Firialta
Ukraina
Spis treści
INSTRUKCJA dotyczÄcâ leczenia medycznego leku FIRIALTA (FIRIALTA)
Skład:
substancja czynna: finerenon;
1 tabletka powlekana zawiera 10 mg finerenonu;
1 tabletka powlekana zawiera 20 mg finerenonu;
substancje pomocnicze:
tabletki powlekane 10 mg: celuloza mikrokrysztaĹ‚yczna, sodowa sol kroskarboksymetelowanej celulozy, hipromeloza 5 cP (hydroksypropylometyloceluloza 2910), laktoza monohydrat, stearyna magnezu, sodowy laurylosiarczan; powĹ‚oka filmowa: lakier jasnoróżowy albo alternatywnie hipromeloza 5 cP (hydroksypropylometyloceluloza 2910), dwutlenek tytanu (E 171), talk, tlenek ÅĽelaza czerwony (E 172);
tabletki powlekane 20 mg: celuloza mikrokrysztaĹ‚yczna, sodowa sol kroskarboksymetelowanej celulozy, hipromeloza 5 cP (hydroksypropylometyloceluloza 2910), laktoza monohydrat, stearyna magnezu, sodowy laurylosiarczan; powĹ‚oka filmowa: lakier jasnożółty albo alternatywnie hipromeloza 5 cP (hydroksypropylometyloceluloza 2910), dwutlenek tytanu (E 171), talk, tlenek ÅĽelaza ÅĽĂłĹ‚ty (E 172).
Postać leku. Tabletka powlekana.
Główne właściwości fizykochemiczne:
tabletki powlekane 10 mg: jasnoróżowa, owalna, wydłużona tabletka powlekana o długości 10 mm i szerokości 5 mm, z oznaczeniem na górnej stronie tabletu „10” oraz oznaczeniem na dolnej stronie tabletu „FI”;
tabletki powlekane 20 mg: jasnożółta, owalna, wydłużona tabletka powlekana o długości 10 mm i szerokości 5 mm, z oznaczeniem na górnej stronie tabletu „20” oraz oznaczeniem na dolnej stronie tabletu „FI”.
Grupa farmakoterapeutyczna. Leki działajÅce na układ sercowo-naczyniowy. Moczopędne. Antagoniści aldosteronu i inne leki zabezpieczajÅce potas. Antagoniści aldosteronu. Finerenon.
Kod ATC C03D A05.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Firialta jest niesteroidowym selektywnym antagonistą receptorów mineralokortykoidowych (MR), które są aktywowane przez aldosteron i kortyzol oraz regulują transkrypcję genów. Wiązanie się firialty z receptorami MR prowadzi do powstania specyficznego kompleksu ligand-receptor, który blokuje rekrutację koaktywatorów transkrypcji genów biorących udział w ekspresji mediatorów prozapalnych i profibrotycznych.
Właściwości farmakodynamiczne
W przebiegu randomizowanego, podwójnie ślepego, placebo kontrolowanego, wieloośrodkowego badania III fazy FIDELIO-DKD oraz badania FIGARO-DKD, w których uczestniczyli dorośli pacjenci z przewlekłą chorobą nerek (PChN) i cukrzycą typu 2 (CT2), względne zmniejszenie skorygowanego względem placebo stosunku albuminu do kreatyniny w moczu (S/A/K) w grupie leczonej firialtą wynosiło odpowiednio 31% i 32% po 4 miesiącach, a wartość S/A/K pozostawała obniżona przez cały okres trwania obu badań.
W przebiegu randomizowanego, podwójnie ślepego, placebo kontrolowanego, wieloośrodkowego badania IIb fazy ARTS-DN, w którym uczestniczyli dorośli pacjenci z PChN i CT2, względne zmniejszenie skorygowanego względem placebo S/A/K po 90 dniach wynosiło 25% i 38% odpowiednio w grupie stosującej firialtę w dawce 10 mg i 20 mg raz dziennie.
Elektrofizjologia serca
Specjalistyczne badanie QT z udziałem 57 zdrowych uczestników wykazało, że firialta nie wpływa na repolaryzację miokardium. Nie stwierdzono żadnych oznak wpływu firialty na wydłużenie interwału QT/QTc po podaniu pojedynczych dawek 20 mg (terapeutycznych) lub 80 mg (nadterapeutycznych).
Skuteczność i bezpieczeństwo kliniczne
W badaniach FIDELIO-DKD i FIGARO-DKD oceniano wpływ firialty w porównaniu z placebo na wskaźniki nerkowe i sercowo-naczyniowe (SN) u dorosłych pacjentów z PChN i CT2.
Pacjenci otrzymywali standardową terapię, w tym maksymalną tolerowaną dawkę inhibitora enzymu konwertującego angiotensynę (IEC) lub blokera receptorów angiotensyny (BRA).
Pacjenci z rozpoznaną niewydolnością serca ze zmniejszoną frakcją wyrzutową (klasy II–IV wg klasyfikacji New York Heart Association (NYHA)) byli wykluczeni z badania ze względu na zalecenie klasy 1A dotyczące leczenia antagonistami receptorów mineralokortykoidowych.
W badaniu FIDELIO-DKD pacjentów włączano przy obecności trwałej albuminurii (> 30 mg/g do 5000 mg/g), GFR w zakresie od 25 do 75 ml/min/1,73 m² oraz stężeniu potasu w surowicy ≤ 4,8 mmol/l podczas skriningu.
Pierwotnym punktem końcowym badania był czas do wystąpienia złożonego zdarzenia: pierwszego wystąpienia niewydolności nerek (określonej jako przewlekła dializoterapia lub przeszczepienie nerki, lub trwałego spadku eGFR do < 15 ml/min/1,73 m² przez co najmniej 4 tygodnie), trwałego spadku eGFR o 40% lub więcej w porównaniu z wartością wyjściową przez co najmniej 4 tygodnie lub śmierci spowodowanej niewydolnością nerek. Kluczowym wtórnym punktem końcowym był czas do pierwszego wystąpienia złożonego zdarzenia: śmierci z przyczyn SN, nieśmiertelnego zawału mięśnia sercowego, nieśmiertelnego udaru mózgu lub hospitalizacji z powodu niewydolności serca.
Łącznie 5674 pacjentów zostało zrandomizowanych do otrzymywania firialty (N = 2833) lub placebo (N = 2841) i włączonych do analizy. Średnia długość obserwacji wyniosła 2,6 roku. W trakcie badania dawkę firialty lub placebo można było dostosować w zakresie od 10 mg do 20 mg raz dziennie, głównie w zależności od stężenia potasu w surowicy. Po 24 miesiącach w grupie otrzymującej firialtę 67% pacjentów przyjmowało 20 mg raz dziennie, 30% – 10 mg raz dziennie, a 3% przerwało leczenie.
Po zakończeniu badania informacje o statusie życiowym uzyskano u 99,7% pacjentów. W populacji badawczej 63% stanowili przedstawiciele rasy białej, 25% – Azjaci, 5% – rasy czarnej. Średni wiek pacjentów przy włączeniu wyniósł 66 lat, 70% stanowili mężczyźni. Na początku badania średnie eGFR wynosiło 44,3 ml/min/1,73 m², przy czym u 55% pacjentów eGFR było < 45 ml/min/1,73 m², mediana S/A/K wynosiła 852 mg/g, a średnie stężenie HbA1c – 7,7%. W wywiadzie u 46% stwierdzono miażdżycę układu sercowo-naczyniowego, u 30% – chorobę wieńcową, u 8% – niewydolność serca, średnie ciśnienie tętnicze wynosiło 138/76 mm Hg. Średnia długość trwania CT2 na początku badania wyniosła 16,6 roku, retinopatię cukrzycową i neuropatię cukrzycową w wywiadzie stwierdzono odpowiednio u 47% i 26% pacjentów. Na początku badania niemal wszyscy pacjenci przyjmowali IEC (34%) lub BRA (66%), a 97% stosowało jeden lub więcej leków przeciwcukrzycowych (insulina [64%], biguanidy [44%], agonisty receptora peptydu podobnego do glukagonu-1 [7%], inhibitory współtransportera sodowego glukozy typu 2 [iNKG2] [5%]). Innymi najczęściej stosowanymi lekami na początku badania były statyny (74%) i blokery kanałów wapniowych (63%).
Wykazano istotną statystycznie różnicę na korzyść firialty dla pierwotnego złożonego punktu końcowego oraz kluczowego wtórnego złożonego punktu końcowego (patrz rys. 1/tabela 1 poniżej).
Efekt leczenia w odniesieniu do pierwotnych i kluczowych wtórnych punktów końcowych był ogólnie jednolity we wszystkich podgrupach, w tym podgrupach podzielonych według eGFR, S/A/K, skurczowego ciśnienia tętniczego i HbA1c na poziomie wyjściowym.
W badaniu FIGARO-DKD pacjentów włączano przy obecności trwałej albuminurii z S/A/K od ≥ 30 mg/g do < 300 mg/g i eGFR od 25 do 90 ml/min/1,73 m² lub S/A/K ≥ 300 mg/g i eGFR ≥ 60 ml/min/1,73 m² podczas skriningu, przy stężeniu potasu w surowicy ≤ 4,8 mmol/l.
Pierwotnym punktem końcowym był czas do wystąpienia zdarzenia: pierwszego przypadku śmierci z przyczyn SN, nieśmiertelnego zawału mięśnia sercowego (ZM), nieśmiertelnego udaru mózgu lub hospitalizacji z powodu niewydolności serca. Wtórny punkt końcowy obejmował czas do wystąpienia zdarzenia: rozwoju niewydolności nerek, trwałego spadku eGFR o 40% lub więcej w porównaniu z wartością wyjściową przez co najmniej 4 tygodnie lub śmierci spowodowanej niewydolnością nerek.
Łącznie zrandomizowano i włączono do analizy 7352 pacjentów do otrzymywania firialty (N = 3686) lub placebo (N = 3666). Średnia długość obserwacji wyniosła 3,4 roku. W trakcie leczenia dawkę firialty lub placebo można było dostosować w zakresie od 10 do 20 mg na dobę, głównie w zależności od stężenia potasu w surowicy. Po 24 miesiącach w grupie otrzymującej firialtę 82% pacjentów przyjmowało 20 mg raz dziennie, 15% – 10 mg raz dziennie, a 3% uczestników badania przerwało leczenie.
Po zakończeniu badania informacje o statusie życiowym uzyskano u 99,8% pacjentów. W populacji badawczej 72% stanowili przedstawiciele rasy białej, 20% – mongoloidalnej, 4% – czarnej. Średni wiek pacjentów przy włączeniu wyniósł 64 lata, 69% stanowili mężczyźni. Na początku badania średnie eGFR wynosiło 67,8 ml/min/1,73 m², przy czym u 62% pacjentów eGFR było ≥ 60 ml/min/1,73 m², mediana S/A/K wynosiła 308 mg/g, a średnie stężenie HbA1c – 7,7%; 45% uczestników miało w wywiadzie miażdżycę układu sercowo-naczyniowego, 8% – niewydolność serca, a średnie ciśnienie tętnicze wynosiło 136/77 mm Hg. Średnia długość trwania CT2 na początku badania wyniosła 14,5 roku, retinopatię cukrzycową i neuropatię cukrzycową w wywiadzie stwierdzono odpowiednio u 31% i 28% pacjentów. Na początku badania niemal wszyscy pacjenci stosowali inhibitory ACE (43%) lub BRA (57%), a 98% pacjentów przyjmowało jeden lub więcej leków przeciwcukrzycowych (insulina [54%], biguanidy [69%], agonisty receptora peptydu podobnego do glukagonu-1 [7%], iNKG2 [8%]). Innymi najczęściej stosowanymi lekami na początku badania były statyny (71%).
Wykazano istotną statystycznie różnicę na korzyść firialty dla pierwotnego złożonego punktu końcowego sercowo-naczyniowego (patrz rysunek 1 i tabela 2 poniżej). Efekt leczenia dla pierwotnego punktu końcowego był podobny w różnych podgrupach, w tym podgrupach wyodrębnionych według regionu, eGFR, S/A/K, skurczowego ciśnienia tętniczego i HbA1c na poziomie wyjściowym.
W grupie firialty w porównaniu z placebo obserwowano niższą częstość osiągnięcia wyników wtórnego złożonego punktu końcowego w postaci rozwoju niewydolności nerek, trwałego spadku eGFR o 40% lub więcej lub śmierci spowodowanej niewydolnością nerek, jednak różnica ta nie osiągnęła istotności statystycznej (patrz tabela 2 poniżej). Efekt leczenia dla wtórnego złożonego punktu końcowego nerkowego był podobny w różnych podgrupach według eGFR na poziomie wyjściowym, jednak dla podgrupy pacjentów z S/A/K < 300 mg/g względne ryzyko (RR) wyniosło 1,16 (95% przedział ufności (PU) 0,91; 1,47), a dla podgrupy pacjentów z S/A/K ≥ 300 mg/g RR wyniosło 0,74 (95% PU 0,62; 0,90).
Dodatkowe, wcześniej zdefiniowane wtórne punkty końcowe czasu do zdarzenia zawarto w tabeli 2.
Tabela 1
Analiza pierwotnych i wtórnych punktów końcowych czasu do zdarzenia (oraz ich pojedynczych składowych) w badaniu III fazy FIDELIO-DKD
| Wskaźnik |
Firialta* (N = 2833) |
Placebo (N = 2841) |
Efekt terapeutyczny |
||
| N (%) |
wypadki/ 100 pacjentów-let |
N (%) |
wypadki/ 100 pacjentów-let |
Stosunek ryzyka (95 % CI) |
|
| Pierwotny nerkowy punkt końcowy złożony i jego składowe |
|||||
| Łączna liczba przypadków niewydolności nerek, trwałego zmniejszenia eGFR ≥ 40 % lub śmierci z powodu niewydolności nerek |
504 (17,8 %) |
7,59 |
600 (21,1 %) |
9,08 |
0,82 (0,73;0,93) p = 0,0014 |
| Niewydolność nerek |
208 (7,3 %) |
2,99 |
235 (8,3 %) |
3,39 |
0,87 (0,72; 1,05) |
| Trwałe zmniejszenie eGFR ≥ 40 % |
479 (16,9 %) |
7,21 |
577 (20,3 %) |
8,73 |
0,81 (0,72; 0,92) |
| Śmierć z powodu niewydolności nerek |
2 (< 0,1 %) |
- |
2 (< 0,1 %) |
- |
- |
| Kluczowy wtórny kardiologiczny punkt końcowy złożony i jego składowe |
|||||
| Łączna liczba zgonów z powodu chorób sercowo-naczyniowych, nieśmiertelnych zawałów serca, nieśmiertelnych udarów mózgu lub hospitalizacji z powodu niewydolności serca |
367 (13,0 %) |
5,11 |
420 (14,8 %) |
5,92 |
0,86 (0,75; 0,99) p = 0,0339 |
| Śmierć z powodu chorób sercowo-naczyniowych |
128 (4,5 %) |
1,69 |
150 (5,3 %) |
1,99 |
0,86 (0,68; 1,08) |
| Nieśmiertelny zawał serca |
70 (2,5 %) |
0,94 |
87 (3,1 %) |
1,17 |
0,80 (0,58; 1,09) |
| Nieśmiertelny udar mózgu |
90 (3,2 %) |
1,21 |
87 (3,1 %) |
1,18 |
1,03 (0,76; 1,38) |
| Hospitalizacja z powodu niewydolności serca |
139 (4,9 %) |
1,89 |
162 (5,7 %) |
2,21 |
0,86 (0,68; 1,08) |
| Wtórny punkt końcowy skuteczności |
|||||
| Śmierć z dowolnej przyczyny |
219 (7,7 %) |
2,90 |
244 (8,6 %) |
3,23 |
0,90 (0,75; 1,07)** |
| Hospitalizacja z dowolnej przyczyny |
1 263 (44,6 %) |
22,56 |
1 321 (46,5 %) |
23,87 |
0,95 (0,88; 1,02)** |
| Łączna liczba przypadków niewydolności nerek, trwałego zmniejszenia eGFR ≥ 57 % lub śmierci z powodu niewydolności nerek |
252 (8,9 %) |
3,64 |
326 (11,5 %) |
4,74 |
0,76 (0,65; 0,90)** |
* Terapia z zastosowaniem 10 lub 20 mg raz dziennie w połączeniu z maksymalnymi dawkami tolerowanymi inhibitorów ACE lub blokerów receptora angiotensyny.
** p nie jest istotne statystycznie po skorygowaniu na wielokrotne porównania.
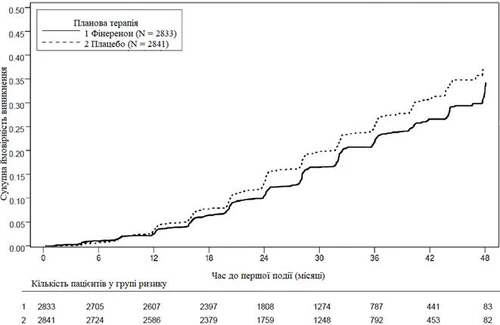
Rys. 1. Czas do pierwszego wystąpienia niewydolności nerek, trwałego obniżenia eGFR ≥ 40 % w stosunku do wartości wyjściowej lub śmierci z powodu niewydolności nerek w badaniu FIDELIO-DKD.
Tabela 2
Analiza punktów końcowych czasu do zdarzenia (oraz ich pojedynczych składowych) w badaniu III fazy FIGARO-DKD
| Wskaźnik |
Firialta* (N = 3686) |
Placebo (N = 3666) |
Skutek terapeutyczny |
||
| N (%) |
wypadki/ 100 pacjentów-roku |
N (%) |
wypadki/ 100 pacjentów-roku |
Stosunek ryzyka (95 % CI) |
|
| Pierwotny punkt końcowy kardiologiczny złożony i jego składowe |
|||||
| Łączna liczba zgonów z przyczyn kardiologicznych, niemortalnego zawału serca, niemortalnego udaru mózgu lub hospitalizacji z powodu niewydolności serca |
458 (12,4) |
3,87 |
519 (14,2) |
4,45 |
0,87 (0,76; 0,98) p = 0,0264 |
| Zgon z przyczyn kardiologicznych |
194 (5,3) |
1,56 |
214 (5,8) |
1,74 |
0,90 (0,74; 1,09) |
| Niemortalny zawał serca |
103 (2,8) |
0,85 |
102 (2,8) |
0,85 |
0,99 (0,76; 1,31) |
| Niemortalny udar mózgu |
108 (2,9) |
0,89 |
111 (3,0) |
0,92 |
0,97 (0,74; 1,26) |
| Hospitalizacja z powodu niewydolności serca |
117 (3,2) |
0,96 |
163 (4,4) |
1,36 |
0,71 (0,56; 0,90) |
| Wtórny punkt końcowy nerkowy złożony i jego składowe |
|||||
| Łączna liczba niewydolności nerek, trwałego spadku eGFR ≥ 40 % lub zgonu z powodu niewydolności nerek |
350 (9,5) |
3,15 |
395 (10,8) |
3,58 |
0,87 (0,76; 1,01) p = 0,0689 ** |
| Niewydolność nerek |
46 (1,2) |
0,40 |
62 (1,7) |
0,54 |
0,72 (0,49;1,05) |
| Trwały spadek eGFR ≥ 40 % |
338 (9,2) |
3,04 |
385 (10,5) |
3,49 |
0,87 (0,75; > 1,00) |
| Zgon z powodu niewydolności nerek |
0 |
- |
2 (< 0,1) |
- |
- |
| Wtórny punkt końcowy skuteczności |
|||||
| Zgon z dowolnej przyczyny |
333 (9,0) |
2,68 |
370 (10,1) |
3,01 |
0,89 (0,77; 1,04)** |
| Hospitalizacja z dowolnej przyczyny |
1573 (42,7) |
16,91 |
1605 (43,8) |
17,52 |
0,97 (0,90; 1,04)** |
| Łączna liczba niewydolności nerek, trwałego spadku eGFR ≥ 57 % lub zgonu z powodu niewydolności nerek |
108 (2,9) |
0,95 |
139 (3,8) |
1,23 |
0,77 (0,60; 0,99)** |
* Terapia z zastosowaniem 10 lub 20 mg raz dziennie w połączeniu z maksymalnymi dawkami inhibitorów ACE lub blokerów receptora angiotensyny (BRA), które mogą być tolerowane.
** p nie jest statystycznie istotne po skorygowaniu pod kątem wielokrotności.
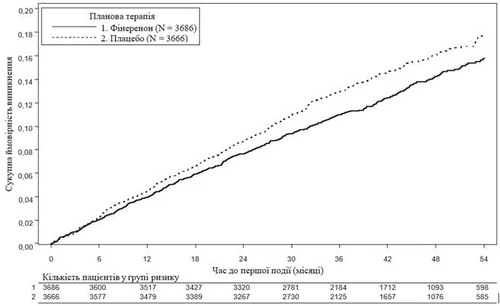
Rys. 2. Czas do pierwszego przypadku śmierci z przyczyn sercowo-naczyniowych, niezgonnego zawału mięśnia sercowego, niezgonnego udaru mózgu lub hospitalizacji z powodu niewydolności serca w badaniu FIGARO-DKD.
Pacjenci pediatryczni
Europejska Agencja Leków odroczyła obowiązek przedłożenia wyników badań leku Firialta w jednej lub kilku podgrupach populacji pediatrycznej w leczeniu przewlekłej choroby nerek (w celu uzyskania informacji dotyczących stosowania w pediatrii, patrz „Sposób stosowania i dawki”).
Farmakokinetyka.
Absorpcja
Finerenon jest niemal całkowicie wchłaniany po doustnym podaniu. Wchłanianie odbywa się szybko, maksymalne stężenie w osoczu (Cₘₐₓ) osiągane jest w ciągu 0,5–1,25 godziny po przyjęciu tabletki na czczo. Bezwzględna biodostępność finerenonu wynosi 43,5% ze względu na metabolizm przy pierwszym przejściu przez ściany jelita i wątrobę. Finerenon in vitro jest substratem białka transportowego wypływu P-glikoproteiny, który jednak nie jest uważany za istotny dla jego wchłaniania in vivo z powodu wysokiej przenikalności finerenonu.
Wpływ posiłku. Przyjmowanie pokarmu o wysokiej zawartości tłuszczu i dużej kaloryczności zwiększało AUC finerenonu o 21%, zmniejszało Cₘₐₓ o 19% i wydłużało czas do osiągnięcia Cₘₐₓ do 2,5 godziny. Ponieważ nie uważa się tego za istotne klinicznie, finerenon można przyjmować niezależnie od posiłku.
Rozkład
Objętość rozkładu w stanie równowagi (Vₛₛ) finerenonu wynosi 52,6 l. Wiązanie finerenonu z białkami osocza krwi człowieka in vitro wynosi 91,7%, przy czym albumina surowicy jest głównym białkiem wiążącym.
Biotransformacja
Około 90% metabolizmu finerenonu jest pośredniczone przez CYP3A4, a 10% – przez CYP2C8. W osoczu wykryto cztery główne metabolity. Wszystkie metabolity są farmakologicznie nieaktywne.
Wydalanie
Wydalanie finerenonu z osocza odbywa się szybko, okres półtrwania (t½) wynosi około 2–3 godziny. Klirens ogólnoustrojowy finerenonu w krwi wynosi około 25 l/h. Około 80% podanej dawki wydalane jest z moczem, a około 20% – z kałem. Wydalanie odbywa się niemal wyłącznie w formie metabolitów, podczas gdy wydalanie niezmienionego finerenonu ma drugorzędne znaczenie (< 1% dawki – z moczem w wyniku filtracji kłębuszkowej, < 0,2% – z kałem).
Liniowość
Farmakokinetyka finerenonu jest liniowa w zakresie dawek badanych od 1,25 do 80 mg tabletek podawanych w dawce pojedynczej.
Grupy specjalne pacjentów
Pacjenci w wieku podeszłym. Spośród 2827 pacjentów, którzy otrzymywali finerenon w badaniu FIDELIO-DKD, 58% miało co najmniej 65 lat, a 15% – co najmniej 75 lat. Spośród 3683 pacjentów, którzy otrzymywali finerenon w badaniu FIGARO-DKD, 52% miało co najmniej 65 lat, a 13% – co najmniej 75 lat. Ogólnie rzecz biorąc, nie zaobserwowano różnic w bezpieczeństwie ani skuteczności finerenonu u tych pacjentów w porównaniu z młodszych pacjentami w obu badaniach.
W badaniu fazy I (N = 48) zdrowi uczestnicy w wieku podeszłym (≥ 65 lat) mieli wyższe stężenia finerenonu w osoczu niż młodszy zdrowi uczestnicy (≤ 45 lat), przy czym średnie wartości AUC i Cₘₐₓ były o 34% i 51% wyższe u pacjentów w wieku podeszłym (patrz sekcja „Sposób stosowania i dawki”). Analiza farmakokinetyki populacyjnej nie wykazała wieku jako kowarianty dla AUC ani Cₘₐₓ finerenonu.
Zaburzenia funkcji nerek. Lekkie zaburzenia funkcji nerek (klirens kreatyniny (KlKr) od 60 do < 90 ml/min) nie wpływały na AUC i Cₘₐₓ finerenonu. W porównaniu z pacjentami z prawidłową funkcją nerek (KlKr ≥ 90 ml/min) wpływ niewydolności nerek umiarkowanego (KlKr od 30 do < 60 ml/min) i ciężkiego (KlKr < 30 ml/min) stopnia na AUC finerenonu był podobny, ze wzrostem o 34–36%. Umiarkowane lub ciężkie zaburzenia funkcji nerek nie wpływały na Cₘₐₓ (patrz sekcja „Sposób stosowania i dawki”).
Z uwagi na wysoki poziom wiązania z białkami osocza nie należy oczekiwać, że finerenon podlega dializie.
Zaburzenia funkcji wątroby. Ekspozycja na finerenon u pacjentów z marskością wątroby i łagodnym uszkodzeniem wątroby nie ulegała zmianie (patrz sekcja „Sposób stosowania i dawki”).
U pacjentów z marskością wątroby i umiarkowanym zaburzeniem funkcji wątroby w porównaniu z uczestnikami grupy kontrolnej stężenie ogólne AUC finerenonu i AUC niezwiązanego finerenonu wzrastały odpowiednio o 38% i 55%, podczas gdy nie zaobserwowano zmian Cₘₐₓ (patrz sekcja „Sposób stosowania i dawki”).
Brak danych dotyczących pacjentów z ciężkim zaburzeniem funkcji wątroby (patrz sekcje „Sposób stosowania i dawki” oraz „Interakcje z innymi lekami i inne rodzaje interakcji”).
Masa ciała. Analiza farmakokinetyki populacyjnej wykazała, że masa ciała jest kowariantą dla Cₘₐₓ finerenonu. U pacjenta o masie ciała 50 kg w porównaniu z pacjentem o masie ciała 100 kg Cₘₐₓ szacuje się na 38–51% wyższe. Korekta dawki w zależności od masy ciała nie jest uzasadniona (patrz sekcja „Sposób stosowania i dawki”).
Zależności farmakokinetyczne/farmakodynamiczne
Zależność „stężenie–efekt” dla wskaźnika stosunku albumin do kreatyniny w moczu (SAC) zależnego od czasu została scharakteryzowana modelem maksymalnego efektu, co wskazuje na nasycenie przy wysokiej ekspozycji. Szacowany modelem czas osiągnięcia pełnego (99%) efektu leku w stanie równowagi na SAC wynosił 138 dni. Farmakokinetyczny (FK) okres półtrwania wynosił 2–3 godziny, a stan równowagi FK osiągany był po 2 dniach, co wskazuje na pośredni i opóźniony wpływ na efekty farmakodynamiczne.
Badania kliniczne bez istotnych interakcji lekowych
Jednoczesne stosowanie gemfibrozyli (600 mg dwa razy dziennie), silnego inhibitora CYP2C8, zwiększało średnią AUC i Cₘₐₓ finerenonu odpowiednio 1,1 i 1,2 razy. Nie uważa się tego za istotne klinicznie.
Wcześniejsze i współbieżne leczenie inhibitorem pompy protonowej omeprazolem (40 mg raz dziennie) nie wpływało na średnią AUC i średnią Cₘₐₓ finerenonu.
Jednoczesne stosowanie leku przeciwwskazowego zawierającego wodorotlenek glinu i wodorotlenek magnezu (70 mVal) nie wpływało na średnią AUC finerenonu i obniżało średnią wartość Cₘₐₓ o 19%. Nie uważa się tego za istotne klinicznie.
In vivo schemat wielokrotnego przyjmowania 20 mg finerenonu raz dziennie przez 10 dni nie wpływał istotnie na AUC midazolamu, substratu CYP3A4. Można zatem wykluczyć klinicznie istotne hamowanie lub indukcję CYP3A4 przez finerenon.
Jednorazowa dawka 20 mg finerenonu nie miała również klinicznie istotnego wpływu na AUC i Cₘₐₓ repaglinidu, substratu CYP2C8. Zatem finerenon nie hamuje CYP2C8.
Wykazano brak interakcji farmakokinetycznej między finerenonem a warfaryną, substratem CYP2C9, oraz między finerenonem a dystalidem, substratem P-gp.
Wielokrotne dawki finerenonu 40 mg raz dziennie nie miały klinicznie istotnego wpływu na AUC i Cₘₐₓ rosuwastatyny, substratu białka oporności na raka piersi i organicznego transportera anionów.
Badania przedkliniczne dotyczące bezpieczeństwa
Badania przedkliniczne nie wykazały szczególnego ryzyka dla człowieka na podstawie ogólnie przyjętych badań farmakologicznej bezpieczności, toksyczności dawki pojedynczej, toksyczności dawek powtarzanych, genotoksyczności, fototoksyczności, potencjału kancerogennego oraz wpływu na płodność u mężczyzn i kobiet.
Dane kliniczne.
Wskazania.
Lek Firialta jest wskazany w leczeniu przewlekłej choroby nerek (z albuminurią) związanej z cukrzycą typu 2 u dorosłych.
Wyniki badań dotyczących zdarzeń nerkowych i sercowo-naczyniowych znajdują się w sekcji „Farmakodynamika”.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych.
Jednoczesne stosowanie silnych inhibitorów CYP3A4 (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”), np. itrakonazolem, ketokonazolem, rytonawirem, nelfinawirem, kobicystatem, klarotromycyną, telitromycyną, nefazodonem. Choroba Addisona.
Interakcje z innymi lekami i inne formy interakcji.
Badania interakcji przeprowadzono wyłącznie u dorosłych pacjentów.
Finerenon jest wydalany niemal wyłącznie poprzez metabolizm oksydacyjny, pośredniczony przez cytochrom P450 (CYP) (głównie CYP3A4 [90 %], z niewielkim udziałem CYP2C8 [10 %]).
Jednoczesne stosowanie jest przeciwwskazane
Silne inhibitory CYP3A4
Jednoczesne stosowanie leku Firialta z itrakonazolem, klarotromycyną oraz innymi silnymi inhibitorami CYP3A4 (np. ketokonazolem, rytonawirem, nelfinawirem, kobicystatem, telitromycyną lub nefazodonem) jest przeciwwskazane (patrz sekcja „Przeciwwskazania”), ponieważ przewiduje się znaczne zwiększenie ekspozycji na finerenon.
Jednoczesne stosowanie nie jest zalecane
Silne i umiarkowane induktory CYP3A4
Nie należy stosować leku Firialta jednoczesnie z ryfampicyną oraz innymi silnymi induktorami CYP3A4 (np. karbamazepiną, fenytoiną, fenylobarbitalą, naparstnikiem) ani z efawirenzem oraz innymi umiarkowanymi induktorami CYP3A4. Przewiduje się, że te induktory CYP3A4 znacząco obniżą stężenie finerenonu w osoczu i prowadzą do zmniejszenia efektu terapeutycznego (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Pewne leki zwiększające stężenie potasu w osoczu
Nie należy stosować leku Firialta jednoczesnie z diuretykami zatrzymującymi potas (np. amiloridem, triamterenem) ani z innymi antagonistami receptorów mineralokortykoidowych (takimi jak eplerenon, esaksyrenon, spironolakton, kanrenon). Przewiduje się, że te leki zwiększają ryzyko wystąpienia hiperkaliemii (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Grapefruit
Nie należy spożywać grejpfruta ani soku grejpfrutowego podczas terapii finerenonem, ponieważ może to prowadzić do zwiększenia stężenia finerenonu w osoczu na skutek hamowania CYP3A4 (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Jednoczesne stosowanie z ostrzeżeniem
Umiarkowane inhibitory CYP3A4
W badaniu klinicznym jednoczesne stosowanie erytromycyny (500 mg trzy razy dziennie) prowadziło do 3,5-krotnego zwiększenia AUC finerenonu oraz 1,9-krotnego zwiększenia jego Cₘₐₓ. W innym badaniu klinicznym werapamil (tabletki z kontrolowanym uwalnianiem, 240 mg raz dziennie) spowodował 2,7-krotne i 2,2-krotne zwiększenie odpowiednio AUC i Cₘₐₓ finerenonu.
Stężenie potasu w osoczu może wzrosnąć, dlatego zaleca się kontrolę stężenia potasu w osoczu, szczególnie na początku lub przy zmianie dawki finerenonu lub inhibitora CYP3A4 (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Słabe inhibitory CYP3A4
Fizjologicznie uzasadnione modelowanie farmakokinetyczne wskazuje, że fluwoksamina (100 mg dwa razy dziennie) zwiększa AUC finerenonu (1,6-krotnie) i Cₘₐₓ (1,4-krotnie).
Stężenie potasu w osoczu może wzrosnąć, dlatego zaleca się kontrolę stężenia potasu w osoczu, szczególnie na początku terapii lub przy zmianie dawki finerenonu lub inhibitora CYP3A4 (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Pewne leki zwiększające stężenie potasu w osoczu (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”)
Oczekuje się, że jednoczesne stosowanie leku Firialta z preparatami potasu oraz trimetoprimem lub trimetoprimem/sulfametoksazolem zwiększy ryzyko hiperkaliemii. Konieczna jest kontrola stężenia potasu w osoczu.
Może być wymagane tymczasowe zaprzestanie przyjmowania leku Firialta podczas leczenia trimetoprimem lub trimetoprimem/sulfametoksazolem.
Leki przeciwhypertensyjne
Ryzyko hipotensji tętniczej zwiększa się przy jednoczesnym stosowaniu wielu innych leków przeciwhypertensyjnych. U takich pacjentów zaleca się kontrolę ciśnienia tętniczego.
Szczególne środki ostrożności.
Hyperkaliemia
U pacjentów leczonych finerenonem obserwowano hyperkaliemię (patrz dział «Działania niepożądane»).
Niektórzy pacjenci są narażeni na większe ryzyko rozwoju hyperkaliemii.
Czynniki ryzyka obejmują niską eGFR, wysoki poziom potasu w surowicy krwi oraz wcześniejsze epizody hyperkaliemii. Należy rozważyć możliwość częstszego monitorowania tych pacjentów.
Wprowadzenie i kontynuacja terapii (patrz dział «Sposób stosowania i dawki»)
Jeśli poziom potasu w surowicy krwi > 5,0 mmol/l, nie należy rozpoczynać terapii finerenonem.
Jeśli poziom potasu w surowicy krwi > 4,8–5,0 mmol/l, można rozważyć rozpoczęcie terapii finerenonem z dodatkową kontrolą stężenia potasu w surowicy krwi w ciągu pierwszych 4 tygodni, w zależności od charakterystyki pacjenta i poziomu potasu w surowicy krwi.
Jeśli poziom potasu w surowicy > 5,5 mmol/l, leczenie finerenonem należy wstrzymać. Należy postępować zgodnie z obowiązującymi zaleceniami dotyczącymi leczenia hyperkaliemii.
Gdy poziom potasu w surowicy krwi spadnie do ≤ 5,0 mmol/l, leczenie finerenonem można wznowić w dawce 10 mg raz dziennie.
Kontrola
Ponownie oznacza się poziom potasu w surowicy krwi oraz eGFR u wszystkich pacjentów po 4 tygodniach od rozpoczęcia terapii, wznowienia terapii lub zwiększenia dawki finerenonu. Następnie należy okresowo i w razie potrzeby oceniać poziom potasu w surowicy krwi, w zależności od charakterystyki pacjenta i poziomu potasu w surowicy krwi (patrz dział «Sposób stosowania i dawki»).
Terapia współistniejąca
Ryzyko hyperkaliemii może również wzrosnąć przy jednoczesnym stosowaniu leków, które mogą podnosić stężenie potasu w surowicy krwi (patrz dział «Interakcje z innymi lekami i inne formy interakcji»). Zobacz również «Jednoczesne stosowanie substancji wpływających na ekspozycję na finerenon».
Nie należy stosować finerenonu jednocześnie z:
- diuretykami zatrzymującymi potas (np. amilorid, triamteren) oraz
- innymi antagonistami receptorów mineralokortykoidowych (ARM), np. eplerenonem, esaxerenonem, spironolaktonem, kanrenonem.
Z ostrożnością należy stosować finerenon i kontrolować poziom potasu w surowicy krwi przy jednoczesnym przyjmowaniu z:
- preparatami potasu;
- trimetoprimem lub trimetoprimem/sulfametoksazolem. Może być konieczne tymczasowe przerwanie przyjmowania finerenonu.
Upośledzenie funkcji nerek
Ryzyko hyperkaliemii wzrasta przy obniżonej funkcji nerek. W razie potrzeby należy prowadzić stałą kontrolę funkcji nerek zgodnie z obowiązującą praktyką kliniczną (patrz dział «Sposób stosowania i dawki»).
Rozpoczęcie terapii
Nie należy rozpoczynać terapii finerenonem u pacjentów z eGFR < 25 ml/min/1,73 m², ponieważ dane kliniczne są ograniczone (patrz działy «Sposób stosowania i dawki» oraz «Farmakokinetyka»).
Kontynuacja terapii
Terapię finerenonem należy przerwać u pacjentów, u których niewydolność nerek postępuje do stadium terminalnego (eGFR < 15 ml/min/1,73 m²), ponieważ dane kliniczne są ograniczone.
Upośledzenie funkcji wątroby
Nie należy rozpoczynać terapii finerenonem u pacjentów z ciężką niewydolnością wątroby (patrz dział «Sposób stosowania i dawki»). Stosowanie leku u tych pacjentów nie było badane (patrz «Farmakokinetyka»), ale oczekuje się istotnego wzrostu ekspozycji na finerenon.
Stosowanie finerenonu u pacjentów z umiarkowaną niewydolnością wątroby może wymagać dodatkowej kontroli ze względu na zwiększoną ekspozycję na finerenon. Należy rozważyć możliwość dodatkowej kontroli poziomu potasu w surowicy krwi i dostosowania monitorowania do charakterystyki pacjenta (patrz działy «Sposób stosowania i dawki» oraz «Farmakokinetyka»).
Niewydolność serca
Pacjenci z rozpoznaną niewydolnością serca z obniżoną frakcją wyrzutową (klasy II–IV według klasyfikacji NYHA – New York Heart Association) byli wykluczeni z badań klinicznych fazy III (patrz «Farmakodynamika»).
Jednoczesne stosowanie substancji wpływających na ekspozycję na finerenon
Umiarkowane i słabe inhibitory CYP3A4
Należy kontrolować poziom potasu w surowicy krwi przy jednoczesnym stosowaniu finerenonu z umiarkowanymi lub słabymi inhibitorami CYP3A4 (patrz działy «Sposób stosowania i dawki» oraz «Interakcje z innymi lekami i inne formy interakcji»).
Silne i umiarkowane induktory CYP3A4
Nie należy stosować finerenonu jednocześnie ze silnymi lub umiarkowanymi induktorami CYP3A4 (patrz dział «Interakcje z innymi lekami i inne formy interakcji»).
Grapefruit
Nie należy spożywać grejpfruta ani soku grejpfrutowego podczas leczenia finerenonem (patrz działy «Sposób stosowania i dawki» oraz «Interakcje z innymi lekami i inne formy interakcji»).
Toxyczność dla zarodka i płodu
Finerenon nie powinien być stosowany w czasie ciąży, z wyjątkiem przypadków, gdy dokładnie rozważono korzyści dla matki i ryzyko dla płodu. Jeśli kobieta zajdzie w ciążę podczas stosowania finerenonu, należy ją poinformować o potencjalnych ryzykach dla płodu.
Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznej antykoncepcji podczas terapii finerenonem.
Kobietom należy zalecić zaprzestanie karmienia piersią podczas terapii finerenonem.
Dodatkowe informacje znajdują się w działach «Stosowanie w czasie ciąży lub karmienia piersią» oraz «Dane przedkliniczne dotyczące bezpieczeństwa».
Informacje dotyczące substancji pomocniczych
Lek Firialta zawiera laktozę
Nie należy podawać tego leku pacjentom z rzadkimi dziedzicznymi schorzeniami, takimi jak nietolerancja galaktozy, niedobór laktoazy lub zaburzenia wchłaniania glukozy-galaktozy.
Lek Firialta zawiera sód
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na tabletkę, co oznacza, że jest praktycznie wolny od sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Antykoncepcja u kobiet
Kobietom w wieku rozrodczym należy stosować skuteczną antykoncepcję podczas terapii finerenonem (patrz dział «Szczególne środki ostrożności»).
Ciąża
Brak danych dotyczących stosowania finerenonu u kobiet w ciąży.
Badania na zwierzętach wykazały toksyczność reprodukcyjną.
Lek Firialta nie powinien być stosowany w czasie ciąży, z wyjątkiem przypadków, gdy stan kliniczny kobiety wymaga leczenia finerenonem. Jeśli kobieta zajdzie w ciążę podczas przyjmowania finerenonu, należy ją poinformować o potencjalnych ryzykach dla płodu (patrz dział «Szczególne środki ostrożności»).
Karmienie piersią
Nie wiadomo, czy finerenon lub jego metabolity wydzielają się z mlekiem matki.
Dostępne dane farmakokinetyczne/toksykologiczne wykazały wydzielanie finerenonu i jego metabolitów do mleka u zwierząt. U szczurów, które były narażone na działanie leku w ten sposób, obserwowano działania niepożądane. Nie można wykluczyć ryzyka dla noworodków/młodzieńców.
Należy podjąć decyzję o zaprzestaniu karmienia piersią lub o rezygnacji z terapii finerenonem, biorąc pod uwagę korzyści wynikające z karmienia piersią dla dziecka oraz korzyści z terapii dla kobiety (patrz dział «Szczególne środki ostrożności»).
Plodność
Brak danych dotyczących wpływu finerenonu na płodność u ludzi.
Badania na zwierzętach wykazały zmniejszenie płodności u samic przy ekspozycji przekraczającej maksymalne ekspozycje u ludzi, co ma niską znaczącość kliniczną.
Wpływ na zdolność prowadzenia pojazdów lub obsługi mechanizmów.
Lek Firialta nie wpływa na szybkość reakcji podczas prowadzenia samochodu lub obsługi innych mechanizmów.
Sposób stosowania i dawki.
Dawkowanie
Zalecana dawka wynosi 20 mg fynerynonu raz dziennie.
Maksymalna zalecana dawka wynosi 20 mg fynerynonu raz dziennie.
Wprowadzenie terapii
Należy zmierzyć stężenie potasu w surowicy i eGFR, aby określić możliwość rozpoczęcia leczenia fynerynonem oraz ustalić dawkę początkową.
Jeśli stężenie potasu w surowicy ≤ 4,8 mmol/l, można rozpocząć terapię fynerynonem. W celu kontroli stężenia potasu w surowicy patrz poniżej „Kontynuacja terapii”.
Jeśli stężenie potasu w surowicy > 4,8–5,0 mmol/l, można rozważyć rozpoczęcie terapii fynerynonem z dodatkową kontrolą stężenia potasu w surowicy w ciągu pierwszych 4 tygodni, w zależności od cech pacjenta i stężenia potasu w surowicy (patrz sekcja „Szczególne wskazania dotyczące stosowania”).
Jeśli stężenie potasu w surowicy > 5,0 mmol/l, nie należy rozpoczynać terapii fynerynonem (patrz sekcja „Szczególne wskazania dotyczące stosowania”).
Zalecana dawka początkowa fynerynonu oparta jest na eGFR i przedstawiona w tabeli 3.
Tabela 3
Rozpoczęcie terapii fynerynonem oraz zalecana dawka
| CLCr (ml/min/1,73 m2) |
Dawka początkowa (raz dziennie) |
| ≥ 60 |
20 mg |
| ≥ 25 do < 60 |
10 mg |
| < 25 |
nie zaleca się |
W dalszym ciągu terapia
Po 4 tygodniach od rozpoczęcia lub wznowienia terapii finerenonem lub zwiększenia dawki należy ponownie oznaczyć stężenie potasu oraz eGFR w surowicy (patrz tabela 4 w celu ustalenia kontynuacji terapii finerenonem i dostosowania dawki).
Następnie należy okresowo oraz w razie potrzeby oznaczać stężenie potasu w surowicy w zależności od cech pacjenta i stężenia potasu w surowicy.
Dodatkowe informacje zawiera sekcja «Szczególne środki ostrożności stosowania» oraz «Interakcje z innymi lekami i inne formy interakcji».
Tabela 4
Kontynuacja terapii finerenonem i dostosowanie dawki
| Bieżąca dawka finerenonu (raz dziennie) |
|||
| 10 mg |
20 mg |
||
| Bieżący poziom potasu w surowicy (mmol/l) |
≤ 4,8 |
Zwiększyć do 20 mg finerenonu raz dziennie* |
Utrzymywać dawkowanie 20 mg raz dziennie |
| > 4,8 do 5,5 |
Utrzymywać dawkowanie |
Utrzymywać dawkowanie 20 mg raz dziennie |
|
| > 5,5 |
Wstrzymać stosowanie finerenonu. Możliwość wznowienia terapii w dawce 10 mg raz dziennie należy rozważyć przy stężeniu potasu w surowicy ≤ 5,0 mmol/l. |
Wstrzymać stosowanie finerenonu. Wznowienie terapii w dawce 10 mg raz dziennie przy stężeniu potasu w surowicy ≤ 5,0 mmol/l. |
|
* Dawkowanie utrzymane 10 mg raz dziennie, jeśli eGFR spadnie > 30 % w porównaniu z poprzednim pomiarem
Zapomniana dawka
Zaleca się podanie pominiętej dawki tak szybko, jak tylko pacjent sobie przypomni, ale wyłącznie w tym samym dniu.
Pacjent nie powinien podawać podwójnej dawki w celu nadrobienia pominiętej dawki.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku
Nie jest wymagana korekta dawki u pacjentów w podeszłym wieku (patrz sekcja „Farmakokinetyka”).
Zaburzenia funkcji nerek
Rozpoczęcie leczenia
U pacjentów z eGFR < 25 ml/min/1,73 m² nie należy rozpoczynać terapii finerenonem ze względu na ograniczone dane kliniczne (patrz sekcje „Szczególne środki ostrożności” i „Farmakokinetyka”).
Kontynuacja leczenia
U pacjentów z eGFR ≥ 15 ml/min/1,73 m² leczenie finerenonem można kontynuować z dostosowaniem dawki w zależności od stężenia potasu w surowicy. eGFR należy zmierzyć po 4 tygodniach od rozpoczęcia terapii, aby określić, czy można zwiększyć dawkę początkową do zalecanej dawki dobowej 20 mg (patrz „Dawkowanie, kontynuacja terapii” i tabela 4).
Ze względu na ograniczone dane kliniczne, terapię finerenonem należy przerwać u pacjentów, u których niewydolność nerek postępowała do stadium zaawansowanego (eGFR < 15 ml/min/1,73 m²) (patrz sekcja „Szczególne środki ostrożności”).
Zaburzenia funkcji wątroby
Pacjenci z
- ciężką niewydolnością wątroby:
nie należy rozpoczynać stosowania finerenonu (patrz sekcje „Szczególne środki ostrożności” i „Farmakokinetyka”). Brak danych dotyczących stosowania.
- umiarkowaną niewydolnością wątroby:
nie jest wymagana korekta dawki początkowej. Należy rozważyć dodatkową kontrolę stężenia potasu w surowicy i dostosować nadzór do indywidualnych cech pacjenta (patrz sekcje „Szczególne środki ostrożności” i „Farmakokinetyka”).
- łagodną niewydolnością wątroby:
nie jest wymagana korekta dawki początkowej.
Terapia współistniejąca
U pacjentów przyjmujących finerenon jednocześnie z umiarkowanymi lub słabymi inhibitorami CYP3A4, lekami zawierającymi potas, trimetoprimem lub trimetoprimem/sulfametokazolem należy rozważyć dodatkową kontrolę stężenia potasu w surowicy i dostosować nadzór do indywidualnych cech pacjenta (patrz sekcja „Szczególne środki ostrożności”). Decyzję o terapii finerenonem należy podejmować zgodnie z wskazaniami w tabeli 4 („Dawkowanie, kontynuacja terapii”).
Może być konieczne tymczasowe przerwanie stosowania finerenonu, jeśli pacjent musi przyjmować trimetoprim lub trimetoprim/sulfametokazol. Dodatkowe informacje znajdują się w sekcjach „Szczególne środki ostrożności” i „Interakcje z innymi lekami i inne formy interakcji”.
Waga ciała
Nie jest wymagana korekta dawki w zależności od masy ciała (patrz sekcja „Farmakokinetyka”).
Sposób podania
Podawać doustnie.
Tabletki można przyjmować z szklanką wody, niezależnie od posiłku (patrz sekcja „Farmakokinetyka”).
Nie należy przyjmować tabletek z grejfrutem ani sokiem grejpfrutowym (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”).
Zmielenie tabletek
Pacjentom, którzy nie mogą połknąć całych tabletek, tabletki leku Firialta można zmiać i zmieszać z wodą lub miękkim pokarmem, np. z puree jabłkowym, bezpośrednio przed podaniem doustnym (patrz sekcja „Farmakokinetyka”).
Dzieci
Bezpieczeństwo i skuteczność stosowania finerenonu u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Przedawkowanie
Najbardziej prawdopodobnym objawem przedawkowania, jak się oczekuje, będzie hiperkaliemia. W przypadku wystąpienia hiperkaliemii należy rozpocząć standardowe leczenie.
Niewielkie prawdopodobieństwo, że finerenon będzie skutecznie usuwany przez hemodializę, ponieważ jego frakcja wiązania z białkami osocza wynosi około 90 %.
Efekty uboczne.
Podsumowanie profilu bezpieczeństwa
Najczęstszą zaobserwowaną reakcją niepożądanej podczas terapii lekiem Firialta była hiperkaliemia (≥ 14 %), zob. „Hiperkaliemia” poniżej oraz sekcja „Szczególne środki ostrożności”.
Bezpieczeństwo stosowania leku Firialta u pacjentów z przewlekłą chorobą nerek i cukrzycą typu 2 oceniano w ramach dwóch kluczowych badań III fazy FIDELIO-DKD (cukrzycowa choroba nerek) oraz FIGARO-DKD. W badaniu FIDELIO-DKD 2827 pacjentów otrzymywało finerenon (10 lub 20 mg raz dziennie), a 2831 – placebo. Średnia długość leczenia wynosiła 2,2 roku. W badaniu FIGARO-DKD 3683 pacjentów otrzymywało finerenon (10 lub 20 mg raz na dobę) przy średniej długości leczenia 2,9 roku.
Zgłoszone reakcje niepożądane przedstawiono w tabeli 5 zgodnie z systemem MedDRA według klas układów narządów i częstości występowania.
Reakcje niepożądane pogrupowano według częstości ich występowania w kolejności malejącej według nasilenia. Częstość określa się jako bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), rzadko (≥ 1/1000 do < 1/100), bardzo rzadko (≥ 1/10000 do < 1/1000), nieznana (nie można oszacować na podstawie dostępnych danych).
Tabela 5
Efekty uboczne
| MedDRA Układ narządów |
Bardzo często |
Często |
Rzadko |
| Zaburzenia przemiany materii i odżywiania |
Hyperkaliemia |
Hyponatremia, hyperurykemia |
|
| Zaburzenia naczyniowe |
Obniżenie ciśnienia tętniczego |
||
| Choroby skóry i tkanek podskórnych |
Zwędzenie |
||
| Wyniki badań |
Obniżenie szybkości filtracji kłębuszkowej |
Obniżenie poziomu hemoglobiny |
Opis poszczególnych działań niepożądanych
Hiperkaliemia
W połączonych danych z badań FIDELIO-DKD i FIGARO-DKD hiperkaliemia wystąpiła u 14% pacjentów przyjmujących finerenon w porównaniu do 6,9% pacjentów przyjmujących placebo. W pierwszym miesiącu leczenia w grupie finerenonu zaobserwowano wzrost średniego stężenia potasu w surowicy o 0,17 mmol/l w stosunku do wartości wyjściowej, które następnie utrzymywało się na stabilnym poziomie. U pacjentów przyjmujących finerenon większość przypadków hiperkaliemii była łagodna lub umiarkowana i była przejściowa. Powikłania seriozne hiperkaliemii zgłaszano częściej przy stosowaniu finerenonu (1,1%) niż przy stosowaniu placebo (0,24%). Stężenia potasu w surowicy > 5,5 mmol/l oraz > 6,0 mmol/l odnotowano odpowiednio u 16,8% i 3,3% pacjentów przyjmujących finerenon oraz u 7,4% i 1,24% pacjentów przyjmujących placebo.
Hiperekaliemia, która doprowadziła do ostatecznego przerwania leczenia, wystąpiła u 1,7% pacjentów przyjmujących finerenon w porównaniu do 0,6% w grupie placebo. Częstość hospitalizacji z powodu hiperkaliemii w grupie finerenonu wyniosła 0,9% w porównaniu do 0,2% w grupie placebo.
Aby uzyskać konkretne zalecenia, należy zapoznać się z rozdziałami „Sposób stosowania i dawki” oraz „Szczególne wskazania dotyczące stosowania”.
Hipotensja tętnicza
W połączonych danych z badań FIDELIO-DKD i FIGARO-DKD hipotensja tętnicza wystąpiła u 4,6% pacjentów przyjmujących finerenon w porównaniu do 3,04% pacjentów przyjmujących placebo. U 3 pacjentów (< 0,1%) leczenie finerenonem zostało ostatecznie przerwane z powodu hipotensji tętniczej. Częstość hospitalizacji z powodu hipotensji tętniczej była taka sama w grupie finerenonu i w grupie placebo (< 0,1%).
U pacjentów przyjmujących finerenon większość przypadków hipotensji tętniczej była łagodna lub umiarkowana i była przejściowa.
Średnie ciśnienie tętnicze skurczowe zmniejszyło się o 2–4 mmHg, a średnie ciśnienie tętnicze rozkurczowe zmniejszyło się o 1–2 mmHg po 1 miesiącu, utrzymując się na stabilnym poziomie dalej.
Hipermocznica
W połączonych danych z badań FIDELIO-DKD i FIGARO-DKD objawy hipermocznicy zaobserwowano u 5,1% pacjentów przyjmujących finerenon w porównaniu do 3,9% pacjentów przyjmujących placebo. Żadne z objawów nie było oceniane jako poważne ani nie prowadziło do ostatecznego przerwania stosowania finerenonu przez pacjentów. W grupie finerenonu w porównaniu z grupą placebo do 16. miesiąca zaobserwowano wzrost średniego stężenia kwasu moczowego o 0,3 mmol/dl w stosunku do wartości wyjściowej, które z czasem uległo zmniejszeniu. Nie zaobserwowano różnic między grupą finerenonu a grupą placebo w zakresie zgłoszeń przypadków podagry (3,0%).
Zmniejszenie szybkości filtracji kłębuszkowej (eGFR)
W połączonych danych z badań FIDELIO-DKD i FIGARO-DKD zmniejszenie eGFR odnotowano u 5,3% pacjentów przyjmujących finerenon w porównaniu do 4,2% pacjentów przyjmujących placebo. U pacjentów przyjmujących finerenon zmniejszenie eGFR prowadzące do ostatecznego przerwania przyjmowania leku było takie samo jak u pacjentów z grupy placebo (0,2%). Częstość hospitalizacji z powodu zmniejszenia eGFR była taka sama w grupie finerenonu i w grupie placebo (< 0,1%).
U pacjentów przyjmujących finerenon większość przypadków zmniejszenia eGFR była łagodna lub umiarkowana i była przejściowa.
U pacjentów przyjmujących finerenon zaobserwowano wstępną redukcję eGFR (średnio o 2 ml/min/1,73 m²), która z czasem się zmniejszyła w porównaniu z odpowiednim wskaźnikiem w grupie placebo. Zmniejszenie to okazało się odwracalne podczas ciągłego leczenia.
Obniżenie poziomu hemoglobiny
W połączonych danych z badań FIDELIO-DKD i FIGARO-DKD stosowanie finerenonu wiązało się ze średnią absolutną redukcją poziomu hemoglobiny skorygowaną względem placebo o 0,154 g/dl oraz ze średnią redukcją hematokrytu o 0,5% po 4 miesiącach terapii. Częstość przypadków anemii była porównywalna u pacjentów przyjmujących finerenon (6,5%) i u pacjentów przyjmujących placebo (6,1%). Częstość poważnych przypadków anemii była niska zarówno u pacjentów przyjmujących finerenon, jak i u pacjentów przyjmujących placebo (0,5%). Zmiany poziomów hemoglobiny i hematokrytu były tymczasowe i po około 24–32 miesiącach osiągnęły wartości zbliżone do tych obserwowanych w grupie przyjmującej placebo.
Zgłaszanie podejrzewanych działań niepożądanych.
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Pozwala to na monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem tego leku. Osoby pracujące w zawodach medycznych i farmaceutycznych, a także pacjenci lub ich prawni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
3 lata.
Warunki przechowywania.
Nie wymaga specjalnych warunków przechowywania. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
Tabletki powlekane, 10 mg: 14 tabletek w opakowaniu blisterowym; 2 lub 7 blisterów z skalą kalendarzową w kartonowej paczce.
Tabletki powlekane, 20 mg: 14 tabletek w opakowaniu blisterowym; 2 lub 7 blisterów z skalą kalendarzową w kartonowej paczce.
Kategoria wydawania.
Na receptę.
Producent.
Bayer AG.
Adres miejsca produkcji i działalności.
Kaiser-Wilhelm-Allee, 51368, Leverkusen, Niemcy.