Lemtrada
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO LEMTRADA (LEMTRADA®)
Composición:
Principio activo: alemtuzumab;
1,2 ml de concentrado contienen 12 mg de alemtuzumab;
1 frasco contiene 12 mg de alemtuzumab;
Excipientes: fosfato de sodio (E 339); edetato disódico dihidrato; cloruro de potasio (E 508); fosfato monopotásico (E 340); polisorbato 80 (E 433); cloruro de sodio; agua para preparaciones inyectables.
Forma farmacéutica. Concentrado para solución para perfusión.
Principales características físico-químicas: concentrado transparente, incoloro o ligeramente amarillo, pH entre 7,0 y 7,4.
Grupo farmacoterapéutico. Inmunosupresores. Inmunosupresores selectivos. Alemtuzumab. Código ATC L04A A34.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción. Alemtuzumab es un anticuerpo monoclonal humanizado producido mediante tecnología de ADN recombinante, dirigido contra la glicoproteína CD52 presente en la superficie celular, con una masa molecular de 21-28 kD. Alemtuzumab es un anticuerpo de la clase IgG1 kappa con regiones variables y constantes humanas, y con regiones determinantes de complementariedad derivadas de un anticuerpo monoclonal murino (producido en ratas). La masa molecular aproximada del anticuerpo es de 150 kD.
Alemtuzumab se une al antígeno CD52 de la superficie celular, presente en altas concentraciones en los linfocitos T (CD3+) y B (CD19+), y en concentraciones más bajas en las células asesinas naturales, monocitos y macrófagos. CD52 se encuentra en concentraciones insignificantes o ausente en neutrófilos, células plasmáticas y células madre de la médula ósea. La acción de alemtuzumab se produce mediante citólisis dependiente de anticuerpos y lisis mediada por complemento tras la unión superficial a linfocitos T y B.
El mecanismo mediante el cual el medicamento Lemtrada ejerce sus efectos terapéuticos en la esclerosis múltiple (EM) aún no se ha esclarecido completamente. Sin embargo, los estudios indican efectos inmunomoduladores a través de la eliminación y repoblación de linfocitos, incluyendo:
- cambios en la cantidad, proporción y características de ciertas subpoblaciones de linfocitos tras la administración del medicamento;
- aumento en el número de subpoblaciones reguladoras de linfocitos T;
- aumento en el número de linfocitos T y B de memoria;
- efecto transitorio sobre componentes de la inmunidad innata (es decir, neutrófilos, macrófagos, células NK).
La reducción en el número de linfocitos T y B circulantes inducida por Lemtrada y su posterior repoblación podrían disminuir el potencial de recaída de la enfermedad, lo que en última instancia ralentiza la progresión de la misma.
Efectos farmacodinámicos. El medicamento Lemtrada reduce los niveles de linfocitos T y B circulantes tras cada ciclo de tratamiento, alcanzando los niveles más bajos un mes después del ciclo (lo que correspondió al punto de evaluación más temprano tras el tratamiento en los estudios de fase III). Con el tiempo, se produce la repoblación de linfocitos, siendo la recuperación de los niveles de células B generalmente dentro de los 6 meses. Los niveles de linfocitos CD3+ y CD4+ aumentan más lentamente y, por lo general, no regresan a los niveles basales a los 12 meses tras el tratamiento. Aproximadamente el 40 % de los pacientes alcanzaron el límite inferior de la normalidad (LIN) en el recuento total de linfocitos a los 6 meses tras cada ciclo de tratamiento, y aproximadamente el 80 % de los pacientes alcanzaron el LIN a los 12 meses tras cada ciclo.
Los neutrófilos, monocitos, eosinófilos, basófilos y células asesinas naturales experimentan únicamente cambios transitorios bajo la acción de Lemtrada.
Eficacia clínica y seguridad. La seguridad y eficacia de alemtuzumab en EM se evaluaron en tres estudios clínicos aleatorizados, ciegos para el evaluador, con comparador activo, y un estudio ampliado no controlado y ciego para el evaluador, que incluyó pacientes con esclerosis múltiple recidivante-remitente (EMRR).
En la tabla 1 se presenta información sobre el diseño de los estudios y los datos demográficos de los pacientes (estudios 1, 2, 3 y 4).
Tabla 1
| Nombre del estudio |
Estudio 1 CAMMS323 |
Estudio 2 CAMMS32400507 |
Estudio 3 CAMMS223 |
| Diseño del estudio |
Controlado, aleatorizado, ciego para el evaluador |
Controlado, aleatorizado, ciego para el evaluador y ciego respecto a la dosis |
Controlado, aleatorizado, ciego para el evaluador |
| Antecedentes de enfermedad |
Pacientes con EM activa definida como al menos 2 brotes en los últimos 2 años |
Pacientes con EM activa definida como al menos 2 brotes en los últimos 2 años o la presencia de una o más lesiones realzadas con contraste |
|
| Duración |
2 años |
3 años‡ |
|
| Población de participantes del estudio |
Pacientes que previamente no habían recibido tratamiento |
Pacientes que previamente habían mostrado una respuesta insatisfactoria a un tratamiento previo* |
Pacientes que previamente no habían recibido tratamiento |
| Características basales |
|||
| Edad media (años) |
33 |
35 |
32 |
| Duración media/mediana de la enfermedad |
2,0/1,6 años |
4,5/3,8 años |
1,5/1,3 años |
| Duración media de la terapia previa para la EM (con uso de ≥ 1 fármaco) |
No realizada |
36 meses |
No realizada |
| % de pacientes que previamente recibieron ≥ 2 cursos de tratamiento para la EM |
No aplicable |
28 % |
No aplicable |
| Puntuación media en la escala EDSS (Escala Ampliada de Discapacidad) al inicio del estudio |
2,0 |
2,7 |
1,9 |
| Estudio 4 |
|||
| Nombre del estudio |
CAMMS03409 |
||
| Diseño del estudio |
Estudio ampliado no controlado y ciego para el evaluador |
||
| Población de participantes del estudio |
Pacientes que participaron en los estudios CAMMS223, CAMMS323 o CAMMS32400507 (ver características basales anteriores) |
||
| Duración de la fase ampliada |
4 años |
||
* Se definieron como pacientes con al menos 1 recidiva durante el tratamiento con interferón beta o acetato de glatiramero tras haber recibido dicha terapia durante un mínimo de 6 meses.
‡ El punto final primario del estudio se evaluó a los 3 años. Una observación adicional proporcionó datos para un periodo con una mediana de duración de 4,8 años (con un periodo máximo de seguimiento de 6,7 años).
En la Tabla 2 se muestran los resultados obtenidos en los estudios 1 y 2.
Tabla 2
| Nombre del estudio |
Estudio 1 CAMMS323 (CARE-MS I) |
Estudio 2 CAMMS32400507 (CARE-MS II) |
||
| Puntos finales clínicos |
Lemtrada 12 mg (N = 376) |
IFN-beta-1a subcutáneo (N = 187) |
Lemtrada 12 mg (N = 426) |
IFN-beta-1a subcutáneo (N = 202) |
| Frecuencia de recaídas1 Frecuencia anual de recaídas (FAR) (IC del 95 %) |
0,18 (0,13; 0,23) |
0,39 (0,29; 0,53) |
0,26 (0,21; 0,33) |
0,52 (0,41; 0,66) |
| Relación de tasas (IC del 95 %) |
0,45 (0,32; 0,63) 54,9 (p < 0,0001) |
0,51 (0,39; 0,65) 49,4 (p < 0,0001) |
||
| Discapacidad1 (empeoramiento confirmado del grado de discapacidad [EGD])2 |
8,0 % (5,7; 11,2) |
11,1 % (7,3; 16,7) |
12,7 % (9,9; 16,3) |
21,1 % (15,9; 27,7) |
| Relación de riesgos (IC del 95 %) |
0,70 (0,40; 1,23) (p = 0,22) |
0,58 (0,38; 0,87) (p = 0,0084) |
||
| Pacientes sin recaídas durante 2 años (IC del 95 %) |
77,6 % (72,9; 81,6) (p < 0,0001) |
58,7 % (51,1; 65,5) |
65,4 % (60,6; 69,7) (p < 0,0001) |
46,7 % (39,5; 53,5) |
| Cambio en la puntuación de la escala EDSS respecto al valor basal a los 2 años3 (IC del 95 %) |
-0,14 (-0,25; -0,02) (p = 0,42) |
-0,14 (-0,29; 0,01) |
-0,17 (-0,29; -0,05) (p < 0,0001) |
0,24 (0,07; 0,41) |
| Puntos finales de RMN (0-2 años) |
||||
| Mediana del cambio en % del volumen de lesiones en imágenes de RMN ponderadas en T2 |
-9,3 (-19,6; -0,2) (p = 0,31) |
-6,5 (-20,7; 2,5) |
-1,3 (p = 0,14) |
-1,2 |
| Número de pacientes con nuevas lesiones o aumento de lesiones en imágenes ponderadas en T2 a los 2 años |
48,5 % (p = 0,035) |
57,6 % |
46,2 % (p < 0,0001) |
67,9 % |
| Número de pacientes con lesiones que captan gadolinio a los 2 años |
15,4 % (p = 0,001) |
27,0 % |
18,5 % (p < 0,0001) |
34,2 % |
| Número de pacientes con nuevas lesiones hipointensas en imágenes ponderadas en T1 a los 2 años |
24,0 % (p = 0,055) |
31,4 % |
19,9 % (p < 0,0001) |
38,0 % |
| Mediana del cambio en % de la fracción parenquimatosa del cerebro |
-0,867 (p < 0,0001) |
-1,488 |
-0,615 (p = 0,012) |
-0,810 |
-
Los componentes del punto final primario fueron la RRP y la DPI. El estudio se consideraba exitoso si se alcanzaba al menos uno de estos dos componentes del punto final primario.
-
La DPI se definía como un incremento de al menos 1,0 punto en la Escala Ampliada del Estado de Discapacidad (EDSS) respecto al valor basal de la puntuación EDSS (para pacientes con una puntuación basal EDSS de 0, se consideraba un incremento de 1,5 puntos), que se mantenía durante 6 meses.
-
Se evaluó utilizando un modelo mixto para mediciones repetidas.
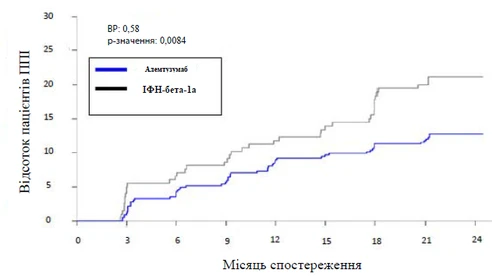
Fig. Tiempo hasta la confirmación del empeoramiento de la discapacidad (durante un período de hasta 6 meses) en el estudio 2
Gravedad de las recaídas
En el estudio sobre el impacto del tratamiento en la frecuencia de recaídas, análisis complementarios realizados en el marco del estudio 1 (CAMMS323) mostraron que el uso del medicamento Lemtrada a una dosis de 12 mg/día resultó en un número estadísticamente significativo menor de pacientes con recaídas graves (reducción del 61 %, p = 0,0056) y un número estadísticamente significativo menor de recaídas que requirieron el uso de medicamentos esteroideos (reducción del 58 %, p < 0,0001), en comparación con el grupo que recibió IFNB-1a.
Análisis complementarios realizados en el marco del estudio 2 (CAMMS32400507) mostraron que el uso del medicamento Lemtrada a una dosis de 12 mg/día resultó en un número estadísticamente significativo menor de pacientes con recaídas graves (reducción del 48 %, p = 0,0121) y un número estadísticamente significativo menor de recaídas que requirieron el uso de medicamentos esteroideos (reducción del 56 %, p < 0,0001) o hospitalización (reducción del 55 %, p = 0,0045), en comparación con el grupo que recibió IFNB-1a.
Mejoría confirmada del grado de discapacidad (MCD)
El tiempo hasta la MCD se definió como una disminución de al menos 2 puntos en la escala EDSS respecto al valor basal de la puntuación EDSS, mantenida durante al menos 6 meses. La MCD es un indicador de mejoría sostenida del grado de discapacidad. En el estudio 2, el 29 % de los pacientes que recibieron Lemtrada alcanzaron la MCD, mientras que solo el 13 % de los pacientes del grupo que recibió IFNB-1a subcutáneo alcanzaron este punto final. Esta diferencia fue estadísticamente significativa (p = 0,0002).
En el estudio 3 (estudio de fase II CAMMS223) se evaluaron la seguridad y eficacia de Lemtrada en pacientes con EMRR durante 3 años. Al momento del reclutamiento, los pacientes tenían una puntuación EDSS entre 0 y 3,0, al menos 2 episodios clínicos de EM en los últimos 2 años y ≥ 1 lesión de captación de gadolinio. Los pacientes no habían recibido previamente ningún tratamiento para la EM. Los pacientes recibieron Lemtrada a una dosis de 12 mg/día (N = 108) o 24 mg/día (N = 108), administrado una vez al día durante 5 días en el mes 0 y durante 3 días en el mes 12, o inyecciones subcutáneas de IFNB-1a a 44 mcg (N = 107), administradas tres veces por semana durante 3 años. Cuarenta y seis pacientes recibieron un tercer ciclo de tratamiento con Lemtrada a 12 mg/día o 24 mg/día durante 3 días en el mes 24.
Tras 3 años de tratamiento con Lemtrada, se observó una reducción del 76 % en el riesgo de DPI mantenida durante 6 meses (razón de riesgos 0,24 [IC del 95 %: 0,110; 0,545], p < 0,0006) y una reducción del 67 % en la TCR (razón de tasas 0,33 [IC del 95 %: 0,196; 0,552], p < 0,0001) en comparación con los valores correspondientes con inyecciones subcutáneas de IFNB-1a. El uso de Lemtrada a una dosis de 12 mg/día resultó en puntuaciones EDSS estadísticamente significativas más bajas (mejoría respecto al nivel basal) durante 2 años de seguimiento en comparación con IFNB-1a (p < 0,0001).
En el subgrupo de pacientes con EMRR con 2 o más recaídas en el año previo y al menos 1 lesión de captación de gadolinio en imágenes ponderadas en T1 al inicio, la tasa anual de recaídas fue de 0,26 (IC del 95 %: 0,20; 0,34) en el grupo tratado con Lemtrada (n = 205) y de 0,51 (IC del 95 %: 0,40; 0,64) en el grupo tratado con IFNB-1a (n = 102) (p < 0,0001). Este análisis incluyó únicamente datos de estudios de fase III (CAMMS324 y CAMMS323) debido a diferencias en los protocolos de lectura de imágenes de RM entre estudios de fase II y estudios de fase III. Estos resultados se obtuvieron en un análisis post hoc y deben interpretarse con precaución.
Datos de eficacia a largo plazo
El estudio 4 fue un estudio de fase III, multicéntrico, abierto, con enmascaramiento para el evaluador, ampliado para evaluar la eficacia y seguridad, que incluyó pacientes con EMRR que participaron en los estudios 1, 2 o 3 (estudios previos de fases II y III), con el objetivo de determinar la eficacia y seguridad a largo plazo del uso de Lemtrada. Este estudio proporcionó datos de eficacia y seguridad durante un período cuya mediana de duración fue de 6 años, a partir del momento del reclutamiento en los estudios 1 y 2. Los pacientes en este estudio ampliado (estudio 4) podían recibir ciclos adicionales de tratamiento con Lemtrada si se documentaba una reactivación de la enfermedad, definida como la aparición de ≥ 1 recaída de EM y/o la aparición de ≥ 2 nuevas o el aumento de ≥ 2 lesiones existentes en el cerebro o médula espinal según imágenes de resonancia magnética (RM). Los ciclos adicionales de tratamiento con Lemtrada se administraban a una dosis de 12 mg/día durante 3 días consecutivos (dosis total: 36 mg), no antes de 12 meses tras el ciclo anterior de tratamiento.
El 91,8 % de los pacientes que recibieron Lemtrada a 12 mg en los estudios 1 y 2 fueron incluidos en el estudio 4. El 82,7 % de estos pacientes completaron el estudio. Aproximadamente la mitad (51,2 %) de los pacientes que inicialmente recibieron Lemtrada a 12 mg/día en los estudios 1 o 2 y que fueron incluidos en el estudio 4, recibieron únicamente esos dos ciclos iniciales de tratamiento con Lemtrada y no recibieron ningún otro medicamento de terapia modificadora de la enfermedad durante 6 años de seguimiento.
El 46,6 % de los pacientes que inicialmente recibieron Lemtrada a 12 mg/día en los estudios 1 o 2 recibieron ciclos adicionales debido a evidencia documentada de reactivación de la EM (aparición de recaídas y/o cambios en RM) y según decisión del médico tratante sobre la necesidad de un nuevo ciclo de tratamiento. Ninguna característica basal permitió identificar a los pacientes que posteriormente recibieron uno o más ciclos adicionales de tratamiento.
Durante los 6 años posteriores al tratamiento inicial con Lemtrada, los pacientes seguidos mostraron tasas similares de recaídas de EM, formación de lesiones en el cerebro según RM y pérdida de tejido cerebral, lo que concuerda con los efectos del tratamiento con Lemtrada observados en los estudios 1 y 2, así como puntuaciones de discapacidad generalmente estables o mejoradas. Considerando el período de seguimiento en el estudio 4, los pacientes que inicialmente recibieron Lemtrada en los estudios 1 y 2 tuvieron una TCR de 0,17 y 0,23, respectivamente, una DPI en el 22,3 % y 29,7 %, y alcanzaron la MCD en el 32,7 % y 42,5 %. Cada año durante el estudio 4, los pacientes de ambos estudios principales continuaron mostrando un bajo riesgo de formación de nuevas lesiones en imágenes ponderadas en T2 (entre 27,4 % y 33,2 %) o lesiones que captan gadolinio (entre 9,4 % y 13,5 %), y la mediana del cambio anual en la fracción parenquimatosa del cerebro en porcentaje osciló entre 0,19 % y -0,09 %.
En los pacientes que recibieron uno o dos ciclos adicionales de tratamiento con Lemtrada, se observó una mejora en la frecuencia de recaídas, la actividad de la enfermedad según RM y la puntuación media en la escala EDSS tras el primer o segundo ciclo de tratamiento adicional con Lemtrada (ciclos 3 y 4) en comparación con los resultados del año anterior. En estos pacientes, la TCR disminuyó de 0,79 en el año previo al ciclo 3 a 0,18 en el año posterior a este ciclo, y la puntuación media en la escala EDSS pasó de 2,89 a 2,69. El porcentaje de pacientes con formación de nuevas lesiones o aumento de lesiones existentes en imágenes ponderadas en T2 disminuyó del 50,8 % en el año previo al ciclo 3 al 35,9 % en el año posterior a este ciclo, y para lesiones que captan gadolinio, la disminución fue del 32,2 % al 11,9 %. Mejoras similares en la TCR, la puntuación media en la escala EDSS, lesiones en imágenes ponderadas en T2 y lesiones que captan gadolinio se observaron también tras el ciclo 4 en comparación con el año anterior. Estas mejoras se mantuvieron posteriormente, aunque no se pueden hacer conclusiones claras sobre la eficacia a largo plazo del tratamiento (por ejemplo, a los 3 y 4 años tras los ciclos adicionales), ya que muchos pacientes finalizaron su participación en el estudio antes de alcanzar estos puntos temporales.
Los beneficios y riesgos de 5 o más ciclos de tratamiento aún no se han establecido.
Inmunogenicidad
Como otros medicamentos proteicos, este producto tiene potencial inmunogénico. Los datos disponibles indican que un cierto porcentaje de pacientes tuvo resultados positivos en pruebas de anticuerpos contra alemtuzumab mediante ensayo inmunoenzimático (ELISA), con confirmación posterior mediante un método competitivo de unión. Las muestras con resultados positivos se evaluaron adicionalmente para detectar inhibición in vitro mediante citometría de flujo. En estudios clínicos de EM, se obtuvieron muestras de suero sanguíneo de pacientes a los 1, 3 y 12 meses tras cada ciclo de tratamiento para determinar la presencia de anticuerpos contra alemtuzumab. Aproximadamente el 85 % de los pacientes que recibieron Lemtrada tuvieron resultados positivos para anticuerpos contra alemtuzumab durante el estudio, y en ≥ 90 % de estos pacientes también se obtuvieron resultados positivos para anticuerpos que inhibieron la unión de alemtuzumab in vitro. En los pacientes que desarrollaron anticuerpos contra alemtuzumab, esto ocurrió dentro de los 15 meses posteriores a la primera administración del medicamento. Durante los 2 ciclos de tratamiento, no se observó ninguna relación entre la presencia de anticuerpos contra alemtuzumab o anticuerpos inhibidores y una disminución de la eficacia del medicamento, cambios en su farmacodinámica o aparición de reacciones adversas, incluidas las asociadas con la infusión. Los títulos elevados de anticuerpos contra alemtuzumab observados en algunos pacientes se asociaron con una eliminación incompleta de linfocitos tras el tercer o cuarto ciclo de tratamiento, aunque no se observó un impacto claro de los anticuerpos contra alemtuzumab sobre la eficacia clínica o el perfil de seguridad de Lemtrada.
Los datos sobre la frecuencia de formación de anticuerpos dependen en gran medida de la sensibilidad y especificidad del ensayo. Además, diversos factores pueden influir en los datos obtenidos sobre la frecuencia de resultados positivos en pruebas de anticuerpos (incluidos anticuerpos inhibidores), tales como la metodología del ensayo, el manejo de las muestras, el momento de la toma de muestras, medicamentos concomitantes y la enfermedad subyacente. Por ello, la comparación de la frecuencia de formación de anticuerpos contra Lemtrada con la de otros medicamentos podría no ser apropiada.
Pacientes pediátricos
La Agencia Europea de Medicamentos eximió al fabricante de la obligación de presentar resultados de estudios sobre el uso de alemtuzumab en niños desde el nacimiento hasta menos de 10 años para el tratamiento de la esclerosis múltiple (la información sobre el uso del medicamento en niños se proporciona en la sección «Posología y forma de administración»).
La Agencia Europea de Medicamentos otorgó al fabricante una prórroga respecto a la obligación de presentar resultados de estudios sobre el uso de Lemtrada en uno o varios subgrupos de la población pediátrica con EMRR (la información sobre el uso del medicamento en niños se proporciona en la sección «Posología y forma de administración»).
Farmacocinética. Las propiedades farmacocinéticas de alemtuzumab se evaluaron en 216 pacientes con EMRR que recibieron el medicamento mediante infusiones intravenosas a una dosis de 12 mg/día o 24 mg/día durante 5 días consecutivos, y posteriormente durante 3 días consecutivos a los 12 meses tras el ciclo inicial de tratamiento. Las concentraciones del medicamento en suero aumentaron con cada dosis sucesiva durante el ciclo de tratamiento, alcanzando concentraciones máximas tras la última infusión del ciclo. La administración del medicamento a 12 mg/día resultó en una Cmáx media de 3014 ng/ml en el día 5 del ciclo inicial de tratamiento y de 2276 ng/ml en el día 3 del segundo ciclo de tratamiento. La fase alfa del periodo de semivida fue de aproximadamente 4-5 días y fue comparable entre ciclos de tratamiento, lo que determinó concentraciones bajas o indetectables en suero aproximadamente a los 30 días tras cada ciclo de tratamiento.
Alemtuzumab es una proteína cuya vía esperada de metabolismo es la degradación en péptidos pequeños y aminoácidos individuales por una amplia gama de enzimas proteolíticas. No se realizaron estudios clásicos de biotransformación.
Dada la información disponible, no se pueden hacer conclusiones definitivas sobre el impacto del origen racial y el sexo del paciente en la farmacocinética de alemtuzumab. La farmacocinética de alemtuzumab en EMRR no se ha estudiado en pacientes de 55 años o más.
Datos preclínicos de seguridad
Carcinogénesis y mutagénesis
Actualmente no se han realizado estudios sobre el potencial carcinogénico o mutagénico de alemtuzumab.
Fertilidad y reproducción
La administración intravenosa de alemtuzumab a dosis de hasta 10 mg/kg/día durante 5 días consecutivos (incremento del nivel de AUC en 7,1 veces respecto a la dosis diaria recomendada) no tuvo ningún efecto sobre la fertilidad ni las funciones reproductivas en ratones transgénicos huCD52 machos. La cantidad de espermatozoides normales disminuyó significativamente (< 10 %) en comparación con los animales control, y el porcentaje de espermatozoides anómalos (con cabezas separadas o sin cabezas) aumentó significativamente (hasta el 3 %). Sin embargo, estos cambios no afectaron la fertilidad y por tanto no se consideraron adversos.
En hembras de ratón que recibieron alemtuzumab por vía intravenosa a dosis de hasta 10 mg/kg/día (incremento del nivel de AUC en 4,7 veces respecto a la dosis diaria recomendada) durante 5 días consecutivos antes del apareamiento con machos de ratón tipo salvaje, el número medio de cuerpos amarillos y de sitios de implantación por animal disminuyó significativamente en comparación con los animales que recibieron placebo. En ratones preñados que recibieron el medicamento a 10 mg/kg/día, se observó una reducción en la ganancia de peso durante el embarazo en comparación con los animales control que recibieron placebo.
En un estudio de toxicidad reproductiva con alemtuzumab en ratones preñados que recibieron el medicamento por vía intravenosa a dosis de hasta 10 mg/kg/día (incremento del nivel de AUC en 2,4 veces respecto a la dosis recomendada de 12 mg/día) durante 5 días consecutivos durante el embarazo, se observó un aumento significativo en el número de hembras con pérdida total de embriones/fetos o reabsorción, junto con una disminución concomitante en el número de hembras con fetos viables. Con dosis de hasta 10 mg/kg/día no se observaron malformaciones ni variaciones anómalas en tejidos externos, blandos o esqueléticos.
Durante el embarazo y tras el nacimiento de la descendencia se observó el paso del medicamento a través de la placenta y la actividad farmacológica potencial de alemtuzumab. En estudios en ratones, la descendencia expuesta a alemtuzumab durante el desarrollo intrauterino con dosis de 3 mg/kg/día durante 5 días consecutivos (incremento del nivel de AUC en 0,6 veces respecto a la dosis recomendada de 12 mg/día) mostró cambios en el número de linfocitos. Con dosis de hasta 10 mg/kg/día no se observaron alteraciones en el desarrollo cognitivo, físico ni sexual de la descendencia expuesta a alemtuzumab durante la lactancia.
Características clínicas.
Indicaciones.
El medicamento Lemtrada está indicado como tratamiento modificador de la enfermedad para la monoterapia de adultos con esclerosis múltiple remitente-recidivante altamente activa (EMRR), pertenecientes a los siguientes grupos:
- pacientes con enfermedad altamente activa a pesar de un tratamiento completo y adecuado con al menos un medicamento modificador de la evolución de la enfermedad, o
- pacientes con esclerosis múltiple remitente-recidivante grave y de rápida progresión, definida como 2 o más brotes incapacitantes en un año, con 1 o más lesiones que realzan con gadolinio en la resonancia magnética (RM) del cerebro, o con un aumento significativo del volumen total de lesiones en secuencias T2 en comparación con una RM previa reciente.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento.
Infección por el virus de la inmunodeficiencia humana (VIH).
Infección grave activa (hasta la recuperación completa del paciente).
Hipertensión arterial no controlada.
Antecedentes de disección de las paredes arteriales de la cabeza o del cuello.
Antecedentes de accidente cerebrovascular (ACV).
Antecedentes de angina de pecho o infarto de miocardio.
Presencia de coagulopatía conocida o tratamiento concomitante con terapia antiagregante o anticoagulante.
Otras enfermedades autoinmunes concomitantes (excepto la EM).
Precauciones especiales de seguridad.
El contenido del frasco debe inspeccionarse antes de la administración para detectar partículas extrañas o cambios de color. No se debe utilizar el medicamento si se observan partículas extrañas o si el color no es adecuado.
No agitar los frascos antes de su uso.
Para la administración intravenosa, se deben extraer 1,2 ml del medicamento Lemtrada del frasco con una técnica aséptica y colocarlos en una jeringa. El medicamento se debe administrar en 100 ml de solución de cloruro de sodio al 9 mg/ml (0,9 %) para perfusión o solución de glucosa al 5 % para perfusión. Este medicamento no debe diluirse con otros disolventes. Para mezclar la solución, se debe girar suavemente el bolsa.
Debe tenerse precaución para garantizar la esterilidad de la solución preparada. Se recomienda utilizar la solución diluida inmediatamente. Cada frasco está destinado únicamente para uso único.
Cualquier resto de medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
Interacción con otros medicamentos y otras formas de interacción.
Hasta la fecha no se han realizado estudios formales sobre interacciones medicamentosas con Lemtrada en las dosis recomendadas en pacientes con EM. En un ensayo clínico controlado en EM, a los pacientes que recientemente habían recibido interferón beta o acetato de glatiramero se les exigió interrumpir el tratamiento con estos medicamentos 28 días antes del inicio del tratamiento con Lemtrada.
Características de uso.
No se recomienda utilizar el medicamento Lemtrada en pacientes con enfermedad inactiva ni en pacientes cuya enfermedad esté estable con su terapia actual.
A los pacientes tratados con Lemtrada se les debe entregar la Hoja informativa para uso médico, la Tarjeta de información para el paciente y las Recomendaciones para el paciente. Antes de iniciar el tratamiento, se debe proporcionar a los pacientes información sobre los riesgos y beneficios del uso del medicamento, así como sobre la necesidad de realizarse exámenes desde el inicio del tratamiento y hasta al menos 48 meses después de la última infusión del segundo ciclo de tratamiento con Lemtrada. Si se prescribe un ciclo adicional de tratamiento, el período de seguimiento para evaluar la seguridad debe prolongarse hasta al menos 48 meses después de la última infusión del medicamento.
Seguimiento
Para mejorar el seguimiento de medicamentos biológicos, el nombre comercial y el número de lote del medicamento administrado deben registrarse claramente en la documentación médica del paciente.
Reacciones autoinmunes
El tratamiento con este medicamento puede provocar la formación de autoanticuerpos y aumentar el riesgo de estados mediados por autoinmunidad, que pueden ser graves y poner en peligro la vida. Las enfermedades autoinmunes notificadas incluyen trastornos de la glándula tiroides, púrpura trombocitopénica inmunitaria (PTI), nefropatías (por ejemplo, enfermedad por anticuerpos contra la membrana basal glomerular), hepatitis autoinmune, hemofilia adquirida A, púrpura trombocitopénica trombótica, sarcoidosis y encefalitis autoinmune. Durante el uso poscomercialización tras el tratamiento con Lemtrada, se han notificado casos de múltiples trastornos autoinmunes. En pacientes con reacciones autoinmunes notificadas, se debe evaluar la posibilidad de otros estados mediados por autoinmunidad (véase la sección «Contraindicaciones»). Los pacientes y médicos deben estar informados sobre la posibilidad de aparición tardía de trastornos autoinmunes después del período de monitoreo de 48 meses.
Nefropatías
Se han observado nefropatías, incluyendo enfermedad por anticuerpos contra la membrana basal glomerular (anti-MBG), en 6 (0,4 %) pacientes durante los estudios clínicos de EM, con una mediana de seguimiento de 6,1 años (máximo de 12 años), y generalmente se desarrollaron dentro de los 39 meses posteriores a la última administración de Lemtrada. Durante los estudios clínicos se registraron 2 casos de enfermedad anti-MBG. Ambos casos fueron graves, se detectaron tempranamente gracias al monitoreo clínico y de laboratorio y tuvieron evolución favorable tras el tratamiento.
Las manifestaciones clínicas de nefropatía pueden incluir aumento de la creatinina sérica, hematuria y/o proteinuria. Aunque no se han observado casos durante los estudios clínicos, en la enfermedad anti-MBG puede ocurrir hemorragia alveolar que se manifiesta como hemoptisis. La hemoptisis también puede indicar PTI o hemofilia adquirida A (véase más arriba), por lo que se debe realizar un diagnóstico diferencial adecuado. Se debe recordar a los pacientes la necesidad de vigilar cualquier síntoma y buscar atención médica inmediata ante cualquier motivo de preocupación. La enfermedad anti-MBG puede provocar insuficiencia renal que requiera diálisis y/o trasplante renal si no se inicia un tratamiento terapéutico inmediato, y puede ser mortal si no se trata.
Antes de iniciar el tratamiento, se debe determinar el nivel de creatinina en suero y repetir este análisis mensualmente durante el tratamiento y hasta al menos 48 meses después de la última infusión del medicamento. Antes de iniciar el tratamiento, se debe realizar un análisis general de orina con microscopía del sedimento y repetirlo mensualmente durante el tratamiento y hasta al menos 48 meses después de la última infusión del medicamento. La detección de cambios clínicamente significativos en los niveles séricos de creatinina en comparación con los valores basales, hematuria de origen desconocido y/o proteinuria requiere una evaluación inmediata para detectar nefropatía, incluyendo derivación urgente a un especialista. La detección y tratamiento tempranos de nefropatías pueden reducir el riesgo de consecuencias clínicas adversas. Posteriormente, los análisis deben realizarse según los datos clínicos que puedan indicar la aparición de nefropatías.
Trastornos de la glándula tiroides
Trastornos endocrinos de la glándula tiroides, incluyendo trastornos autoinmunes de la tiroides, se observaron en 36,8 % de los pacientes que recibieron Lemtrada a una dosis de 12 mg en estudios clínicos de EM, con una mediana de seguimiento de 6,1 años (máximo de 12 años) tras la primera administración de Lemtrada. La frecuencia de eventos adversos de la tiroides fue mayor en pacientes con antecedentes de trastornos tiroideos, tanto en el grupo que recibió Lemtrada como en el grupo que recibió interferón beta 1a (IFNB-1a). Los trastornos autoinmunes tiroideos registrados incluyeron hipertiroidismo e hipotiroidismo. La mayoría de los casos fueron de gravedad leve o moderada. Los eventos adversos endocrinos graves se desarrollaron en 4,4 % de los pacientes, con hipertiroidismo, hipotiroidismo, tiroiditis autoinmune, bocio y enfermedad de Basedow (también conocida como enfermedad de Graves) en más de un paciente. La mayoría de los eventos adversos tiroideos se trataron con intervenciones medicamentosas estándar, aunque algunos pacientes requirieron cirugía. En pacientes con tiroiditis autoinmune confirmada por biopsia durante el período poscomercialización, previamente se habían registrado trastornos autoinmunes tiroideos.
Antes de iniciar el tratamiento, se deben realizar análisis para evaluar la función tiroidea, como la determinación de niveles de hormona estimulante de la tiroides (TSH), y repetirlos cada 3 meses durante el tratamiento y hasta al menos 48 meses después de la última infusión del medicamento. Posteriormente, los análisis deben realizarse según los datos clínicos que puedan indicar la aparición de trastornos de la función tiroidea o en caso de embarazo.
Las enfermedades de la glándula tiroides representan un riesgo especial para las mujeres embarazadas (véase la sección «Uso durante el embarazo o la lactancia»).
En estudios clínicos, los eventos adversos tiroideos ocurrieron en 74 % de los pacientes con resultados positivos en la determinación de anticuerpos contra la peroxidasa tiroidea (TPO) en el valor basal, en comparación con 38 % de los pacientes con resultados negativos en la determinación de estos anticuerpos en el valor basal. La mayoría abrumadora (aproximadamente 80 %) de los pacientes que desarrollaron eventos adversos tiroideos tras el tratamiento tenían resultados negativos en la determinación de anticuerpos anti-TPO en el valor basal. Por lo tanto, las reacciones adversas tiroideas pueden ocurrir independientemente del estado de anticuerpos anti-TPO antes del tratamiento con el medicamento, por lo que se deben realizar periódicamente todos los análisis adecuados, como se describe anteriormente.
Citopenias
Durante los estudios clínicos de EM se observaron casos infrecuentes de citopenias autoinmunes sospechosas, como neutropenia, anemia hemolítica y pancitopenia. Se deben realizar análisis sanguíneos completos (véase más arriba en la sección sobre PTI) para monitorear citopenias, incluyendo neutropenia. Si se confirma el desarrollo de citopenia, se recomienda tratamiento inmediato y adecuado, incluyendo derivación al especialista.
Hepatitis autoinmune y afectación hepática
En pacientes que recibieron Lemtrada se han observado casos de hepatitis autoinmune (incluyendo casos fatales o que requirieron trasplante hepático) y afectación hepática relacionada con infecciones (véase la sección «Contraindicaciones»). Se debe evaluar la función hepática antes de iniciar la terapia con el medicamento y mensualmente hasta al menos 48 meses después de la última infusión del medicamento. Los pacientes deben informarse sobre el riesgo de hepatitis autoinmune, afectación hepática y los síntomas correspondientes.
Linfohistiocitosis hemofagocítica (LHH)
Durante el uso poscomercialización del medicamento en pacientes que recibieron Lemtrada, se han observado casos de LHH (incluyendo casos fatales). La LHH es un síndrome potencialmente mortal de activación inmune patológica, caracterizado por signos y síntomas clínicos de inflamación sistémica extremadamente marcada. La LHH se manifiesta con fiebre, hepatomegalia y citopenias. Está asociada con una alta mortalidad si no se diagnostica y trata precozmente. Se ha observado que los síntomas aparecen entre varios meses y cuatro años después del inicio del tratamiento. Los pacientes deben informarse sobre los síntomas de LHH y su momento de aparición. En pacientes con signos tempranos de activación inmune patológica, se debe realizar una evaluación inmediata y considerar el diagnóstico probable de LHH.
Reacciones asociadas a la infusión (RAI)
En estudios clínicos, las reacciones asociadas a la infusión (RAI) se definieron como cualquier evento adverso que ocurre durante o dentro de las 24 horas posteriores a la infusión de Lemtrada. La mayoría de estos eventos pueden deberse a la liberación de citoquinas durante la infusión. La mayoría de los pacientes que recibieron Lemtrada durante estudios clínicos de EM experimentaron RAI de gravedad leve o moderada durante o dentro de las 24 horas posteriores a la administración de Lemtrada a una dosis de 12 mg. La frecuencia de RAI fue mayor durante el primer ciclo de tratamiento en comparación con ciclos posteriores. Considerando todos los datos disponibles de seguimiento, incluyendo pacientes que recibieron ciclos adicionales de tratamiento, las RAI más frecuentes fueron cefalea, erupción cutánea, pirexia, náuseas, urticaria, prurito, insomnio, escalofríos, hiperemia, fatiga, disnea, disgeusia, molestias en el pecho, erupción generalizada, taquicardia, bradicardia, dispepsia, mareo y dolor. Las reacciones graves ocurrieron en 3 % de los pacientes e incluyeron cefalea, pirexia, urticaria, taquicardia, fibrilación auricular, náuseas, molestias en el pecho e hipotensión arterial. Las manifestaciones clínicas de anafilaxia pueden ser similares a las asociadas con reacciones a la infusión, pero tienden a ser más graves o potencialmente mortales. Las reacciones anafilácticas se han notificado raramente en comparación con las reacciones asociadas a la infusión.
Para reducir los efectos de las reacciones a la infusión, se recomienda la premedicación en los pacientes (véase la sección «Posología y forma de administración»).
La mayoría de los pacientes en estudios clínicos controlados recibieron antihistamínicos y/o antipiréticos antes de al menos una infusión de Lemtrada. Las RAI pueden ocurrir a pesar de la premedicación. Se recomienda observar a los pacientes durante y al menos 2 horas después de la infusión de Lemtrada para detectar reacciones a la infusión. Si es necesario, puede ser conveniente un período de observación más prolongado (hospitalización). En caso de reacciones graves a la infusión, la infusión intravenosa debe interrumpirse inmediatamente. Deben estar disponibles medios para tratar anafilaxia y otras reacciones graves (véase más abajo).
Enfermedad de Still del adulto (ESA)
Durante el uso poscomercialización, se han notificado casos de enfermedad de Still del adulto (ESA) en pacientes tratados con Lemtrada. La ESA es una enfermedad inflamatoria rara que requiere evaluación y tratamiento inmediatos. Los pacientes con ESA pueden presentar combinación de fiebre, artritis, erupción cutánea y leucocitosis en ausencia de infecciones, neoplasias malignas u otras enfermedades reumáticas. Debe considerarse la posibilidad de interrumpir o suspender el tratamiento con Lemtrada si no se identifica una etiología alternativa para estos signos o síntomas.
Otras reacciones graves relacionadas temporalmente con la infusión de Lemtrada
Durante el uso poscomercialización del medicamento, se han notificado casos raros, graves, a veces fatales e impredecibles de eventos adversos graves en múltiples sistemas orgánicos. En la mayoría de los casos, el tiempo hasta el inicio de la reacción fue de 1 a 3 días tras la infusión de Lemtrada. Las reacciones ocurrieron tras cualquier dosis del medicamento y tras ciclos posteriores de terapia. Los pacientes deben informarse sobre los signos y síntomas de estas afecciones y su momento de aparición. Se debe recomendar a los pacientes buscar atención médica inmediata si presentan alguno de estos síntomas. Los pacientes deben informarse sobre la posibilidad de aparición tardía de estos eventos.
Accidente cerebrovascular hemorrágico
Varios pacientes con casos documentados de accidente cerebrovascular hemorrágico tenían menos de 50 años, no tenían antecedentes de hipertensión arterial, trastornos hemorrágicos ni uso de anticoagulantes o inhibidores plaquetarios. En algunos pacientes, antes del desarrollo de hemorragia se observó un aumento de la presión arterial en comparación con sus niveles basales.
Isquemia miocárdica e infarto de miocardio
Varios pacientes con casos documentados de estos trastornos tenían menos de 40 años y no tenían factores de riesgo para enfermedad isquémica cardiaca. Se observó que en algunos de estos pacientes, durante la infusión del medicamento, hubo cambios temporales respecto a los valores normales en la presión arterial y/o frecuencia cardíaca.
Desgarro de las arterias de la cabeza y el cuello
Se han notificado casos de desgarro de las arterias de la cabeza y el cuello, incluyendo desgarros múltiples, tanto en los primeros días tras la infusión de Lemtrada como en momentos posteriores dentro del primer mes tras la infusión.
Hemorragia pulmonar alveolar
Se han documentado casos de este trastorno relacionado temporalmente con el uso del medicamento, sin asociación con la enfermedad anti-MBG (síndrome de Goodpasture).
Trombocitopenia
Se han documentado casos de trombocitopenia que ocurrieron en los primeros días tras la infusión del medicamento (a diferencia de la PTI). La trombocitopenia desapareció frecuentemente espontáneamente y fue relativamente leve, aunque en muchos casos la gravedad y consecuencias clínicas fueron desconocidas.
Pericarditis
Se han notificado casos raros de pericarditis, derrame pericárdico y otros eventos pericárdicos, tanto como parte de una reacción aguda a la infusión como posteriormente.
Neumonitis
Se han notificado casos de neumonitis en pacientes que recibieron infusión de Lemtrada. La mayoría de estos casos ocurrieron durante el primer mes tras el tratamiento con Lemtrada. Los pacientes deben informarse sobre la necesidad de notificar síntomas de neumonitis, que pueden incluir disnea, tos, sibilancias, dolor o sensación de opresión en el pecho y hemoptisis.
Instrucciones para la infusión destinadas a reducir el riesgo de reacciones graves relacionadas temporalmente con la infusión de Lemtrada
- Evaluaciones previas a la infusión:
- Se debe realizar una evaluación de los valores basales del ECG y signos vitales principales, incluyendo frecuencia cardíaca y presión arterial.
- Se deben realizar análisis de laboratorio (hemograma completo con recuento diferencial de leucocitos, determinación de niveles de transaminasas en suero, determinación de niveles de creatinina en suero, análisis para evaluar función tiroidea y análisis general de orina con microscopía del sedimento).
- Durante la infusión:
- Se debe realizar un control continuo o frecuente (al menos cada hora) de la frecuencia cardíaca, presión arterial y estado clínico general de los pacientes.
- La infusión debe interrumpirse:
- Se debe realizar un control continuo o frecuente (al menos cada hora) de la frecuencia cardíaca, presión arterial y estado clínico general de los pacientes.
- En caso de evento adverso grave.
- Si el paciente presenta síntomas clínicos que sugieran el desarrollo de un evento adverso grave relacionado con la infusión (isquemia miocárdica, accidente cerebrovascular hemorrágico, desgarro de las arterias de la cabeza y el cuello o hemorragia pulmonar alveolar).
- Después de la infusión:
- Se recomienda observar al paciente durante al menos 2 horas tras la infusión de Lemtrada para detectar reacciones a la infusión. Los pacientes con síntomas clínicos que sugieran el desarrollo de un evento adverso grave relacionado con la infusión (isquemia miocárdica, accidente cerebrovascular hemorrágico, desgarro de las arterias de la cabeza y el cuello o hemorragia pulmonar alveolar) deben ser vigilados estrechamente hasta la desaparición completa de tales síntomas. Si es necesario, puede ser conveniente un período de observación más prolongado (hospitalización). Se debe informar a los pacientes sobre la posibilidad de aparición tardía de reacciones asociadas a la infusión y se les debe instruir sobre la necesidad de notificar sus síntomas y buscar atención médica adecuada.
Los niveles de plaquetas deben determinarse inmediatamente tras la infusión, en el día 3 y día 5 del primer ciclo de infusión, así como inmediatamente tras la infusión en el día 3 de cualquier ciclo posterior. En caso de trombocitopenia clínicamente significativa, se debe vigilar al paciente hasta su resolución. Debe considerarse la derivación del paciente a un hematólogo.
Infecciones
En estudios clínicos controlados de EM de hasta 2 años de duración, las infecciones ocurrieron en 71 % de los pacientes que recibieron Lemtrada a una dosis de 12 mg, en comparación con 53 % de los pacientes que recibieron inyecciones subcutáneas de interferón beta 1a [IFNB-1a] (44 mcg 3 veces por semana), y fueron principalmente de gravedad leve o moderada. Las infecciones que ocurrieron con mayor frecuencia en pacientes que recibieron Lemtrada que en los que recibieron IFNB-1a incluyeron nasofaringitis, infección del tracto urinario, infección de las vías respiratorias superiores, sinusitis, herpes oral, gripe y bronquitis. Las infecciones graves en estudios clínicos controlados de EM ocurrieron en 2,7 % de los pacientes que recibieron Lemtrada, en comparación con 1 % de los que recibieron IFNB-1a. Las infecciones graves en el grupo que recibió Lemtrada incluyeron apendicitis, gastroenteritis, neumonía, herpes zóster y infección dental. Las infecciones generalmente tuvieron duración típica y se curaron con tratamientos medicamentosos estándar.
La frecuencia anual acumulada de infecciones fue de 0,99 con una mediana de seguimiento de 6,1 años (máximo de 12 años) tras la primera administración de Lemtrada, en comparación con 1,27 en estudios clínicos controlados.
En estudios clínicos, las infecciones graves por el virus de la varicela-zóster, incluyendo varicela primaria y reactivación del virus, ocurrieron con mayor frecuencia (0,4 %) en pacientes que recibieron Lemtrada a una dosis de 12 mg que en los que recibieron IFNB-1a (0 %). También se registraron casos (2 %) de infección del cuello uterino por el virus del papiloma humano (VPH) en pacientes que recibieron Lemtrada a una dosis de 12 mg, incluyendo displasia cervical y verrugas genitales. Se recomienda a las pacientes mujeres realizarse cribado anual para VPH.
Se han notificado infecciones por citomegalovirus (CMV), incluyendo casos de reactivación de CMV, en pacientes que recibieron Lemtrada. La mayoría de los casos ocurrieron dentro de los 2 meses tras la administración de alemtuzumab. Antes del inicio del tratamiento, puede evaluarse el estado inmunológico (serológico) según las recomendaciones locales.
En pacientes que recibieron Lemtrada se han notificado casos de infección, incluyendo reactivación del virus de Epstein-Barr (VEB), hepatitis grave y a veces fatal causada por VEB.
En estudios clínicos controlados se notificaron casos de tuberculosis en pacientes que recibieron Lemtrada o IFNB-1a. Casos de tuberculosis activa y latente, incluyendo varios casos de tuberculosis diseminada, se observaron en 0,3 % de los pacientes que recibieron Lemtrada, más frecuentemente en regiones endémicas para esta enfermedad. Antes de iniciar el tratamiento, todos los pacientes deben evaluarse para detectar infección tuberculosa activa o inactiva (latente) según las guías locales.
En pacientes que recibieron Lemtrada se han notificado casos de listeriosis/meningitis por listeria, generalmente dentro del mes tras la infusión de Lemtrada. Para reducir el riesgo de infección, los pacientes que reciben Lemtrada deben evitar el consumo de carne cruda o poco cocida, quesos blandos y productos lácteos no pasteurizados durante 2 semanas antes, durante y al menos 1 mes después de la infusión de Lemtrada.
En estudios clínicos controlados de EM, las infecciones fúngicas superficiales, especialmente candidiasis oral y vaginal, ocurrieron con mayor frecuencia (12 %) en pacientes que recibieron Lemtrada que en los que recibieron IFNB-1a (3 %).
El inicio del tratamiento con Lemtrada debe posponerse en pacientes con infecciones graves activas hasta su recuperación. Los pacientes que reciben Lemtrada deben instruirse sobre la necesidad de notificar síntomas de infección al médico.
Debe iniciarse el uso profiláctico de un antiviral oral contra el herpes desde el primer día del tratamiento con Lemtrada y continuar al menos 1 mes tras cada ciclo de tratamiento. Durante estudios clínicos, los pacientes recibieron aciclovir a 200 mg dos veces al día o un fármaco equivalente.
Lemtrada no se indicó para el tratamiento de EM simultáneamente con medicamentos antineoplásicos o inmunosupresores ni tras su uso. Como con otros inmunomoduladores, debe considerarse el posible impacto de la combinación de estos medicamentos sobre el sistema inmune del paciente al considerar la prescripción de Lemtrada. La administración concomitante de Lemtrada con cualquiera de estos medicamentos puede aumentar el riesgo de inmunosupresión.
Actualmente no hay datos sobre la relación entre el uso de Lemtrada y la reactivación del virus de la hepatitis B (VHB) o del virus de la hepatitis C (VHC), ya que los pacientes con signos de infección activa o crónica por estos virus se excluyeron de los estudios clínicos. Debe considerarse la posibilidad de cribado en pacientes con alto riesgo de infección por VHB y/o VHC antes del inicio del tratamiento con Lemtrada, y debe tenerse precaución al prescribir Lemtrada a pacientes identificados como portadores de VHB y/o VHC, ya que estos pacientes pueden tener un riesgo aumentado de daño hepático irreversible relacionado con la posible reactivación del virus debido a su estado de infección preexistente.
Leucoencefalopatía multifocal progresiva (LMP)
Se han notificado casos raros de LMP (incluyendo casos fatales) en pacientes con EM tras el tratamiento con alemtuzumab. El estado de los pacientes que reciben alemtuzumab debe controlarse para detectar signos que puedan indicar LMP. Un factor de riesgo particularmente importante es la terapia inmunosupresora previa, especialmente otros tratamientos para EM con factores de riesgo conocidos para LMP.
Antes de la aparición de signos o síntomas clínicos, la presencia de LMP puede sospecharse mediante resonancia magnética (RM). Antes del tratamiento inicial y posterior con alemtuzumab, debe realizarse una exploración por RM y evaluarse sus resultados para detectar signos de LMP. Si es necesario, deben realizarse estudios adicionales, incluyendo análisis del líquido cefalorraquídeo para detectar ADN del virus JC, y evaluaciones neurológicas repetidas. El médico debe enfatizar especialmente los síntomas que indican LMP, que el paciente puede no notar (por ejemplo, síntomas cognitivos, neurológicos o psiquiátricos). También se debe recomendar a los pacientes informar a sus familiares o cuidadores sobre su tratamiento, ya que ellos podrían notar tales síntomas que el paciente desconoce. En todos los pacientes con EM que reciben alemtuzumab y presentan síntomas neurológicos y/o signos de lesiones focales cerebrales en RM, debe considerarse LMP como diagnóstico diferencial.
Si se establece el diagnóstico de LMP, no debe iniciarse ni reanudarse el tratamiento con alemtuzumab.
Colecistitis aguda acalculosa
Lemtrada puede aumentar el riesgo de desarrollar colecistitis aguda acalculosa (sin formación de cálculos). En estudios clínicos controlados, el 0,2 % de los pacientes con EM que recibieron Lemtrada desarrollaron colecistitis aguda acalculosa, en comparación con 0 % de los que recibieron IFNB-1a. Durante el uso poscomercialización del medicamento se han notificado casos adicionales de colecistitis aguda acalculosa en pacientes que recibieron Lemtrada. El tiempo hasta la aparición de síntomas varió desde menos de 24 horas hasta 2 meses tras la infusión de Lemtrada. La mayoría de los pacientes recibieron tratamiento conservador con antibióticos y se recuperaron sin cirugía, aunque algunos requirieron colecistectomía. Los síntomas de colecistitis aguda acalculosa incluyen dolor abdominal, dolor a la palpación abdominal, fiebre, náuseas y vómitos. La colecistitis aguda acalculosa es una condición que puede asociarse con alta morbilidad y mortalidad si no se diagnostica y trata precozmente. En caso de sospecha de colecistitis aguda acalculosa, se debe evaluar inmediatamente al paciente y prescribir el tratamiento adecuado.
Neoplasias malignas
Como con otros inmunomoduladores, debe tenerse precaución al iniciar el tratamiento con Lemtrada en pacientes con neoplasias malignas preexistentes o actuales. Actualmente no se sabe si el uso de Lemtrada aumenta el riesgo de desarrollar neoplasias malignas de la glándula tiroides, ya que las reacciones autoinmunes tiroideas por sí mismas pueden ser un factor de riesgo para neoplasias malignas tiroideas.
Anticoncepción
En ratones, durante la gestación y tras el nacimiento de la descendencia, se observó el paso de Lemtrada a través de la placenta y actividad farmacológica potencial. Las mujeres en edad fértil deben usar métodos anticonceptivos eficaces durante el tratamiento y durante 4 meses tras cada ciclo de tratamiento con Lemtrada (véase la sección «Uso durante el embarazo o la lactancia»).
Vacunas
Se recomienda que los pacientes completen todos los procedimientos de inmunización necesarios según los requisitos locales al menos 6 semanas antes del inicio del tratamiento con Lemtrada. La capacidad del organismo para responder inmunológicamente a cualquier vacuna tras el tratamiento con Lemtrada no ha sido estudiada.
La seguridad de la inmunización con vacunas virales vivas tras un ciclo de tratamiento con Lemtrada no ha sido estudiada formalmente en estudios clínicos controlados en pacientes con EM, por lo que no deben administrarse estas vacunas a pacientes con EM que recientemente hayan recibido un ciclo de tratamiento con Lemtrada.
Análisis de anticuerpos contra el virus de la varicela-zóster/vacunación contra el virus de la varicela-zóster. Como con cualquier medicamento inmunomodulador, antes del inicio del tratamiento con Lemtrada en pacientes que no hayan tenido varicela ni hayan sido vacunados contra el virus de la varicela-zóster (VVZ), debe realizarse un análisis para determinar anticuerpos contra el VVZ. En pacientes con resultados negativos en este análisis, debe considerarse la posibilidad de vacunación contra el VVZ antes del inicio del tratamiento con Lemtrada. Para asegurar el efecto completo de la vacunación contra el VVZ, el tratamiento con Lemtrada debe posponerse 6 semanas tras la vacunación.
Análisis de laboratorio recomendados para el monitoreo de pacientes
Los exámenes clínicos y análisis de laboratorio que deben realizarse regularmente durante al menos 48 meses tras el último ciclo de tratamiento con Lemtrada para monitorear signos tempranos de enfermedades autoinmunes:
- Hemograma completo con recuento diferencial de leucocitos, determinación de niveles de transaminasas en suero y determinación de niveles de creatinina en suero (antes del inicio del tratamiento y mensualmente después);
- Análisis general de orina con microscopía del sedimento (antes del inicio del tratamiento y mensualmente después);
- Análisis para evaluar función tiroidea, por ejemplo, determinación del nivel de hormona estimulante de la tiroides (antes del inicio del tratamiento y posteriormente cada 3 meses).
Información sobre el uso de alemtuzumab antes de la autorización comercial de Lemtrada fuera de estudios patrocinados por la compañía
Las siguientes reacciones adversas se identificaron antes de la autorización de Lemtrada con el uso de alemtuzumab para el tratamiento de la leucemia linfocítica crónica B (LLC-B), así como para el tratamiento de otros trastornos, generalmente en dosis más altas (por ejemplo, 30 mg) y con mayor frecuencia de administración que la recomendada para el tratamiento de EM. Dado que estos eventos se notificaron de forma voluntaria y la población de pacientes era de tamaño indefinido, no siempre es posible estimar con precisión su frecuencia o establecer una relación causal con el uso de alemtuzumab.
Enfermedad autoinmune
En pacientes que recibieron alemtuzumab se han registrado trastornos autoinmunes, como neutropenia, anemia hemolítica (incluyendo un caso fatal), hemofilia adquirida, enfermedad anti-MBG y trastornos tiroideos. En pacientes sin EM que recibieron alemtuzumab se han notificado trastornos autoinmunes graves y a veces fatales, como anemia hemolítica autoinmune, trombocitopenia autoinmune, anemia aplásica, síndrome de Guillain-Barré y polirradiculoneuropatía desmielinizante inflamatoria crónica. En pacientes oncológicos que recibieron alemtuzumab se han notificado casos de resultados positivos en la prueba de Coombs. En un paciente oncológico que recibió alemtuzumab se registró una reacción fatal de "injerto contra huésped" asociada a transfusión.
Reacciones asociadas a la infusión
En pacientes sin EM que recibieron alemtuzumab en dosis más altas y con mayor frecuencia de administración que la usada en EM, se observaron RAI graves y a veces fatales, incluyendo broncoespasmo, hipoxia, síncope, infiltrados pulmonares, síndrome de dificultad respiratoria aguda, paro respiratorio, infarto de miocardio, arritmias, insuficiencia cardíaca aguda y paro cardíaco. También se han notificado casos de anafilaxia grave y otras reacciones de hipersensibilidad, incluyendo shock anafiláctico y angioedema.
Enfermedades infecciosas y parasitarias
En pacientes sin EM que recibieron alemtuzumab en dosis más altas y con mayor frecuencia de administración que la usada en EM, se observaron infecciones virales, bacterianas, protozoarias y fúngicas graves y a veces fatales, incluyendo reactivación de infecciones latentes.
Trastornos de la sangre y sistema linfático
En pacientes sin EM se observaron reacciones hemorrágicas graves.
Trastornos cardíacos
En pacientes sin EM que recibieron alemtuzumab y que previamente fueron tratados con agentes potencialmente cardiotoxícos, se observaron casos de insuficiencia cardíaca congestiva, cardiomiopatía y disminución de la fracción de eyección.
Trastornos linfoproliferativos asociados con el virus de Epstein-Barr
Fuera de estudios patrocinados por la compañía, se observaron casos de trastornos linfoproliferativos asociados con el virus de Epstein-Barr.
Lemtrada contiene sodio y potasio
Este medicamento contiene menos de 1 mmol de potasio (39 mg) por infusión, es decir, prácticamente carece de potasio.
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por infusión, es decir, prácticamente carece de sodio.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil
Aproximadamente 30 días tras cada ciclo de tratamiento, las concentraciones del medicamento en suero fueron bajas o no detectables. Debido a esto, las mujeres en edad fértil deben usar métodos anticonceptivos eficaces durante el ciclo de tratamiento con Lemtrada y durante 4 meses tras cada ciclo de tratamiento.
Embarazo
Actualmente los datos sobre el uso de alemtuzumab en mujeres embarazadas son limitados. Lemtrada debe administrarse a mujeres embarazadas únicamente si el beneficio esperado para la mujer supera el riesgo potencial para el feto.
Se sabe que las IgG humanas atraviesan la barrera placentaria; alemtuzumab también puede atravesar esta barrera y por lo tanto potencialmente puede representar un riesgo para el feto. En estudios en animales se demostró un efecto tóxico sobre la función reproductiva (véase la sección «Datos preclínicos de seguridad»). No se sabe si alemtuzumab puede dañar al feto si se administra a mujeres embarazadas o si puede afectar la función reproductiva.
Las enfermedades de la glándula tiroides (véase la sección «Trastornos de la glándula tiroides») representan riesgos especiales para las mujeres embarazadas. Sin tratamiento, el hipotiroidismo durante el embarazo se asocia con un mayor riesgo de abortos espontáneos y alteraciones en el desarrollo fetal, como retraso en el desarrollo mental y enanismo. En mujeres con enfermedad de Graves, los anticuerpos contra los receptores de la hormona estimulante de la tiroides de la madre embarazada pueden pasar al feto en desarrollo y provocar enfermedad de Graves transitoria en el recién nacido.
Lactancia
Alemtuzumab se detectó en la leche de ratas hembra durante el período de lactancia y en la descendencia.
No se sabe si alemtuzumab se excreta en la leche materna humana. No puede descartarse el riesgo para recién nacidos/lactantes amamantados. Por ello, la lactancia debe suspenderse durante cada ciclo de tratamiento con Lemtrada y durante 4 meses tras la última infusión de cada ciclo de tratamiento. Sin embargo, el beneficio de la inmunidad transmitida a través de la leche materna puede superar los riesgos potenciales de exposición al alemtuzumab en recién nacidos/lactantes amamantados.
Fertilidad
Actualmente no hay datos clínicos adecuados sobre la seguridad del efecto de Lemtrada sobre la fertilidad. En un subestudio con 13 pacientes masculinos que recibieron Lemtrada (a dosis de 12 mg o 24 mg), no se encontraron evidencias de aspermia, azoospermia, disminución sostenida del recuento espermático, alteraciones en la movilidad espermática o aumento de alteraciones morfológicas espermáticas.
Se sabe que la proteína CD52 está presente en los tejidos de los órganos reproductivos en humanos y roedores. Los datos de estudios en animales mostraron ciertos efectos del medicamento sobre la fertilidad en ratones humanizados (véase la sección «Datos preclínicos de seguridad»), pero el impacto potencial sobre la fertilidad en humanos durante el período de uso del medicamento es actualmente desconocido según los datos disponibles.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
Lemtrada tiene un efecto insignificante sobre la capacidad para conducir vehículos o manejar maquinaria. La mayoría de los pacientes experimentan RAI que se desarrollan durante o dentro de las 24 horas tras la administración de Lemtrada. Algunas de las RAI (por ejemplo, mareo) pueden afectar temporalmente la capacidad del paciente para conducir vehículos o manejar maquinaria, por lo que se debe tener precaución hasta que estos efectos desaparezcan.
Vía de administración y dosis
El tratamiento con Lemtrada debe iniciarse y realizarse únicamente bajo la supervisión de un neurólogo con experiencia en el tratamiento de pacientes con esclerosis múltiple (EM), en un hospital donde se disponga de cuidados intensivos.
Además, deben estar disponibles especialistas y equipos necesarios para el diagnóstico oportuno y la asistencia médica en caso de reacciones adversas, especialmente isquemia miocárdica e infarto de miocardio, reacciones adversas cerebrovasculares, estados autoinmunes e infecciones.
Debe haber disponibles medios para el tratamiento del síndrome de liberación de citocinas, reacciones de hipersensibilidad y/o reacciones anafilácticas.
A los pacientes que reciban tratamiento con Lemtrada se les debe entregar una Tarjeta de alerta para el paciente y Recomendaciones para el paciente, así como proporcionarles información sobre los riesgos asociados al uso de Lemtrada (véase también el prospecto del medicamento).
Dosificación
La dosis recomendada de alemtuzumab es de 12 mg/día, administrada mediante infusión intravenosa en dos cursos iniciales de tratamiento, pudiéndose utilizar hasta dos cursos adicionales si fuera necesario.
Tratamiento inicial con dos cursos
- Primer curso de tratamiento: 12 mg/día durante 5 días consecutivos (dosis total: 60 mg).
- Segundo curso de tratamiento: 12 mg/día durante 3 días consecutivos (dosis total: 36 mg), administrados 12 meses después del primer curso de tratamiento.
Si fuera necesario, puede considerarse la conveniencia de utilizar hasta dos cursos adicionales de tratamiento (véase la sección «Propiedades farmacológicas / Farmacodinamia»).
- Tercer o cuarto curso: 12 mg/día durante 3 días consecutivos (dosis total: 36 mg), administrados 12 meses después del curso de tratamiento anterior (véanse las secciones «Propiedades farmacológicas / Farmacodinamia» y «Indicaciones»).
Si se omite alguna dosis, no debe administrarse el mismo día que la dosis programada para ese día.
Seguimiento del paciente
El tratamiento con este medicamento se recomienda realizar con dos cursos iniciales de tratamiento y, si fuera necesario, hasta dos cursos adicionales (véase el apartado «Dosificación»). Durante este periodo y al menos durante 48 meses tras la última infusión del segundo curso de tratamiento, debe realizarse un seguimiento del paciente para evaluar la seguridad. Si se administra un tercer o cuarto curso adicional de tratamiento, el seguimiento para evaluar la seguridad debe continuar durante al menos 48 meses tras la última infusión del medicamento (véase la sección «Precauciones de uso»).
Premedicación
Los pacientes deben recibir premedicación con corticosteroides inmediatamente antes de la administración de Lemtrada durante cada uno de los primeros 3 días de cualquier curso de tratamiento. En los estudios clínicos, los pacientes recibieron premedicación con metilprednisolona a una dosis de 1000 mg durante los primeros 3 días de cada curso de tratamiento con Lemtrada.
También puede considerarse la premedicación con antihistamínicos y/o agentes antipiréticos antes de la administración de Lemtrada.
A todos los pacientes se debe recetar un tratamiento oral profiláctico contra el herpes, que debe comenzar el primer día de cada curso de tratamiento y continuar durante al menos 1 mes tras finalizar el tratamiento con Lemtrada (véase también la sección «Precauciones de uso / Infecciones»). En los estudios clínicos, se recetó aciclovir a una dosis de 200 mg dos veces al día o un fármaco equivalente.
Categorías especiales de pacientes
Pacientes de edad avanzada
Los estudios clínicos de este medicamento no incluyeron pacientes mayores de 61 años. Actualmente no se sabe si estos pacientes presentan diferencias en la respuesta al medicamento en comparación con pacientes más jóvenes.
Disfunción renal o hepática
No se ha estudiado la administración de Lemtrada en pacientes con alteraciones de la función renal o hepática.
Pacientes pediátricos
La seguridad y eficacia del uso de Lemtrada en niños con EM de 0 a 18 años de edad aún no han sido establecidas. Actualmente no existe experiencia significativa en el uso de alemtuzumab en niños desde el nacimiento hasta menos de 10 años para el tratamiento de la esclerosis múltiple. No hay datos disponibles al respecto.
Vía de administración
Lemtrada debe reconstituirse inmediatamente antes de la infusión. La solución reconstituida se administra mediante infusión intravenosa de aproximadamente 4 horas de duración.
Las instrucciones para la reconstitución de este medicamento antes de su administración se describen en la sección «Precauciones especiales de manipulación».
Niños. La seguridad y eficacia del uso de Lemtrada en niños con EM de 0 a 18 años aún no han sido establecidas. Actualmente no existe experiencia significativa en el uso de alemtuzumab en niños desde el nacimiento hasta menos de 10 años para el tratamiento de la esclerosis múltiple. No hay datos disponibles al respecto.
Sobredosificación.
En estudios clínicos controlados, 2 pacientes con EM recibieron accidentalmente hasta 60 mg de Lemtrada (es decir, la dosis completa del curso inicial de tratamiento) en una sola infusión, presentando reacciones graves (cefalea, erupción cutánea, y ya sea hipotensión arterial o taquicardia sinusal). Dosis superiores a las evaluadas en los estudios clínicos podrían aumentar la intensidad de las reacciones adversas asociadas a la infusión y/o los efectos inmunitarios del medicamento.
No se conoce antídoto para el tratamiento de la sobredosificación con alemtuzumab. El tratamiento consiste en la suspensión del medicamento y la administración de terapia de soporte.
Reacciones adversas.
Perfil de seguridad resumido a partir de los resultados de los estudios clínicos. La población de pacientes utilizada para evaluar la seguridad en el análisis conjunto de datos de estudios clínicos de EM, con una mediana de duración de seguimiento de 6,1 años (y un periodo máximo de seguimiento de 12 años), incluyó en total 1486 pacientes que recibieron Lemtrada (a una dosis de 12 mg o 24 mg), lo que proporcionó 8635 pacientes-año de exposición para la evaluación de la seguridad.
Las reacciones adversas más importantes son las reacciones autoinmunes (TPI, trastornos tiroideos, nefropatías, citopenias), las reacciones de infusión (AIR) y las infecciones (véase la sección «Precauciones de uso»).
Las reacciones adversas más frecuentes con Lemtrada (observadas en ≥ 20 % de los pacientes) fueron erupción cutánea, cefalea, fiebre y infecciones del tracto respiratorio.
Lista tabulada de reacciones adversas. La tabla 3 que se muestra a continuación se ha elaborado a partir de datos agregados de seguridad procedentes de todos los pacientes que recibieron Lemtrada a una dosis de 12 mg, basándose en todos los datos disponibles de seguimiento de los estudios clínicos. Las reacciones adversas se han clasificado según las categorías de Clasificación por Sistema de Órganos (SOC) y los términos preferentes del Diccionario Médico para Actividades Regulatorias (MedDRA). La frecuencia de los eventos adversos se determinó según los siguientes criterios: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raros (≥ 1/10.000 a < 1/1.000); muy raros (< 1/10.000); frecuencia desconocida (no puede determinarse a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 3
Reacciones adversas observadas en los estudios 1, 2, 3 y 4 en pacientes que recibieron Lemtrada a una dosis de 12 mg, y durante la vigilancia poscomercialización
| Sistema-Organo-Clase |
Muy frecuente |
Frecuente |
No frecuente |
Raro |
Frecuencia desconocida |
| Enfermedades infecciosas y parasitarias |
Infecciones de las vías respiratorias superiores, infecciones del tracto urinario, infección causada por el virus del herpes simple1 |
Infecciones causadas por el virus de la varicela2, infecciones de las vías respiratorias inferiores, gastroenteritis, candidiasis oral, candidiasis vulvovaginal, gripe, infección del oído, neumonía, infección vaginal, infección dental |
Onicomicosis, gingivitis, infección fúngica de la piel, amigdalitis, sinusitis aguda, flemona, tuberculosis, infecciones por citomegalovirus |
Listeriosis / meningitis por listeria, infección (incluida reactivación) por el virus de Epstein-Barr (VEB) |
|
| Neoplasias benignas, malignas e inespecíficas (incluidas quistes y pólipos) |
Papiloma cutáneo |
||||
| Alteraciones de la sangre y del sistema linfático |
Linfopenia, leucopenia, incluyendo neutropenia |
Linfadenopatía, púrpura trombocitopénica inmune, trombocitopenia, anemia, disminución del hematocrito, leucocitosis |
Pancitopenia, anemia hemolítica, hemofilia adquirida A |
Hemofagocitosis linfohistiocítica (HLH), púrpura trombocitopénica trombótica (PTT) |
|
| Alteraciones del sistema inmunitario |
Síndrome de liberación de citocinas*, hipersensibilidad (incluida anafilaxia)* |
Sarcoidosis |
|||
| Alteraciones endocrinas |
Enfermedad de Basedow, hipertiroidismo, hipotiroidismo |
Tiroiditis autoinmune (incluida tiroiditis subaguda), bocio, resultados positivos en el análisis de anticuerpos frente a componentes de la glándula tiroides |
|||
| Alteraciones metabólicas y nutricionales |
Pérdida de apetito |
||||
| Alteraciones psiquiátricas |
Insomnio*, ansiedad, depresión |
||||
| Alteraciones del sistema nervioso |
Cefalea* |
Recidiva de EM, vértigo*, hipestesia, parestesia, temblor, disgeusia*, migraña* |
Alteraciones sensitivas, hiperestesia, cefalea tensional, encefalitis autoinmune |
Accidente cerebrovascular hemorrágico**, disección de la pared de las arterias de la cabeza y del cuello** |
|
| Alteraciones oculares |
Conjuntivitis, oftalmopatía endocrina, visión borrosa |
Diplopía |
|||
| Alteraciones del oído y del equilibrio |
Vértigo |
Dolor de oído |
|||
| Alteraciones cardíacas |
Taquicardia* |
Bradicardia*, palpitaciones* |
Fibrilación auricular* |
Isquemia miocárdica**, infarto de miocardio** |
|
| Alteraciones vasculares |
Hiperemia* |
Hipotensión arterial*, hipertensión arterial* |
|||
| Alteraciones del aparato respiratorio, del tórax y del mediastino |
Disnea*, tos, epistaxis, hipo, dolor en la orofaringe, asma bronquial |
Sensación de opresión en la garganta*, irritación de la garganta, neumonitis |
Hemorragia pulmonar alveolar** |
||
| Alteraciones gastrointestinales |
Náuseas* |
Dolor abdominal, vómitos, diarrea, dispepsia*, estomatitis |
Estreñimiento, enfermedad por reflujo gastroesofágico, sangrado de encías, sequedad bucal, disfagia, trastornos gastrointestinales, hematochecia |
||
| Alteraciones hepatobiliares |
Aumento de los niveles de aspartato aminotransferasa, aumento de los niveles de alanina aminotransferasa |
Colecistitis (incluida colecistitis litiásica y colecistitis litiásica aguda) |
Hepatitis autoinmune, hepatitis (asociada a infección por el virus de Epstein-Barr (VEB)) |
||
| Alteraciones de la piel y del tejido subcutáneo |
Urticaria*, erupción cutánea*, prurito*, erupción generalizada* |
Eritema*, equimosis, alopecia, hiperhidrosis, acné, lesiones cutáneas, dermatitis |
Ampollas, sudoración nocturna, edema facial, eccema, vitíligo, alopecia areata |
||
| Alteraciones del sistema musculoesquelético y del tejido conectivo |
Mialgia, debilidad muscular, artralgia, dolor de espalda, dolor en las extremidades, espasmos musculares, dolor de cuello, dolor musculoesquelético |
Rigidez del sistema musculoesquelético, molestias en las extremidades |
Enfermedad de Still del adulto (ESA) |
||
| Alteraciones renales y del tracto urinario |
Proteinuria, hematuria |
Nefrolitiasis, cetónuria, nefropatías (incluidas enfermedades con formación de anticuerpos anti-MBC) |
|||
| Alteraciones del sistema reproductor y de las glándulas mamarias |
Menorragia, menstruaciones irregulares |
Displasia del cuello uterino, amenorrea |
|||
| Alteraciones generales y reacciones en el lugar de administración |
Pirexia*, fatiga aumentada*, escalofríos* |
Malestar en el pecho*, dolor*, edemas periféricos, debilidad general, enfermedad tipo gripe, malestar general, dolor en el lugar de la infusión |
|||
| Alteraciones en las pruebas analíticas |
Aumento de los niveles de creatinina en sangre |
Pérdida de peso, aumento de peso, disminución del contenido de eritrocitos, resultados positivos en la prueba bacteriana, aumento de la glucosa en sangre, aumento del volumen corpuscular medio de los eritrocitos |
|||
| Lesiones, envenenamientos y complicaciones procedimentales |
Contusión, reacciones relacionadas con la infusión |
-
Entre las infecciones causadas por el virus del herpes se incluyen (según los términos preferidos): herpes oral; herpes simple; herpes genital; infección por virus del herpes; herpes genital simple; dermatitis herpética; oftalmoherpes simple; resultados positivos de serología para herpes simple.
-
Entre las infecciones causadas por el virus de la varicela se incluyen (según los términos preferidos): herpes zóster; herpes zóster cutáneo diseminado; herpes zóster oftálmico; oftalmoherpes; infección neurológica causada por el virus del herpes zóster; meningitis causada por el virus del herpes zóster.
Descripción de reacciones adversas individuales. Los términos marcados con un asterisco (*) en la tabla 3 incluyen reacciones adversas notificadas como asociadas con reacciones a la infusión.
Los términos marcados con dos asteriscos (**) en la tabla 3 incluyen reacciones adversas observadas durante el período poscomercialización y que en la mayoría de los casos comenzaron entre 1 y 3 días después de la infusión de Lemtrada, tras la administración de cualquier dosis durante el curso del tratamiento.
Neutropenia
Se han notificado casos de neutropenia grave (incluyendo casos fatales) que ocurrieron dentro de los 2 meses posteriores a la infusión de Lemtrada.
Perfil de seguridad según los resultados del seguimiento a largo plazo. El tipo de reacciones adversas, incluyendo su gravedad e intensidad, observadas en los pacientes tratados con Lemtrada según todos los datos disponibles de seguimiento, incluidos los pacientes que recibieron cursos adicionales de tratamiento con el medicamento, fue similar al de las reacciones adversas observadas en los estudios con control activo. La frecuencia de reacciones adversas inmunitarias (RAI) durante el primer curso de tratamiento fue mayor en comparación con los cursos posteriores.
En los pacientes que continuaron bajo observación tras los estudios clínicos controlados y que no recibieron ningún curso adicional de Lemtrada después de los dos primeros cursos iniciales de tratamiento, la frecuencia (número de eventos por paciente-año) de la mayoría de las reacciones adversas durante los años 3 a 6 fue comparable o inferior a la observada durante los años 1 y 2. La frecuencia de reacciones adversas relacionadas con la glándula tiroides fue más alta durante el tercer año y posteriormente disminuyó.
Notificación de reacciones adversas sospechosas.
Este medicamento está sujeto a un seguimiento adicional. Esto permitirá identificar rápidamente nueva información sobre su seguridad. La notificación de reacciones adversas tras la comercialización del medicamento es de gran importancia, ya que permite realizar un seguimiento continuo de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar todos los casos de reacciones adversas sospechosas y de falta de eficacia del medicamento a través del sistema automatizado de información de farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Período de validez.
Concentrado: 4 años.
Solución reconstituida: se ha demostrado que el medicamento listo para su uso mantiene su estabilidad química y física durante 8 horas a una temperatura de 2-8 °C. Desde el punto de vista microbiológico, se recomienda utilizar el medicamento inmediatamente. Si no se utiliza inmediatamente, el usuario es responsable del tiempo y las condiciones de almacenamiento antes de su uso, que no deben exceder las 8 horas a una temperatura de 2-8 °C, siempre que se garantice la protección frente a la luz.
Condiciones de almacenamiento. Conservar en nevera (a una temperatura de 2-8 °C). No congelar. No agitar. Conservar el frasco en su envase exterior de cartón para protegerlo de la luz. Mantener fuera del alcance y de la vista de los niños.
Incompatibilidades. No se han realizado estudios de compatibilidad; por lo tanto, no se recomienda mezclar este medicamento con otros medicamentos, excepto los indicados en la sección «Precauciones especiales de seguridad».
Envase.
Nº1: 1,2 ml en un frasco; 1 frasco por caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante.
Genzyme Ireland Limited.
Dirección del fabricante y lugar de actividad.
IDA Industrial Park, Old Kilmeaden Road, Waterford, Irlanda.
Titular de la autorización de comercialización.
S.A. «Sanofi-Aventis Ucrania», Ucrania.
Dirección del titular de la autorización de comercialización.
Ucrania, 01033, Kiev, calle Zhilyanska, 48-50A.