Firialta
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT FIRIALTA (FIRIALTA)
Composition:
Active substance: finerenone;
One film-coated tablet contains 10 mg of finerenone;
One film-coated tablet contains 20 mg of finerenone;
Excipients:
Film-coated tablets of 10 mg: microcrystalline cellulose, sodium croscarmellose, hypromellose 5 cP (hydroxypropylmethylcellulose 2910), lactose monohydrate, magnesium stearate, sodium lauryl sulfate; film coating: light pink lake or alternatively hypromellose 5 cP (hydroxypropylmethylcellulose 2910), titanium dioxide (E 171), talc, iron oxide red (E 172);
Film-coated tablets of 20 mg: microcrystalline cellulose, sodium croscarmellose, hypromellose 5 cP (hydroxypropylmethylcellulose 2910), lactose monohydrate, magnesium stearate, sodium lauryl sulfate; film coating: light yellow lake or alternatively hypromellose 5 cP (hydroxypropylmethylcellulose 2910), titanium dioxide (E 171), talc, iron oxide yellow (E 172).
Pharmaceutical form. Film-coated tablets.
Main physico-chemical properties:
Film-coated tablets of 10 mg: pink, oval-shaped film-coated tablet, 10 mm in length and 5 mm in width, with "10" marked on the upper side and "FI" marked on the lower side of the tablet;
Film-coated tablets of 20 mg: pale yellow, oval-shaped film-coated tablet, 10 mm in length and 5 mm in width, with "20" marked on the upper side and "FI" marked on the lower side of the tablet.
Pharmacotherapeutic group. Drugs affecting the cardiovascular system. Diuretics. Aldosterone antagonists and other potassium-sparing agents. Aldosterone antagonists. Finerenone.
ATC code C03D A05.
Pharmacological Properties
Pharmacodynamics
Mechanism of Action
Finerenone is a non-steroidal, selective mineralocorticoid receptor (MR) antagonist that binds to MRs activated by aldosterone and cortisol and regulates gene transcription. Its binding to MRs results in the formation of a specific receptor-ligand complex that blocks recruitment of transcriptional coactivators involved in the expression of pro-inflammatory and pro-fibrotic mediators.
Pharmacodynamic Effects
In the randomized, double-blind, placebo-controlled, multicenter phase III FIDELIO-DKD and FIGARO-DKD trials involving adult patients with chronic kidney disease (CKD) and type 2 diabetes (T2D), the relative reduction in placebo-adjusted urinary albumin-to-creatinine ratio (UACR) in the finerenone treatment group was 31% and 32%, respectively, at month 4, and UACR remained reduced throughout both studies.
In the randomized, double-blind, placebo-controlled, multicenter phase IIb ARTS-DN trial involving adult patients with CKD and T2D, the relative reduction in placebo-adjusted UACR on day 90 was 25% and 38% in the groups receiving finerenone 10 mg and 20 mg once daily, respectively.
Cardiac Electrophysiology
A dedicated QT study in 57 healthy subjects showed that finerenone does not affect myocardial repolarization. There was no evidence of an effect of finerenone on QT/QTc interval prolongation following single doses of 20 mg (therapeutic) or 80 mg (supratherapeutic).
Clinical Efficacy and Safety
The effects of finerenone versus placebo on renal and cardiovascular (CV) outcomes were evaluated in adult patients with CKD and T2D in the FIDELIO-DKD and FIGARO-DKD trials.
Patients received standard of care treatment, including the maximum tolerated dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB).
Patients with diagnosed heart failure with reduced ejection fraction (NYHA functional classes II–IV) were excluded from the study due to Class 1A recommendations for treatment with mineralocorticoid receptor antagonists.
In the FIDELIO-DKD trial, patients were enrolled if they had persistent albuminuria (>30 mg/g to 5000 mg/g), eGFR between 25 and 75 mL/min/1.73 m², and serum potassium ≤4.8 mmol/L at screening.
The primary endpoint was time to first occurrence of a composite renal outcome: onset of kidney failure (defined as chronic dialysis or kidney transplantation, or sustained eGFR <15 mL/min/1.73 m² for at least 4 weeks), sustained ≥40% reduction in eGFR from baseline for at least 4 weeks, or death due to kidney disease. The key secondary endpoint was time to first occurrence of a composite cardiovascular outcome: CV death, non-fatal myocardial infarction (MI), non-fatal stroke, or hospitalization for heart failure.
A total of 5,674 patients were randomized to receive finerenone (N = 2,833) or placebo (N = 2,841) and included in the analysis. The mean duration of follow-up was 2.6 years. During the study, the dose of finerenone or placebo could be adjusted between 10 mg and 20 mg once daily, primarily based on serum potassium concentration. At month 24, 67% of patients in the finerenone group were receiving 20 mg once daily, 30% were receiving 10 mg once daily, and 3% had discontinued treatment.
Vital status information was obtained for 99.7% of patients after study completion. In the study population, 63% were White, 25% were Asian, and 5% were Black. The mean age at enrollment was 66 years, and 70% of patients were male. At baseline, the mean eGFR was 44.3 mL/min/1.73 m², with 55% of patients having eGFR <45 mL/min/1.73 m², median UACR was 852 mg/g, and mean HbA1c was 7.7%. A history of atherosclerotic cardiovascular disease was present in 46%, ischemic heart disease in 30%, and heart failure in 8%. Mean blood pressure was 138/76 mmHg. The mean duration of T2D at study initiation was 16.6 years, and diabetic retinopathy and diabetic neuropathy were recorded in 47% and 26% of patients, respectively. At baseline, nearly all patients were receiving an ACEi (34%) or ARB (66%), and 97% were receiving one or more antidiabetic agents (insulin [64%], biguanides [44%], glucagon-like peptide-1 receptor agonists [7%], sodium-glucose cotransporter-2 inhibitors [SGLT2i] [5%]). Other commonly used medications at baseline included statins (74%) and calcium channel blockers (63%).
Statistically significant benefit in favor of finerenone was demonstrated for both the primary composite endpoint and the key secondary composite endpoint (see Figure 1/Table 1 below).
The treatment effect on the primary and key secondary endpoints was consistent across all subgroups, including baseline eGFR, UACR, systolic blood pressure, and HbA1c.
In the FIGARO-DKD trial, patients were enrolled if they had persistent albuminuria with UACR ≥30 mg/g to <300 mg/g and eGFR 25–90 mL/min/1.73 m², or UACR ≥300 mg/g and eGFR ≥60 mL/min/1.73 m² at screening, and serum potassium ≤4.8 mmol/L.
The primary endpoint was time to first occurrence of a composite cardiovascular outcome: CV death, non-fatal MI, non-fatal stroke, or hospitalization for heart failure. The secondary endpoint was time to first occurrence of a composite renal outcome: onset of kidney failure, sustained ≥40% reduction in eGFR from baseline for at least 4 weeks, or death due to kidney disease.
A total of 7,352 patients were randomized and included in the analysis to receive finerenone (N = 3,686) or placebo (N = 3,666). The mean duration of follow-up was 3.4 years. During treatment, the dose of finerenone or placebo could be adjusted between 10 mg and 20 mg once daily, primarily based on serum potassium concentration. At month 24, 82% of patients in the finerenone group were receiving 20 mg once daily, 15% were receiving 10 mg once daily, and 3% of study participants had discontinued treatment.
Vital status information was obtained for 99.8% of patients after study completion. In the study population, 72% were White, 20% were East Asian, and 4% were Black. The mean age at enrollment was 64 years, and 69% of patients were male. At baseline, the mean eGFR was 67.8 mL/min/1.73 m², with 62% of patients having eGFR ≥60 mL/min/1.73 m², median UACR was 308 mg/g, and mean HbA1c was 7.7%. A history of atherosclerotic cardiovascular disease was present in 45%, and heart failure in 8%. Mean blood pressure was 136/77 mmHg. The mean duration of T2D at study initiation was 14.5 years, and diabetic retinopathy and diabetic neuropathy were recorded in 31% and 28% of patients, respectively. At baseline, nearly all patients were receiving an ACEi (43%) or ARB (57%), and 98% were receiving one or more antidiabetic agents (insulin [54%], biguanides [69%], glucagon-like peptide-1 receptor agonists [7%], SGLT2i [8%]). Other commonly used medications at baseline included statins (71%).
Statistically significant benefit in favor of finerenone was demonstrated for the primary composite cardiovascular endpoint (see Figure 1 and Table 2 below). The treatment effect for the primary endpoint was consistent across subgroups, including region, baseline eGFR, UACR, systolic blood pressure, and HbA1c.
In the finerenone group compared to placebo, a lower incidence of the secondary composite renal endpoint (onset of kidney failure, sustained ≥40% reduction in eGFR, or death due to kidney disease) was observed, although this difference did not reach statistical significance (see Table 2 below). The treatment effect for the secondary composite renal endpoint was consistent across subgroups defined by baseline eGFR. However, for the subgroup of patients with UACR <300 mg/g, the relative risk (RR) was 1.16 (95% confidence interval [CI] 0.91; 1.47), while for patients with UACR ≥300 mg/g, the RR was 0.74 (95% CI 0.62; 0.90).
Additional pre-specified secondary time-to-event endpoints are included in Table 2.
Table 1
Analysis of primary and secondary time-to-event endpoints (and their individual components) in the phase III FIDELIO-DKD trial
| Parameter |
Firialta* (N = 2833) |
Placebo (N = 2841) |
Treatment effect |
||
| N (%) |
events/ 100 patient-years |
N (%) |
events/ 100 patient-years |
Relative risk (95% CI) |
|
| Primary renal composite endpoint and its components |
|||||
| Composite of kidney failure, sustained reduction in eGFR ≥ 40%, or death due to kidney failure |
504 (17.8%) |
7.59 |
600 (21.1%) |
9.08 |
0.82 (0.73; 0.93) p = 0.0014 |
| Kidney failure |
208 (7.3%) |
2.99 |
235 (8.3%) |
3.39 |
0.87 (0.72; 1.05) |
| Sustained reduction in eGFR ≥ 40% |
479 (16.9%) |
7.21 |
577 (20.3%) |
8.73 |
0.81 (0.72; 0.92) |
| Death due to kidney failure |
2 (< 0.1%) |
- |
2 (< 0.1%) |
- |
- |
| Key secondary CV composite endpoint and its components |
|||||
| Composite of death due to CV disease, non-fatal MI, non-fatal stroke, or hospitalization due to heart failure |
367 (13.0%) |
5.11 |
420 (14.8%) |
5.92 |
0.86 (0.75; 0.99) p = 0.0339 |
| Death due to CV disease |
128 (4.5%) |
1.69 |
150 (5.3%) |
1.99 |
0.86 (0.68; 1.08) |
| Non-fatal MI |
70 (2.5%) |
0.94 |
87 (3.1%) |
1.17 |
0.80 (0.58; 1.09) |
| Non-fatal stroke |
90 (3.2%) |
1.21 |
87 (3.1%) |
1.18 |
1.03 (0.76; 1.38) |
| Hospitalization due to heart failure |
139 (4.9%) |
1.89 |
162 (5.7%) |
2.21 |
0.86 (0.68; 1.08) |
| Secondary efficacy endpoint |
|||||
| Death from any cause |
219 (7.7%) |
2.90 |
244 (8.6%) |
3.23 |
0.90 (0.75; 1.07)** |
| Hospitalization due to any cause |
1,263 (44.6%) |
22.56 |
1,321 (46.5%) |
23.87 |
0.95 (0.88; 1.02)** |
| Composite of kidney failure, sustained reduction in eGFR ≥ 57%, or death due to kidney failure |
252 (8.9%) |
3.64 |
326 (11.5%) |
4.74 |
0.76 (0.65; 0.90)** |
* Therapy with 10 or 20 mg once daily in addition to maximally tolerated doses of ACE inhibitors or ARBs.
** p is not statistically significant after adjustment for multiplicity.
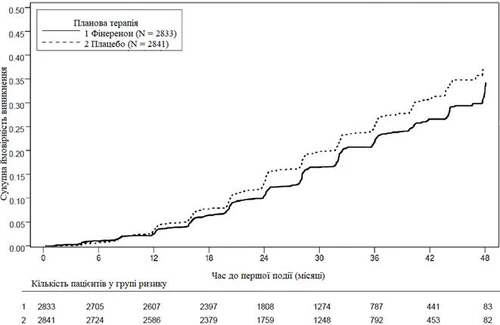
Figure 1. Time to first occurrence of kidney failure, sustained reduction in eGFR ≥ 40% from baseline, or death due to kidney failure in the FIDELIO-DKD study.
Table 2
Analysis of primary and secondary time-to-event endpoints (and their individual components) in the Phase III FIGARO-DKD study
| Parameter |
Firialta* (N = 3686) |
Placebo (N = 3666) |
Treatment effect |
||
| N (%) |
events/ 100 patient-years |
N (%) |
events/ 100 patient-years |
Hazard ratio (95 % CI) |
|
| Primary CV composite endpoint and its components |
|||||
| Composite of death from CV causes, non-fatal MI, non-fatal stroke, or hospitalization due to heart failure |
458 (12.4) |
3.87 |
519 (14.2) |
4.45 |
0.87 (0.76; 0.98) p = 0.0264 |
| Death from CV causes |
194 (5.3) |
1.56 |
214 (5.8) |
1.74 |
0.90 (0.74; 1.09) |
| Non-fatal MI |
103 (2.8) |
0.85 |
102 (2.8) |
0.85 |
0.99 (0.76; 1.31) |
| Non-fatal stroke |
108 (2.9) |
0.89 |
111 (3.0) |
0.92 |
0.97 (0.74; 1.26) |
| Hospitalization due to heart failure |
117 (3.2) |
0.96 |
163 (4.4) |
1.36 |
0.71 (0.56; 0.90) |
| Secondary renal composite endpoint and its components |
|||||
| Composite of renal failure, sustained reduction in eGFR ≥ 40%, or death from renal failure |
350 (9.5) |
3.15 |
395 (10.8) |
3.58 |
0.87 (0.76; 1.01) p = 0.0689 ** |
| Renal failure |
46 (1.2) |
0.40 |
62 (1.7) |
0.54 |
0.72 (0.49; 1.05) |
| Sustained reduction in eGFR ≥ 40% |
338 (9.2) |
3.04 |
385 (10.5) |
3.49 |
0.87 (0.75; > 1.00) |
| Death from renal failure |
0 |
- |
2 (< 0.1) |
- |
- |
| Secondary efficacy endpoint |
|||||
| Death from any cause |
333 (9.0) |
2.68 |
370 (10.1) |
3.01 |
0.89 (0.77; 1.04)** |
| Hospitalization due to any cause |
1573 (42.7) |
16.91 |
1605 (43.8) |
17.52 |
0.97 (0.90; 1.04)** |
| Composite of renal failure, sustained reduction in eGFR ≥ 57%, or death from renal failure |
108 (2.9) |
0.95 |
139 (3.8) |
1.23 |
0.77 (0.60; 0.99)** |
* Therapy with 10 or 20 mg once daily in addition to maximally tolerated doses of ACE inhibitors or ARBs.
** p is not statistically significant after correction for multiplicity.
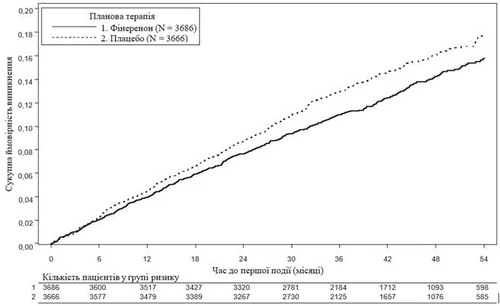
Fig. 2. Time to first event of cardiovascular death, non-fatal MI, non-fatal stroke, or hospitalization due to heart failure in the FIGARO-DKD study.
Pediatric patients
The European Medicines Agency has deferred the obligation to submit the results of studies with Firialta in one or several subgroups of the pediatric population for the treatment of chronic kidney disease (for information on pediatric use, see "Posology and method of administration").
Pharmacokinetics.
Absorption
Finerenone is almost completely absorbed after oral administration. Absorption occurs rapidly, with maximum plasma concentration (Cₘₐₓ) reached within 0.5–1.25 hours after administration of a tablet on an empty stomach. Absolute bioavailability of finerenone is 43.5% due to first-pass metabolism in the intestinal wall and liver. In vitro, finerenone is a substrate of the P-glycoprotein efflux transporter; however, this is not considered relevant for its in vivo absorption due to the high permeability of finerenone.
Effect of food intake. Administration with a high-fat, high-calorie meal increased AUC of finerenone by 21%, decreased Cₘₐₓ by 19%, and prolonged the time to reach Cₘₐₓ to 2.5 hours. As this is not considered clinically significant, finerenone may be taken independently of food intake.
Distribution
The steady-state volume of distribution (Vₛₛ) of finerenone is 52.6 L. In vitro binding of finerenone to human plasma proteins is 91.7%, with serum albumin being the primary binding protein.
Biotransformation
Approximately 90% of finerenone metabolism is mediated by CYP3A4 and 10% by CYP2C8. Four major metabolites were detected in plasma. All metabolites are pharmacologically inactive.
Elimination
Plasma elimination of finerenone is rapid, with a half-life (t½) of approximately 2 to 3 hours. Systemic clearance of finerenone in blood is approximately 25 L/h. About 80% of the administered dose was excreted in urine and approximately 20% in feces. Elimination occurred almost exclusively in the form of metabolites, whereas excretion of unchanged finerenone is a minor pathway (< 1% of dose in urine via glomerular filtration, < 0.2% in feces).
Linearity
The pharmacokinetics of finerenone are linear within the studied dose range of 1.25 to 80 mg tablets administered as single doses.
Special patient groups
Elderly patients. Of the 2827 patients who received finerenone in the FIDELIO-DKD study, 58% were aged 65 years and older, and 15% were aged 75 years and older. Of the 3683 patients who received finerenone in the FIGARO-DKD study, 52% were aged 65 years and older, and 13% were aged 75 years and older. Overall, no differences in safety or efficacy of finerenone were observed between these patients and younger patients in both studies.
In a Phase I study (N = 48), healthy elderly participants (≥ 65 years) had higher plasma concentrations of finerenone than younger healthy participants (≤ 45 years), with mean AUC and Cₘₐₓ values 34% and 51% higher, respectively, in elderly patients (see section "Posology and method of administration"). Population pharmacokinetic analysis did not identify age as a covariate for AUC or Cₘₐₓ of finerenone.
Renal impairment. Mild renal impairment (creatinine clearance (CrCl) 60 to < 90 mL/min) did not affect AUC and Cₘₐₓ of finerenone. Compared to patients with normal renal function (CrCl ≥ 90 mL/min), moderate (CrCl 30 to < 60 mL/min) and severe (CrCl < 30 mL/min) renal impairment had a similar effect on finerenone AUC, with increases of 34–36%. Moderate or severe renal impairment did not affect Cₘₐₓ (see section "Posology and method of administration").
Due to the high degree of plasma protein binding, dialysis is not expected to remove finerenone.
Hepatic impairment. Exposure to finerenone was not altered in patients with cirrhosis and mild hepatic impairment (see section "Posology and method of administration").
In patients with liver cirrhosis and moderate hepatic impairment, compared to healthy control participants, total AUC and AUC of unbound finerenone increased by 38% and 55%, respectively, while no changes in Cₘₐₓ were observed (see section "Posology and method of administration").
There are no data available for patients with severe hepatic impairment (see sections "Posology and method of administration" and "Interaction with other medicinal products and other forms of interaction").
Body weight. Population pharmacokinetic analysis identified body weight as a covariate for Cₘₐₓ of finerenone. In a patient with a body weight of 50 kg compared to a patient with a body weight of 100 kg, Cₘₐₓ is estimated to be 38–51% higher. Dose adjustment based on body weight is not justified (see section "Posology and method of administration").
Pharmacokinetic/pharmacodynamic relationships
The concentration-effect relationship over time for the urinary albumin-to-creatinine ratio (UACR) was characterized by a maximum effect model, indicating saturation at high exposure. The model-predicted time to achieve full (99%) drug effect on UACR at steady state was 138 days. The pharmacokinetic (PK) half-life was 2–3 hours, and PK steady state was reached within 2 days, indicating an indirect and delayed effect on pharmacodynamic outcomes.
Clinical studies without significant drug-drug interactions
Concomitant administration of gemfibrozil (600 mg twice daily), a strong CYP2C8 inhibitor, increased the mean AUC and Cₘₐₓ of finerenone by 1.1 and 1.2 times, respectively. This is not considered clinically significant.
Prior and concomitant therapy with the proton pump inhibitor omeprazole (40 mg once daily) did not affect the mean AUC or mean Cₘₐₓ of finerenone.
Concomitant administration of the antacid aluminum hydroxide and magnesium hydroxide (70 mVal) did not affect the mean AUC of finerenone and reduced the mean Cₘₐₓ by 19%. This is not considered clinically significant.
In vivo, repeated administration of 20 mg finerenone once daily for 10 days did not significantly affect the AUC of midazolam, a CYP3A4 substrate. Thus, clinically significant inhibition or induction of CYP3A4 by finerenone can be ruled out.
A single 20 mg dose of finerenone also had no clinically significant effect on the AUC and Cₘₐₓ of repaglinide, a CYP2C8 substrate. Therefore, finerenone does not inhibit CYP2C8.
Lack of pharmacokinetic interaction has been demonstrated between finerenone and warfarin, a CYP2C9 substrate, and between finerenone and digoxin, a P-gp substrate.
Repeated doses of 40 mg finerenone once daily had no clinically significant effect on the AUC and Cₘₐₓ of rosuvastatin, a substrate of breast cancer resistance protein and organic anion-transporting polypeptide.
Preclinical safety data
Preclinical data revealed no special risk to humans based on conventional studies of pharmacological safety, single-dose toxicity, repeated-dose toxicity, genotoxicity, phototoxicity, carcinogenic potential, and effects on fertility in males and females.
Clinical characteristics.
Indications.
Firialta is indicated for the treatment of chronic kidney disease (with albuminuria) associated with type 2 diabetes in adults.
For study results on renal and cardiovascular outcomes, see section "Pharmacodynamics".
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
Concomitant use with strong CYP3A4 inhibitors (see section "Interaction with other medicinal products and other forms of interactions"), e.g. itraconazole, ketoconazole, ritonavir, nelfinavir, cobicistat, clarithromycin, telithromycin, nefazodone. Addison's disease.
Interaction with other medicinal products and other forms of interactions.
Interaction studies have been conducted only in adult patients.
Finerenone is eliminated almost exclusively via oxidative metabolism mediated by cytochrome P450 (CYP) enzymes (mainly CYP3A4 [90%], with a minor contribution from CYP2C8 [10%]).
Concomitant use is contraindicated
Strong CYP3A4 inhibitors
Concomitant use of Firialta with itraconazole, clarithromycin, and other strong CYP3A4 inhibitors (e.g. ketoconazole, ritonavir, nelfinavir, cobicistat, telithromycin, or nefazodone) is contraindicated (see section "Contraindications") due to the expected significant increase in finerenone exposure.
Concomitant use is not recommended
Strong and moderate CYP3A4 inducers
Firialta should not be used concomitantly with rifampicin and other strong CYP3A4 inducers (e.g. carbamazepine, phenytoin, phenobarbital, St. John’s wort) or with efavirenz and other moderate CYP3A4 inducers. These CYP3A4 inducers are expected to substantially reduce plasma concentrations of finerenone, leading to diminished therapeutic effect (see section "Special precautions for use").
Certain medicinal products that increase serum potassium levels
Firialta should not be used concomitantly with potassium-sparing diuretics (e.g. amiloride, triamterene) and other mineralocorticoid receptor antagonists (such as eplerenone, esaxerenone, spironolactone, canrenone). These medicinal products are expected to increase the risk of hyperkalaemia (see section "Special precautions for use").
Grapefruit
Grapefruit or grapefruit juice should not be consumed during treatment with finerenone, as this is expected to increase plasma concentrations of finerenone due to inhibition of CYP3A4 (see sections "Dosage and administration" and "Special precautions for use").
Concomitant use with caution
Moderate CYP3A4 inhibitors
In a clinical study, concomitant administration of erythromycin (500 mg three times daily) resulted in a 3.5-fold increase in finerenone AUC and a 1.9-fold increase in Cₘₐₓ. In another clinical study, verapamil (controlled-release tablets 240 mg once daily) increased finerenone AUC and Cₘₐₓ by 2.7- and 2.2-fold, respectively.
Serum potassium levels may increase; therefore, monitoring of serum potassium is recommended, particularly at the initiation or dose adjustment of finerenone or the CYP3A4 inhibitor (see sections "Dosage and administration" and "Special precautions for use").
Weak CYP3A4 inhibitors
Physiologically based pharmacokinetic modelling indicates that fluvoxamine (100 mg twice daily) increases finerenone AUC by 1.6-fold and Cₘₐₓ by 1.4-fold.
Serum potassium levels may increase; therefore, monitoring of serum potassium is recommended, particularly at the start of therapy or upon dose adjustment of finerenone or the CYP3A4 inhibitor (see sections "Dosage and administration" and "Special precautions for use").
Certain medicinal products that increase serum potassium levels (see section "Special precautions for use")
Concomitant use of Firialta with potassium supplements, trimethoprim, or trimethoprim/sulfamethoxazole is expected to increase the risk of hyperkalaemia. Monitoring of serum potassium levels is required.
Temporary discontinuation of Firialta may be necessary during treatment with trimethoprim or trimethoprim/sulfamethoxazole.
Antihypertensive medicinal products
The risk of arterial hypotension increases with concomitant use of multiple antihypertensive agents. Blood pressure monitoring is recommended in such patients.
Special precautions for use.
Hyperkalemia
Hyperkalemia has been observed in patients receiving finerenone therapy (see section "Adverse reactions").
Some patients are at higher risk of developing hyperkalemia.
Risk factors include low eGFR, elevated serum potassium levels, and previous episodes of hyperkalemia. More frequent monitoring of these patients should be considered.
Initiation and continuation of therapy (see section "Dosage and administration")
If serum potassium level is > 5.0 mmol/L, initiation of finerenone therapy is not recommended.
If serum potassium level is > 4.8–5.0 mmol/L, initiation of finerenone therapy may be considered with additional monitoring of serum potassium levels during the first 4 weeks, depending on patient characteristics and baseline serum potassium levels.
If serum potassium level is > 5.5 mmol/L, finerenone treatment should be discontinued. Standard guidelines for the management of hyperkalemia should be followed.
Once serum potassium level is ≤ 5.0 mmol/L, finerenone therapy may be resumed at a dose of 10 mg once daily.
Monitoring
Serum potassium and eGFR should be re-measured in all patients 4 weeks after initiation, resumption, or dose escalation of finerenone. Thereafter, serum potassium levels should be periodically and as needed assessed based on patient characteristics and serum potassium levels (see section "Dosage and administration").
Concomitant therapy
The risk of hyperkalemia may also be increased when finerenone is used concomitantly with medicinal products that may elevate serum potassium levels (see section "Interaction with other medicinal products and other forms of interaction"). See also "Concomitant use of substances affecting finerenone exposure".
Finerenone should not be used concomitantly with:
- potassium-sparing diuretics (e.g., amiloride, triamterene), and
- other mineralocorticoid receptor antagonists (MRA) (e.g., eplerenone, esaxerenone, spironolactone, canrenone).
Finerenone should be used with caution and serum potassium levels should be monitored when administered concomitantly with:
- potassium-containing supplements;
- trimethoprim or trimethoprim/sulfamethoxazole. Temporary discontinuation of finerenone may be required.
Renal impairment
The risk of hyperkalemia increases with declining renal function. Ongoing monitoring of renal function should be performed as appropriate according to standard practice (see section "Dosage and administration").
Initiation of therapy
Finerenone therapy should not be initiated in patients with eGFR < 25 mL/min/1.73 m² due to limited clinical data (see sections "Dosage and administration" and "Pharmacokinetics").
Continuation of therapy
Finerenone therapy should be discontinued in patients whose renal function has progressed to end-stage renal disease (eGFR < 15 mL/min/1.73 m²) due to limited clinical data.
Hepatic impairment
Finerenone therapy should not be initiated in patients with severe hepatic impairment (see section "Dosage and administration"). The use of finerenone in these patients has not been studied (see "Pharmacokinetics"), but a substantial increase in finerenone exposure is expected.
The use of finerenone in patients with moderate hepatic impairment may require additional monitoring due to increased finerenone exposure. Additional monitoring of serum potassium levels and adaptation of monitoring frequency according to patient characteristics should be considered (see sections "Dosage and administration" and "Pharmacokinetics").
Heart failure
Patients with diagnosed heart failure with reduced ejection fraction (NYHA functional class II–IV) were excluded from phase III clinical trials (see "Pharmacodynamics").
Concomitant use of substances affecting finerenone exposure
Moderate and weak CYP3A4 inhibitors
Serum potassium levels should be monitored when finerenone is used concomitantly with moderate or weak CYP3A4 inhibitors (see sections "Dosage and administration" and "Interaction with other medicinal products and other forms of interaction").
Strong and moderate CYP3A4 inducers
Concomitant use of finerenone with strong or moderate CYP3A4 inducers is not recommended (see section "Interaction with other medicinal products and other forms of interaction").
Grapefruit
Grapefruit or grapefruit juice should not be consumed during finerenone therapy (see sections "Dosage and administration" and "Interaction with other medicinal products and other forms of interaction").
Embryo-fetal toxicity
Finerenone should not be used during pregnancy unless the potential benefit to the mother clearly outweighs the potential risk to the fetus. If a woman becomes pregnant while taking finerenone, she should be informed of the potential risks to the fetus.
Women of childbearing potential should be advised to use effective contraception during finerenone therapy.
Women should be advised to discontinue breastfeeding during finerenone therapy.
For additional information, see sections "Pregnancy and breastfeeding" and "Preclinical safety data".
Information on excipients
Firialta contains lactose
Patients with rare hereditary conditions such as galactose intolerance, Lapp lactase deficiency, or glucose-galactose malabsorption should not take this medicinal product.
Firialta contains sodium
This medicinal product contains less than 1 mmol sodium (23 mg) per tablet, i.e., essentially "sodium-free".
Use during pregnancy or breastfeeding.
Contraception in women
Women of childbearing potential should use effective contraception during finerenone therapy (see section "Special precautions for use").
Pregnancy
There are no data on the use of finerenone in pregnant women.
Animal studies have shown reproductive toxicity.
Firialta should not be used during pregnancy unless the woman's clinical condition necessitates treatment with finerenone. If a woman becomes pregnant while taking finerenone, she should be informed of the potential risks to the fetus (see section "Special precautions for use").
Breastfeeding
It is unknown whether finerenone or its metabolites are excreted in human breast milk.
Available pharmacokinetic/toxicological data show excretion of finerenone and its metabolites into milk in animals. Adverse effects were observed in rat pups exposed via this route. A risk to newborns/infants cannot be excluded.
A decision should be made whether to discontinue breastfeeding or to discontinue finerenone therapy, taking into account the benefit of breastfeeding for the child and the benefit of therapy for the woman (see section "Special precautions for use").
Fertility
There are no data on the effect of finerenone on fertility in humans.
Animal studies showed reduced fertility in females at exposures exceeding the maximum human exposures, which is considered to have low clinical relevance.
Ability to influence the speed of reactions when driving or operating machinery.
Firialta has no influence on the ability to drive or operate machinery.
Method of Administration and Dosage
Dosage
The recommended dose is 20 mg of finerenone once daily.
The maximum recommended dose is 20 mg of finerenone once daily.
Initiation of Therapy
Serum potassium and eGFR should be measured to determine whether treatment with finerenone can be initiated and to calculate the initial dose.
If serum potassium level is ≤ 4.8 mmol/L, therapy with finerenone may be initiated. For monitoring of serum potassium levels, see below "Continuation of Therapy".
If serum potassium level is > 4.8–5.0 mmol/L, initiation of finerenone therapy may be considered with additional monitoring of serum potassium levels during the first 4 weeks, depending on patient characteristics and serum potassium levels (see section "Special Warnings and Precautions").
If serum potassium level is > 5.0 mmol/L, finerenone therapy should not be initiated (see section "Special Warnings and Precautions").
The recommended initial dose of finerenone is based on eGFR and is presented in Table 3.
Table 3
Initiation of Finerenone Therapy and Recommended Dose
| eGFR (ml/min/1.73 m2) |
Initial dose (once daily) |
| ≥ 60 |
20 mg |
| ≥ 25 to < 60 |
10 mg |
| < 25 |
not recommended |
Continuation of therapy
Four weeks after initiating or resuming finerenone therapy or increasing the dose, serum potassium and eGFR should be re-measured (see Table 4 for guidance on continuing finerenone therapy and dose adjustment).
Thereafter, serum potassium levels should be monitored periodically and as clinically indicated, depending on patient characteristics and serum potassium levels.
For additional information, see sections «Special precautions for use» and «Interaction with other medicinal products and other forms of interaction».
Table 4
Continuation of finerenone therapy and dose adjustment
| Current finerenone dose (once daily) |
|||
| 10 mg |
20 mg |
||
| Current serum potassium level (mmol/L) |
≤ 4.8 |
Increase to 20 mg finerenone once daily* |
Maintain dose of 20 mg once daily |
| > 4.8 to 5.5 |
Maintain dose of |
Maintain dose of 20 mg once daily |
|
| > 5.5 |
Discontinue finerenone. |
Discontinue finerenone. |
|
* Maintain the dose of 10 mg once daily if eGFR decreases by > 30% compared to the previous measurement.
Missed dose
A missed dose should be taken as soon as the patient remembers, but only on the same day.
The patient should not take two doses to make up for a missed dose.
Special patient groups
Elderly patients
Dose adjustment in elderly patients is not required (see section "Pharmacokinetics").
Renal impairment
Initiation of treatment
Finerenone therapy should not be initiated in patients with eGFR < 25 mL/min/1.73 m² due to limited clinical data (see sections "Special precautions" and "Pharmacokin游戏副本
Adverse reactions.
Summary of safety profile
The most commonly observed adverse reaction during treatment with the medicinal product Firialta was hyperkalaemia (≥ 14%), see "Hyperkalaemia" below and section "Special warnings and precautions for use".
The safety of Firialta in patients with chronic kidney disease and type 2 diabetes was evaluated in two pivotal Phase III trials: FIDELIO-DKD (diabetic kidney disease) and FIGARO-DKD. In the FIDELIO-DKD study, 2827 patients received finerenone (10 or 20 mg once daily), and 2831 received placebo. The mean duration of treatment was 2.2 years. In the FIGARO-DKD study, 3683 patients received finerenone (10 or 20 mg once daily) with a mean treatment duration of 2.9 years.
Reported adverse reactions are listed in Table 5 below, classified by MedDRA system organ class and frequency of occurrence.
Adverse reactions are grouped according to their frequency in descending order of severity. Frequency is defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10000 to < 1/1000), very rare (< 1/10000), and not known (cannot be estimated from available data).
Table 5
Adverse Reactions
| MedDRA System organ class |
Very common |
Common |
Uncommon |
| Metabolism and nutrition disorders |
Hyperkalaemia |
Hyponatraemia, hyperuricaemia |
|
| Vascular disorders |
Arterial hypotension |
||
| Skin and subcutaneous tissue disorders |
Pruritus |
||
| Investigations |
Decreased glomerular filtration rate |
Decreased haemoglobin levels |
Description of individual adverse reactions
Hyperkalaemia
In the combined data from the FIDELIO-DKD and FIGARO-DKD studies, hyperkalaemia was reported in 14% of patients receiving finerenone compared with 6.9% of patients receiving placebo. During the first month of treatment, an increase in mean serum potassium level of 0.17 mmol/L from baseline was observed in the finerenone group compared to the placebo group, after which levels remained stable. Most cases of hyperkalaemia in patients receiving finerenone were mild to moderate in severity and transient. Serious cases of hyperkalaemia were reported more frequently with finerenone (1.1%) than with placebo (0.24%). Serum potassium concentrations > 5.5 mmol/L and > 6.0 mmol/L were recorded in 16.8% and 3.3% of patients receiving finerenone, and in 7.4% and 1.24% of patients receiving placebo, respectively.
Hyperkalaemia leading to permanent discontinuation of treatment occurred in 1.7% of patients receiving finerenone compared to 0.6% in the placebo group. The rate of hospitalisation due to hyperkalaemia was 0.9% in the finerenone group versus 0.2% in the placebo group.
For specific recommendations, see sections "Posology and method of administration" and "Special warnings and precautions for use".
Arterial hypotension
In the combined data from the FIDELIO-DKD and FIGARO-DKD studies, arterial hypotension was reported in 4.6% of patients receiving finerenone compared to 3.04% of patients receiving placebo. Treatment with finerenone was permanently discontinued due to arterial hypotension in 3 patients (< 0.1%). The rate of hospitalisation due to arterial hypotension was the same in the finerenone and placebo groups (< 0.1%).
In patients receiving finerenone, most cases of arterial hypotension were mild or moderate in severity and transient.
Mean systolic blood pressure decreased by 2–4 mm Hg and mean diastolic blood pressure decreased by 1–2 mm Hg after 1 month, remaining stable thereafter.
Hyperuricaemia
In the combined data from the FIDELIO-DKD and FIGARO-DKD studies, hyperuricaemia was observed in 5.1% of patients receiving finerenone compared to 3.9% of patients receiving placebo. None of these events were considered serious or led to permanent discontinuation of finerenone. In the finerenone group compared to the placebo group, an increase in mean serum uric acid level of 0.3 mg/dL from baseline was observed up to month 16, after which levels declined over time. There was no difference between the finerenone and placebo groups in the incidence of gout events (3.0%).
Decreased glomerular filtration rate (GFR)
In the combined data from the FIDELIO-DKD and FIGARO-DKD studies, decreased GFR was reported in 5.3% of patients taking finerenone compared to 4.2% of patients receiving placebo. In patients receiving finerenone, the incidence of decreased GFR leading to permanent discontinuation of the drug was the same as in the placebo group (0.2%). The rate of hospitalisation due to decreased GFR was similar in the finerenone and placebo groups (< 0.1%).
In patients receiving finerenone, most cases of decreased GFR were mild or moderate in severity and transient.
In patients receiving finerenone, an initial decrease in GFR (on average by 2 mL/min/1.73 m²) was observed, which attenuated over time compared to the placebo group. This decrease was reversible during continuous treatment.
Decreased haemoglobin levels
In the combined data from the FIDELIO-DKD and FIGARO-DKD studies, treatment with finerenone was associated with a placebo-corrected mean absolute decrease in haemoglobin level of 0.154 g/dL and in haematocrit of 0.5% after 4 months of therapy. The incidence of anaemia was comparable in patients receiving finerenone (6.5%) and those receiving placebo (6.1%). The rate of serious anaemia cases was low in both the finerenone and placebo groups (0.5%). Changes in haemoglobin and haematocrit levels were transient and returned to levels similar to those observed in the placebo group after approximately 24–32 months.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after marketing authorisation is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals and patients, as well as their legal representatives, are encouraged to report any suspected adverse reactions and lack of efficacy through the Automated Information System for Pharmacovigilance at: https://aisf.dec.gov.ua.
Shelf life
3 years.
Storage conditions
No special storage conditions required. Keep out of the reach and sight of children.
Packaging
Tablets, film-coated, 10 mg: 14 tablets in a blister; 2 or 7 blisters with calendar strip in a cardboard carton.
Tablets, film-coated, 20 mg: 14 tablets in a blister; 2 or 7 blisters with calendar strip in a cardboard carton.
Prescription status
Prescription only.
Manufacturer
Bayer AG.
Manufacturer's address
Kaiser-Wilhelm-Allee, 51368 Leverkusen, Germany