VZV. U pacjentów z ujemnymi wynikami badań na te przeciwciała należy rozważyć możliwość szczepienia przeciwko VZV przed rozpoczęciem leczenia lekiem Lemtrada. Aby zapewnić pełny efekt szczepienia przeciwko VZV, leczenie lekiem Lemtrada należy odłożyć na okres 6 tygodni po szczepieniu.
Zalecane badania laboratoryjne do monitorowania stanu pacjentów
Badania kliniczne i laboratoryjne, które należy regularnie wykonywać przez co najmniej 48 miesięcy po ostatnim cyklu leczenia lekiem Lemtrada w celu monitorowania wcześniejszych objawów chorób autoimmunologicznych:
- rozwinięty morfologiczny badanie krwi z liczeniem formuły leukocytów, oznaczenie stężenia transaminaz w surowicy i oznaczenie stężenia kreatyniny w surowicy (przed rozpoczęciem leczenia lekiem i co miesiąc później);
- ogólne badanie moczu z mikroskopią osadu moczu (przed rozpoczęciem leczenia lekiem i co miesiąc później);
- badanie oceniające funkcję tarczycy, np. oznaczenie stężenia hormonu tyreotropowego (przed rozpoczęciem leczenia lekiem i następnie co 3 miesiące).
Informacje dotyczące stosowania alemtuzumabu przed uzyskaniem licencji handlowej na lek Lemtrada poza badaniami sponsorowanymi przez firmę
Poniżej wymienione niepożądane reakcje zostały zidentyfikowane przed rejestracją leku Lemtrada przy stosowaniu alemtuzumabu do leczenia chłoniaka B-klasowego przewlekłego limfocytarnego (B-CLL), a także do leczenia innych zaburzeń, zazwyczaj w wyższych dawkach (np. 30 mg) i z większą częstotliwością podawania leku niż zalecane do leczenia RR. Ponieważ o tych reakcjach zgłaszano dobrowolnie, a populacja pacjentów była nieokreślonej liczby, nie zawsze można wiarygodnie oszacować ich częstotliwość ani ustalić istnienia związku przyczynowo-skutkowego ze stosowaniem alemtuzumabu.
Choroba autoimmunologiczna
U pacjentów, którzy otrzymywali alemtuzumab, rejestrowano zaburzenia autoimmunologiczne, takie jak neutropenia, anemia hemolityczna (w tym jeden przypadek śmiertelny), nabyte hemofilii, choroby z przeciwciałami przeciwko anty-BMK i choroby tarczycy. U pacjentów bez RR, którzy otrzymywali alemtuzumab, rejestrowano ciężkie i czasem śmiertelne zaburzenia autoimmunologiczne, takie jak autoimmunologiczna anemia hemolityczna, autoimmunologiczny zespół małopłytkowy, anemia aplastyczna, zespół Guillaina-Barré i przewlekła zapalna polineuropatia demielinizacyjna. U pacjentów onkologicznych, którzy otrzymywali alemtuzumab, rejestrowano przypadki uzyskania dodatnich wyników testu Coombsa. U jednego pacjenta onkologicznego, który otrzymywał alemtuzumab, zarejestrowano śmiertelną reakcję „przeszczep przeciwko gospodarzowi” związaną z transfuzją.
Reakcje związane z infuzją
U pacjentów bez RR, którzy otrzymywali alemtuzumab w wyższych dawkach i z większą częstotliwością podawania leku niż stosowane przy RR, obserwowano ciężkie i czasem śmiertelne RIA, w tym skurcz oskrzeli, hipoksję, omdlenia, infiltrowanie płuc, ostre zespół ostrej niewydolności oddechowej, zatrzymanie oddechu, zawał mięśnia sercowego, zaburzenia rytmu serca, ostre niewydolność serca i zatrzymanie serca. Zgłaszano również przypadki wystąpienia ciężkiej anafilaksji i innych reakcji nadwrażliwości, w tym szoku anafilaktycznego i obrzęku naczynioruchowego.
Choroby zakaźne i pasożytnicze
U pacjentów bez RR, którzy otrzymywali alemtuzumab w wyższych dawkach i z większą częstotliwością podawania leku niż stosowane przy RR, obserwowano ciężkie i czasem śmiertelne wirusowe, bakteryjne, pierwotniakowe i grzybicze infekcje, w tym spowodowane reaktywacją utajonych infekcji.
Zaburzenia układu krwi i limfatycznego
U pacjentów bez RR obserwowano ciężkie reakcje krwotoczne.
Zaburzenia serca
U pacjentów bez RR, którzy otrzymywali alemtuzumab i którzy wcześniej leczono potencjalnie kardiotoksycznymi środkami, obserwowano przypadki rozwoju niewydolności serca, kardiomiopatii i zmniejszenia frakcji wyrzutowej.
Zaburzenia limfoproliferacyjne związane z wirusem Epsteina-Barr
Poza badaniami sponsorowanymi przez firmę obserwowano przypadki zaburzeń limfoproliferacyjnych związanych z wirusem Epsteina-Barr.
Lek Lemtrada zawiera sod i potas
Ten lek zawiera mniej niż 1 mmol potasu (39 mg) na jedną infuzję, co oznacza, że praktycznie nie zawiera potasu.
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na jedną infuzję, co oznacza, że praktycznie nie zawiera sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym
Około 30 dni po każdym cyklu terapii stężenia leku w surowicy były niskie lub nie wykrywano. Z tego względu kobiety w wieku rozrodczym powinny stosować skuteczne środki antykoncepcyjne podczas cyklu leczenia lekiem Lemtrada i przez 4 miesiące po każdym cyklu leczenia.
Ciąża
Dane dotyczące stosowania alemtuzumabu u ciężarnych kobiet są obecnie ograniczone. Leku Lemtrada należy przepisywać ciężarnym wyłącznie w przypadkach, gdy oczekiwana korzyść dla kobiety przewyższa potencjalne ryzyko dla płodu.
Wiadomo, że IgG człowieka przenika przez barierę łożyskową; alemtuzumab może również przenikać przez barierę łożyskową i w związku z tym potencjalnie może stanowić ryzyko dla płodu. W badaniach na zwierzętach wykazano toksyczne działanie na funkcję rozrodczą (patrz podsekcja „Dane przedkliniczne dotyczące bezpieczeństwa”). Nie wiadomo, czy alemtuzumab może szkodzić płodowi przy stosowaniu u ciężarnych kobiet lub czy może wpływać na funkcję rozrodczą.
Choroby tarczycy (patrz podsekcja „Zaburzenia tarczycy”) wiążą się z szczególnymi ryzykami dla ciężarnych kobiet. Przy nieleczonym niedoczynności tarczycy w czasie ciąży występuje zwiększone ryzyko poronień i zaburzeń rozwoju płodu, takich jak opóźnienie rozwoju umysłowego i karłowatość. U kobiet z chorobą Gravesa przeciwciała przeciwko receptorom hormonu tyreotropowego ciężarnej mogą przenikać do rozwijającego się płodu i prowadzić do przejściowej choroby Gravesa u noworodka.
Karmienie piersią
Alemtuzumab wykrywano w mleku samic myszy w okresie laktacji i w organizmie ich potomstwa.
Nie wiadomo, czy alemtuzumab wydzielany jest do mleka matki. Ryzyko dla noworodków/niemowląt karmionych piersią nie można wykluczyć. Z tego względu karmienie piersią należy przerwać podczas każdego cyklu leczenia lekiem Lemtrada i przez 4 miesiące po ostatniej infuzji każdego cyklu leczenia. Jednak korzyści wynikające z odporności przekazywanej z mlekiem matki mogą przeważać nad ryzykiem potencjalnego przeniknięcia alemtuzumabu do organizmu noworodków/niemowląt karmionych piersią.
Niepłodność
Brak odpowiednich danych klinicznych dotyczących bezpieczeństwa wpływu leku Lemtrada na płodność. W jednym podbadaniu, w którym wzięło udział 13 pacjentów męskich, którzy otrzymywali lek Lemtrada (w dawce 12 mg lub 24 mg), nie stwierdzono żadnych dowodów azoospermii, trwałego zmniejszenia liczby plemników, zaburzeń ruchomości plemników lub zwiększenia zaburzeń morfologii plemników.
Wiadomo, że białko CD52 jest obecne w tkankach narządów rozrodczych u ludzi i gryzoni. Dane badań na zwierzętach wykazały pewne efekty leku na płodność u zhumanizowanych myszy (patrz podsekcja „Dane przedkliniczne dotyczące bezpieczeństwa”), jednak potencjalny wpływ na płodność u ludzi w okresie stosowania leku jest obecnie nieznany ze względu na dostępne dane.
Wpływ na zdolność do prowadzenia pojazdów i obsługi maszyn.
Lek Lemtrada wywołuje nieistotny wpływ na zdolność do prowadzenia pojazdów i obsługi maszyn. U większości pacjentów występują RIA, które rozwijają się podczas podania leku Lemtrada lub w ciągu 24 godzin po tym. Niektóre z RIA (np. zawroty głowy) mogą tymczasowo wpływać na zdolność pacjenta do prowadzenia pojazdów lub obsługi maszyn, dlatego należy zachować ostrożność, dopóki te zjawiska nie ustąpią.
Sposób stosowania i dawki
Leczenie lekiem Lemtrada należy rozpoczynać i prowadzić wyłącznie pod nadzorem neurologa z doświadczeniem w leczeniu pacjentów ze stwardnieniem rozsianym (SR), w szpitalu, gdzie zapewniona jest możliwość prowadzenia intensywnej terapii.
Powinny być dostępne specjaliści oraz wyposażenie niezbędne do szybkiej diagnostyki i udzielenia pomocy medycznej w przypadku wystąpienia niepożądanych reakcji, szczególnie w przypadku niedokrwienia mięśnia sercowego i zawału mięśnia sercowego, niepożądanych zdarzeń naczyniowych mózgu, stanów autoimmunologicznych oraz infekcji.
Powinny być dostępne środki do leczenia zespołu uwalniania cytokin, reakcji nadwrażliwości i/lub reakcji anafilaktycznych.
Pacjentom otrzymującym leczenie lekiem Lemtrada należy wydać Kartę Pacjenta oraz Instrukcję dla Pacjenta, a także dostarczyć informacje o ryzykach stosowania leku Lemtrada (patrz także ulotka dla pacjenta dołączona do opakowania leku).
Dawkowanie
Zalecana dawka alemtuzumabu wynosi 12 mg/doba, podawana w formie wlewów dożylnych w dwóch początkowych cyklach leczenia oraz, w razie potrzeby, do dwóch dodatkowych cykli leczenia.
Początkowe leczenie – dwa cykle
- Pierwszy cykl leczenia: 12 mg/doba przez 5 kolejnych dni (dawka całkowita – 60 mg).
- Drugi cykl leczenia: 12 mg/doba przez 3 kolejne dni (dawka całkowita – 36 mg), podawane 12 miesięcy po pierwszym cyklu leczenia.
W razie potrzeby można rozważyć stosowanie do dwóch dodatkowych cykli leczenia (patrz sekcja „Właściwości farmakologiczne / Farmakodynamika”).
- Trzeci lub czwarty cykl: 12 mg/doba przez 3 kolejne dni (dawka całkowita – 36 mg), podawane 12 miesięcy po poprzednim cyklu leczenia (patrz sekcje „Właściwości farmakologiczne / Farmakodynamika”, „Wskazania”).
Jeśli jakaś dawka została pominięta, nie należy jej podawać w tym samym dniu, co zaplanowaną dawkę na ten dzień.
Obserwacja pacjenta
Zaleca się leczenie lekiem Lemtrada w dwóch początkowych cyklach oraz, w razie potrzeby, do dwóch dodatkowych cykli leczenia (patrz podsekcja „Dawkowanie”), przy jednoczesnej obserwacji pacjentów w celu oceny bezpieczeństwa od momentu rozpoczęcia leczenia aż do co najmniej 48 miesięcy po ostatniej infuzji drugiego cyklu leczenia. Jeśli przepisano dodatkowy trzeci lub czwarty cykl leczenia, należy kontynuować obserwację w celu oceny bezpieczeństwa przez co najmniej 48 miesięcy od ostatniej infuzji leku (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Premeodykacja
Pacjenci powinni otrzymać premedykację kortykosteroidami bezpośrednio przed podaniem leku Lemtrada w każdy z pierwszych 3 dni każdego cyklu leczenia. W badaniach klinicznych pacjenci otrzymywali premedykację metyloprednizolonem w dawce 1000 mg w pierwsze 3 dni każdego cyklu leczenia lekiem Lemtrada.
Można również rozważyć premedykację lekami przeciwhistaminowymi i/lub lekami przeciwgorączkowymi przed podaniem leku Lemtrada.
Wszystkim pacjentom należy przepisać doustne środki zapobiegające reaktywacji wirusa opryszczki, które należy zaczynać stosować od pierwszego dnia każdego cyklu leczenia i kontynuować przez co najmniej 1 miesiąc po zakończeniu leczenia lekiem Lemtrada (patrz także sekcja „Szczególne ostrzeżenia i środki ostrożności / Infekcje”). W badaniach klinicznych pacjentom podawano acyklowir w dawce 200 mg dwa razy na dobę lub lek równoważny.
Osobliwe kategorie pacjentów
Pacjenci w wieku podeszłym
W badaniach klinicznych tego leku nie włączono pacjentów w wieku powyżej 61 roku życia. Obecnie nie wiadomo, czy u tych pacjentów występują różnice w odpowiedzi na lek w porównaniu z młodszymi pacjentami.
Dysfunkcja nerek lub wątroby
Nie badano stosowania leku Lemtrada u pacjentów z zaburzeniami funkcji nerek lub wątroby.
Pacjenci w wieku dziecięcym
Bezpieczeństwo i skuteczność stosowania leku Lemtrada u dzieci z SR w wieku od 0 do 18 lat nie są obecnie ustalone. Brak istotnego doświadczenia w stosowaniu alemtuzumabu u dzieci w wieku od urodzenia do poniżej 10 roku życia w leczeniu stwardnienia rozsianego. Dane w tej kwestii są niedostępne.
Sposób stosowania
Lek Lemtrada należy rozcieńczyć bezpośrednio przed infuzją. Rozcieńczony roztwór podaje się w formie wlewu dożylnego trwającego około 4 godziny.
Instrukcje dotyczące rozcieńczania tego leku przed podaniem znajdują się w sekcji „Szczególne środki ostrożności”.
Dzieci. Bezpieczeństwo i skuteczność stosowania leku Lemtrada u dzieci z SR w wieku od 0 do 18 lat nie są obecnie ustalone. Brak istotnego doświadczenia w stosowaniu alemtuzumabu u dzieci w wieku od urodzenia do 10 roku życia w leczeniu stwardnienia rozsianego. Dane w tej kwestii są niedostępne.
Przedawkowanie.
W kontrolowanych badaniach klinicznych dwóch pacjentów z SR przypadkowo otrzymało do 60 mg leku Lemtrada (czyli pełną dawkę pierwszego cyklu leczenia) w jednej infuzji i u tych pacjentów wystąpiły poważne reakcje (ból głowy, wysypka oraz albo hipotensja tętnicza, albo tachykardia zatokowa). Dawkowanie leku Lemtrada wyższe niż to stosowane w badaniach klinicznych może nasilić nasilenie reakcji związanych z infuzją i/lub efekty immunologiczne leku.
Nieznany jest antydotum w leczeniu przedawkowania alemtuzumabem. Leczenie polega na odstawieniu leku oraz podaniu terapii wspierającej.
Niepożądane reakcje.
Uogólniony profil bezpieczeństwa na podstawie wyników badań klinicznych. Populacja pacjentów wykorzystana do oceny bezpieczeństwa w analizie złożonej danych z badań klinicznych nad SM, z medianą czasu obserwacji 6,1 roku (i maksymalnym czasem obserwacji 12 lat), obejmowała łącznie 1486 pacjentów otrzymujących lek Lemtrada (w dawce 12 mg lub 24 mg), co zapewniło 8635 pacjentolat obserwacji do oceny bezpieczeństwa.
Najważniejszymi niepożądanymi reakcjami są reakcje autoimmunologiczne (ITP, zaburzenia tarczycy, nefropatia, cytopenia), AIR oraz infekcje (patrz punkt „Szczególne ostrzeżenia i środki ostrożności”).
Najczęstszymi niepożądanymi reakcjami podczas stosowania leku Lemtrada (obserwowanymi u ≥ 20% pacjentów) były wysypka, ból głowy, gorączka oraz infekcje dróg oddechowych.
Wykaz niepożądanych reakcji w formie tabeli. Poniższa tabela 3 została sporządzona na podstawie uogólnionych danych dotyczących bezpieczeństwa, uzyskanych od wszystkich pacjentów otrzymujących lek Lemtrada w dawce 12 mg, na podstawie wszystkich dostępnych danych obserwacyjnych z badań klinicznych. Niepożądane reakcje wymieniono według kategorii „Klasa Układ-Organ” (SOC) i preferowanych terminów Medycznego Słownika do Celów Regulacyjnych (MedDRA). Częstotliwość występowania niepożądanych zjawisk określono według następujących kryteriów: bardzo często (≥ 1/10); często (od ≥ 1/100 do < 1/10); rzadko (od ≥ 1/1000 do < 1/100); rzadko (od ≥ 1/10 000 do < 1/1000); bardzo rzadko (< 1/10 000); częstotliwość nieznana (nie można oszacować na podstawie dostępnych danych). W ramach każdej grupy częstotliwości niepożądane reakcje wymieniono w kolejności malejącej według nasilenia.
Tabela 3
Niepożądane reakcje obserwowane w badaniach 1, 2, 3 i 4 u pacjentów otrzymujących lek Lemtrada w dawce 12 mg oraz w okresie nadzoru pozarejestracyjnego
| Układ-Organy-Klasa |
Bardzo często |
Często |
Nieczęsto |
Rzadko |
Częstotliwość nieznana |
| Choroby zakaźne i pasożytnicze |
Zakażenia dróg oddechowych górnych, zakażenia dróg moczowych, zakażenie wywołane wirusem herpesu1 |
Zakażenia wywołane wirusem ospy wietrznej2, zakażenia dróg oddechowych dolnych, gastroenterocolitis, kandydoza jamy ustnej, kandydoza pochwy i sromu, grypa, zakażenie ucha, zapalenie płuc, zakażenie pochwy, zakażenie zęba |
Onychomycosis, zapalenie dziąseł, grzybica skóry, zapalenie migdałków, zatkanie ostre, flegmona, gruźlica, infekcje cytomegalowirusowe |
Listerioza / listeriowy zapalenie opon mózgowo-rdzeniowych, infekcja (w tym reaktywacja) wirusem Epsteina-Barr (EBV) |
|
| Nowotwory łagodne, złośliwe i niejednoznaczne (w tym torbiele i polipy) |
Brodawczak skóry |
||||
| Zaburzenia układu krwi i chłonnego |
Lymfopenia, leukopenia, w tym neutropenia |
Lymfadenopatia, immunologiczna plamica małopłytkowa, trombocytopenia, anemia, obniżenie hematokrytu, leukocytoza |
Pancytopenia, anemia hemolityczna, nabyta hemofilia A |
Hemofagocytowy lymfohistiocytoza (HLH), zespół hemolityczno-uremiczny (TTP) |
|
| Zaburzenia układu odpornościowego |
Zespół uwalniania cytokin*, nadwrażliwość (w tym anafilaksja)* |
Sarkoidoza |
|||
| Zaburzenia endokrynologiczne |
Choroba Basedowa, nadczynność tarczycy, niedoczynność tarczycy |
Autoimmunologiczne zapalenie tarczycy (w tym podostre zapalenie tarczycy), wole, pozytywne wyniki badań na obecność przeciwciał przeciwko składnikom tarczycy |
|||
| Zaburzenia metaboliczne i odżywienie |
Obniżenie apetytu |
||||
| Zaburzenia psychiczne |
Bezsenność*, lęk, depresja |
||||
| Zaburzenia neurologiczne |
Ból głowy* |
Przezbrojenie SM, zawroty głowy*, hipestezja, parestezja, drżenie, dysgezja*, migrena* |
Zaburzenia czucia, hiperestezja, ból głowy napięciowy, autoimmunologiczne zapalenie mózgu |
Udar mózgu krwotoczny**, rozwarstwienie ścian tętnic głowy i szyi** |
|
| Zaburzenia oka |
Zapalenie spojówek, ophthalmopatia endokrynna, nieostrość widzenia |
Diplopia |
|||
| Zaburzenia narządu słuchu i równowagi |
Zawroty głowy |
Ból ucha |
|||
| Zaburzenia serca |
Tachykardia* |
Bradykardia*, kołatanie serca* |
Migotanie przedsionków* |
Ischemia mięśnia sercowego**, zawał mięśnia sercowego** |
|
| Zaburzenia naczyniowe |
Zaczerwienienie* |
Obniżone ciśnienie tętnicze*, podwyższone ciśnienie tętnicze* |
|||
| Zaburzenia układu oddechowego, narządów klatki piersiowej i jamy opłucnowej |
Duszność*, kaszel, krwawienie z nosa, dudnienie, ból gardła, astma oskrzelowa |
Odczucie ucisku w gardle*, podrażnienie gardła, zapalenie płuc |
Krwawienie płuca** |
||
| Zaburzenia przewodu pokarmowego |
Nudności* |
Ból brzucha, wymioty, biegunka, dyspepsja*, zapalenie błony śluzowej jamy ustnej |
Wstyd, choroba refluksowa przełyku, krwawienie dziąseł, suchość w ustach, dysfagia, zaburzenia przewodu pokarmowego, krwawienie z odbytu |
||
| Zaburzenia wątroby i dróg żółciowych |
Podwyższenie poziomu aminotransferazy asparaginianowej, podwyższenie poziomu aminotransferazy alaninowej |
Zapalenie pęcherzyka żółciowego (w tym kamica żółciowa i ostre zapalenie pęcherzyka żółciowego) |
Autoimmunologiczne zapalenie wątroby, zapalenie wątroby (związane z infekcją wirusem Epsteina-Barr (EBV)) |
||
| Zaburzenia skóry i tkanki podskórnej |
Krzypa*, wysypka*, świąd*, ogólna wysypka* |
Zapalenie czerwone*, siniaki, łysienie, nadpotliwość, trądzik, zmiany skórne, zapalenie skóry |
Pęcherze, nocne pocenie się, obrzęk twarzy, egzema, bielactwo, plackowate łysienie |
||
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
Ból mięśni, osłabienie mięśni, ból stawów, ból pleców, ból kończyn, skurcze mięśni, ból szyi, ból mięśniowo-szkieletowy |
Stiffness układu mięśniowo-szkieletowego, dyskomfort kończyn |
Choroba Still'a u dorosłych (AOSD) |
||
| Zaburzenia nerek i dróg moczowych |
Białkomocz, krwiomocz |
Kamica nerkowa, ciałka ketonowe w moczu, nefropatie (w tym choroby z tworzeniem się przeciwciał przeciwko błonie podstawnej) |
|||
| Zaburzenia układu rozrodczego i gruczołów mlekowych |
Miastenia, nieregularne miesiączkowanie |
Dysplazja szyjki macicy, amenorhea |
|||
| Zaburzenia ogólne i reakcje w miejscu podania leku |
Podwyższenie temperatury ciała*, zwiększone zmęczenie*, dreszcze* |
Dyskomfort w klatce piersiowej*, ból*, obrzęki obwodowe, osłabienie, grypopodobne zachorowanie, niedobrze, ból w miejscu infuzji |
|||
| Zmiany wyników badań |
Podwyższenie poziomu kreatyniny we krwi |
Spadek masy ciała, przyrost masy ciała, zmniejszenie zawartości czerwonych krwinek, pozytywne wyniki testu bakteryjnego, podwyższenie poziomu glukozy we krwi, zwiększenie średniej objętości czerwonych krwinek |
|||
| Urazy, zatrucia i powikłania proceduralne |
Siniaki, reakcje związane z infuzją |
1 Do infekcji wywołanych wirusem herpesa należą (według preferowanych terminów): opryszcz ustny; opryszcz zwykły; opryszcz narządów płciowych; infekcja wywołana wirusem herpesa; zwykły opryszcz narządów płciowych; zapalenie skóry herpetyczne; zwykły opryszcz oczny; dodatni wynik serologiczny na opryszcz zwykły.
2 Do infekcji wywołanych wirusem ospy wietrznej należą (według preferowanych terminów): opryszcz półpasiec; uogólniony opryszcz półpasiec skórny; opryszcz półpasiec oczny; opryszcz oczny; infekcja neurologiczna wywołana wirusem opryszczu półpasca; zapalenie opon mózgowo-rdzeniowych wywołane wirusem opryszczu półpasca.
Opis poszczególnych niepożądanych reakcji. Terminy oznaczone gwiazdką (*) w tabeli 3 obejmują niepożądane reakcje zgłaszane jako związane z infuzjami.
Terminy oznaczone dwiema gwiazdkami (**) w tabeli 3 obejmują niepożądane reakcje obserwowane w okresie pozarejestracyjnym, które w większości przypadków pojawiały się w ciągu 1–3 dni po podaniu infuzji leku Lemtrada po wprowadzeniu jakiejkolwiek dawki w trakcie cyklu leczenia.
Neutropenia
Zgłaszano przypadki ciężkiej (w tym śmiertelnej) neutropenii, która występowała w ciągu 2 miesięcy po infuzji leku Lemtrada.
Profil bezpieczeństwa na podstawie wyników długotrwałej obserwacji po leczeniu. Typ niepożądanych reakcji, w tym ich poważność i ciężkość, obserwowanych u pacjentów z grup leczonych lekiem Lemtrada na podstawie wszystkich dostępnych danych z dalszej obserwacji, w tym u pacjentów otrzymujących dodatkowe cykle leczenia, był podobny do niepożądanych reakcji obserwowanych w badaniach z aktywnym kontrolowaniem. Częstość reakcji związanej z infuzją (RIA) w trakcie pierwszego cyklu leczenia była wyższa w porównaniu z kolejnymi cyklami.
U pacjentów, których obserwacja była kontynuowana po zakończeniu kontrolowanych badań klinicznych i którzy nie otrzymali żadnego dodatkowego cyklu leczenia lekiem Lemtrada po dwóch początkowych cyklach, częstość (liczba zdarzeń na pacjenta-rok) większości niepożądanych reakcji w latach 3–6 była porównywalna lub niższa niż w latach 1 i 2. Częstość niepożądanych reakcji ze strony tarczycy była najwyższa w trzecim roku i następnie malała.
Zgłaszanie podejrzewanych niepożądanych reakcji.
Ten lek podlega dodatkowemu monitorowaniu. Umożliwia to szybkie wykrywanie nowych informacji dotyczących bezpieczeństwa. Zgłaszanie niepożądanych reakcji po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich ustawowieni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych niepożądanych reakcji oraz braku skuteczności leku za pośrednictwem zautomatyzowanego systemu informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
Koncentrat: 4 lata.
Rozcieńczony roztwór: wykazano, że gotowy do użycia lek pozostaje chemicznie i fizycznie stabilny przez 8 godzin w temperaturze 2–8 °C. Z mikrobiologicznego punktu widzenia zaleca się natychmiastowe użycie leku. Jeśli nie zostanie użyty natychmiast, użytkownik odpowiada za okres i warunki przechowywania przed użyciem, które nie powinny przekraczać 8 godzin w temperaturze 2–8 °C, przy warunku zapewnienia ochrony przed światłem.
Warunki przechowywania. Przechowywać w lodówce (przy temperaturze 2–8 °C). Nie zamrażać. Nie wstrząsać. Przechowywać fiolkę w zewnętrznej tekturowej opakowaniu w celu ochrony przed światłem. Przechowywać w miejscu niedostępnym dla dzieci i poza zasięgiem ich wzroku.
Niezgodność. Badania zgodności nie były prowadzone, dlatego nie zaleca się mieszania tego leku z innymi lekami, z wyjątkiem tych wymienionych w sekcji „Specjalne środki ostrożności”.
Opakowanie.
Nr 1: po 1,2 ml w fiolce; po 1 fiolce w tekturowym pudełku.
Kategoria wydawania. Na receptę.
Producent.
Genzyme Ireland Limited.
Adres producenta i miejsce prowadzenia działalności.
IDA Industrial Park, Old Kilmeaden Road, Waterford, Irlandia.
Wnioskodawca.
Spółka z o.o. „Sanofi-Aventis Ukraina”, Ukraina.
Adres wnioskodawcy.
Ukraina, 01033, miasto Kijów, ul. Żyliańska 48-50A.