Esperoct
UkrainaSpis treści
INSTRUKCJA stosowania leku Esperoct (Esperoct)
Skład:
substancja czynna: turkoctocog alfa pegol;
500 MI
Jedno fiolka z proszkiem zawiera nominalną ilość 500 MI turkoctocog alfa pegolu*.
Po odtworzeniu leku, 1 ml roztworu zawiera około 125 MI turkoctocog alfa pegolu.
1000 MI
Jedno fiolka z proszkiem zawiera nominalną ilość 1000 MI turkoctocog alfa pegolu*.
Po odtworzeniu leku, 1 ml roztworu zawiera około 250 MI turkoctocog alfa pegolu.
1500 MI
Jedno fiolka z proszkiem zawiera nominalną ilość 1500 MI turkoctocog alfa pegolu*.
Po odtworzeniu leku, 1 ml roztworu zawiera około 375 MI turkoctocog alfa pegolu.
2000 MI
Jedno fiolka z proszkiem zawiera nominalną ilość 2000 MI turkoctocog alfa pegolu*.
Po odtworzeniu leku, 1 ml roztworu zawiera około 500 MI turkoctocog alfa pegolu.
3000 MI
Jedno fiolka z proszkiem zawiera nominalną ilość 3000 MI turkoctocog alfa pegolu*.
Po odtworzeniu leku, 1 ml roztworu zawiera około 750 MI turkoctocog alfa pegolu.
Aktywność (w jednostkach międzynarodowych, MI) oznaczana jest metodą chromogenną zgodnie z Europejską Farmakopeą. Specyficzna aktywność turkoctocog alfa pegolu wynosi około 9500 MI/mg białka.
Substancja czynna turkoctocog alfa pegol jest kowalencyjnym koniugatem białka turkoctocog alfa* z polietylenoglikolem (PEG) o masie 40 kDa.
* Czynnik VIII ludzki wytwarzany metodą rekombinowaną technologii DNA w linii komórkowej jajnika chomika chińskiego, przy czym żadne składniki pochodzenia ludzkiego lub zwierzęcego nie są stosowane w tej linii komórkowej, ani w trakcie oczyszczania, koniugacji ani wytwarzania składników leku Esperoct.
substancje pomocnicze:
proszek: L-histydyna; sacharoza; polisorbat 80; chlorek sodu; L-metionina; chlorek wapnia, dwuwodny; wodorotlenek sodu (do regulacji pH); kwas chlorowodorowy (do regulacji pH);
rozpuszczalnik: chlorek sodu; woda do wstrzykiwań.
Postać leku. Proszek i rozpuszczalnik do roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: proszek liofilizowany ma postać liofilizatu od białego do praktycznie białego.
Rozpuszczalnik jest klarowny i bezbarwny; pH 6,9; osmolalność 590 mOsmol/kg.
Grupa farmakoterapeutyczna. Środki hemostatyczne. Czynnik krzepnięcia VIII.
Kod ATC B02B D02.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Turkotokog alfa pegol to oczyszczony rekombinowany czynnik VIII człowieka (rFVIII) z cząsteczką polietylenglikolu (PEG) o wielkości 40 kDa, skonjugowaną z białkiem. PEG jest przyłączony do wiązanego O-glikanu w obciętym domenie B rFVIII (turkotokog alfa). Mechanizm działania turkotokogu alfa pegolu opiera się na zastępowaniu niedoborowego lub brakującego czynnika VIII u pacjentów z hemofilią typu A.
Gdy turkotokog alfa pegol jest aktywowany przez trombinę w miejscu uszkodzenia, domena B zawierająca fragment PEG oraz region a3 są odcinane, w wyniku czego powstaje aktywowany rekombinowany czynnik VIII (rFVIIIa), który strukturalnie przypomina naturalny czynnik VIIIa.
Zespół czynnika VIII z czynnikiem von Willebranda składa się z dwóch cząsteczek (czynnika VIII i czynnika von Willebranda), które charakteryzują się różnymi funkcjami fizjologicznymi. Po podaniu leku pacjentowi z hemofilią czynnik VIII wiąże się z czynnikiem von Willebranda w krążeniu. Aktywowany czynnik VIII działa jako kofaktor aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w aktywowany czynnik X.
Aktywowany czynnik X przekształca protrombinę w trombinę. Następnie trombina przekształca fibrynogen w fibrynę, co umożliwia powstanie skrzepu. Hemofilia typu A to sprzężone z chromosomem X dziedziczne zaburzenie krzepnięcia krwi spowodowane obniżonym poziomem czynnika VIII:C, prowadzące do nasilonych krwawień do stawów, mięśni lub narządów wewnętrznych, zarówno spontanicznie, jak i w wyniku urazu lub zabiegu chirurgicznego. Dzięki terapii zastępczej czynnikiem VIII poziomy czynnika VIII w osoczu krwi wzrastają, umożliwiając tymczasową korekcję niedoboru i przeciwdziałanie skłonności do krwawień.
Skuteczność kliniczna w profilaktyce i leczeniu epizodów krwawień
Skuteczność kliniczną leku Esperoct w profilaktyce i leczeniu krwawień badano w siedmiu wieloośrodkowych, prospektywnych badaniach klinicznych. Wszyscy pacjenci mieli ciężką hemofilię typu A.
Należy zaznaczyć, że nie należy porównywać wskaźników częstotliwości krwawień w ciągu roku (CKR), uzyskanych przy stosowaniu różnych koncentratów czynników i w różnych badaniach klinicznych.
Profilaktyka u dorosłych/dzieci w wieku od 12 lat
Skuteczność stosowania leku Esperoct w profilaktyce i leczeniu krwawień oceniano w badaniu otwartym, niekontrolowanym z udziałem dorosłych pacjentów i dzieci w wieku od 12 lat z ciężką hemofilią typu A. Efekt profilaktyczny leku Esperoct wykazano przy dawce 50 MI na 1 kg masy ciała co 4 dni lub co 3–4 dni (dwa razy w tygodniu) u 175 pacjentów. Średni wskaźnik częstotliwości krwawień w ciągu roku (CKR) u dorosłych i dzieci w wieku od 12 lat otrzymujących lek Esperoct wyniósł 1,18 (międzykwartylowy zakres (MŻ): 0,00; 4,25), podczas gdy wskaźnik CKR dla przypadków spontanicznych wyniósł 0,00 (MŻ: 0,00; 1,82), dla przypadków urazowych – 0,00 (MŻ: 0,00; 1,74), a wskaźnik CKR krwawień do stawów wyniósł 0,85 (MŻ: 0,00; 2,84). Po dodaniu warunkowych wartości (zastępujących brakujące dane pacjentów, którzy zakończyli udział w badaniu) obliczony średni wskaźnik CKR dla wszystkich przypadków krwawień wyniósł 3,70 (95 % CI: 2,94; 4,66). Spośród 175 dorosłych/dzieci w wieku od 12 lat, którzy otrzymywali profilaktykę, u 70 (40 %) pacjentów nie odnotowano żadnej krwawienia. Średnioroczne zastosowanie leku w celu profilaktyki wyniosło 4 641 MI/kg.
Dorośli/dzieci w wieku od 12 lat, u których częstotliwość krwawień była niska i wynosiła 0–2 epizody krwawień w ciągu ostatnich 6 miesięcy oraz którzy otrzymali co najmniej 50 dawek leku Esperoct, mieli możliwość uczestnictwa w randomizacji do grup leczenia profilaktycznego – z podawaniem leku co 7 dni (75 MI/kg co 7 dni) lub co 4 dni (50 MI/kg co 4 dni). Ogółem 55 spośród 120 pacjentów spełniających kryteria badania wyraziło zgodę na randomizację do grup (17 osób do grupy dawki 75 MI/kg co 4 dni oraz 38 osób do grupy co 7 dni). Wskaźnik CKR u pacjentów poddanych randomizacji wyniósł 1,77 (0,59; 5,32) przy podawaniu leku co 4 dni oraz 3,57 (2,13; 6,00) – przy profilaktyce według schematu raz w tygodniu. W trakcie fazy randomizowanego badania dziewięciu z tych pacjentów powróciło do profilaktyki co 4 dni. Ogółem, biorąc pod uwagę wszystkie części przedłużonego badania, 31 spośród 61 pacjentów, którzy otrzymywali profilaktykę co 7 dni, powróciło do schematu leczenia z podawaniem leku co 4 dni.
Profilaktyka u wcześniejszych leczonych pacjentów (do 12 roku życia)
Skuteczność i bezpieczeństwo stosowania leku Esperoct w profilaktyce oraz leczeniu epizodów krwawień oceniano w otwartym, jednogrupowym, niekontrolowanym badaniu z udziałem 68 dzieci do 12 roku życia z ciężką hemofilią typu A.
Efekt profilaktyczny leku Esperoct wykazano przy dawce profilaktycznej 64,7 MI na 1 kg masy ciała dwa razy w tygodniu. Mediana i obliczona średnia roczna częstotliwość krwawień u dzieci do 12 roku życia otrzymujących Esperoct dwa razy w tygodniu wyniosła odpowiednio 1,95 i 2,13 (95 % CI: 1,48; 3,06), podczas gdy dla przypadków spontanicznych wskaźnik CKR wyniósł 0,00 i 0,58 (95 % CI: 0,24; 1,40), dla przypadków urazowych – 0,00 i 1,52 (95 % CI: 1,07; 2,17), dla krwawień do stawów – 0,00 i 1,03 (95 % CI: 0,59; 1,81). W grupie 68 dzieci do 12 roku życia u 29 pacjentów (42,6 %), którzy otrzymywali Esperoct profilaktycznie, nie odnotowano żadnej krwawienia.
Średnioroczne zapotrzebowanie na lek w profilaktyce wyniosło 6 475 MI/kg.
Ze względu na znaczną długość trwania badania kilku pacjentów przekroczyło granice wiekowe grupy, do której pierwotnie zostali zakwalifikowani: niektórzy pacjenci < 6 roku życia przeszli również do kategorii wiekowej 6–11 lat, a niektórzy pacjenci z grupy 6–11 lat przeszli do kategorii wiekowej nastolatków. Główne wyniki oceny skuteczności u pacjentów w wieku < 12 lat, uzyskane na etapie podstawowym i przedłużonym badania, podsumowano w tabeli 1.
Tabela 1. Częstotliwość krwawień w ciągu roku (CKR) w badaniu z udziałem wcześniejszych leczonych dzieci według aktualnych grup wiekowych (etapy podstawowy i przedłużony badania) – pełna populacja badawcza
| Etap podstawowy |
Etap przedłużony |
|||
| Wiek pacjenta* |
0–5 lat (N = 34) |
6–11 lat (N = 34) |
0–5 lat (N = 27) |
6–11 lat (N = 53) |
| Liczba krwawień |
30 |
32 |
41 |
134 |
| Okres podstawowego leczenia (lata) |
0,46 |
0,51 |
4,79 |
4,86 |
| Całkowita roczna częstość krwawień (CRF) |
||||
| Średnia wartość rozkładu Poissona |
1,94 |
1,84 |
0,32 |
0,52 |
| Mediana |
1,94 |
1,94 |
0,22 |
0,21 |
* Niektórzy pacjenci należeli do dwóch grup wiekowych.
Profilaktyka u wcześniejszych nieleczonych pacjentów (PUPs) (dzieci do 6 roku życia)
Skuteczność i bezpieczeństwo stosowania leku Esperoct oceniano w trakcie wielonarodowego, nierandomizowanego, otwartego badania fazy 3. Wcześniejszą profilaktykę (profilaktyka opcjonalna, leczenie potrzebującego się epizodów krwawień i/lub dawkowanie 60 J/kg w odstępach dłuższych niż tydzień, aż pacjent osiągnie 20 dni ekspozycji (DE) lub wiek 24 miesięcy) oraz profilaktykę krwawień oceniano u 81 wcześniejszych nieleczonych pacjentów w wieku do 6 lat z ciężką hemofilią A. Spośród 81 pacjentów, 55 pacjentów rozpoczęło otrzymywanie wcześniejszej profilaktyki, z czego 42 pacjentów przeszło następnie na profilaktykę. Ogółem 69 pacjentów otrzymywało profilaktykę w dawce profilaktycznej 68,9 J/kg masy ciała dwa razy w tygodniu.
Efekt profilaktyczny leku Esperoct u wcześniejszych nieleczonych dzieci w wieku do 6 lat z ciężką hemofilią A wykazano na podstawie średniej rocznej częstości krwawień, wynoszącej medianę 1,35 oraz średnią obliczoną 1,76 (95 % CI: 1,26; 2,46).
Średnie roczne zapotrzebowanie dla 69 wcześniejszych nieleczonych pacjentów otrzymujących profilaktykę wynosiło 5 395 J/kg.
Główne wyniki oceny skuteczności u wcześniejszych nieleczonych pacjentów otrzymujących profilaktykę, podzielone na etap główny i etap przedłużony badania, podsumowano w tabeli 2.
Tabela 2. Częstość krwawień w ciągu roku (CKR) w badaniu z udziałem wcześniejszych nieleczonych pacjentów dziecięcych (etapy główne i przedłużone) – pełna próba pacjentów
| Etapa podstawowa |
Etapa przedłużona |
|
| Liczba krwawień |
124 |
223 |
| Okres podstawowego leczenia (lat) |
0,60 |
2,83 |
| Całkowita częstotliwość krwawień na rok (CCK) |
||
| Średnia wartość rozkładu Poissona (95% przedział ufności) |
2,98(2,16; 4,10) |
1,43 (0,98; 2,10) |
| Mediana |
2,49(0,00; 5,22) |
0,73 (0,00; 2,57) |
W trakcie badania zgłoszono 56 przypadków działań niepożądanych u 43 z 81 pacjentów oraz łącznie 80 poważnych działań niepożądanych u 48 pacjentów po podaniu leku Esperoct.
U 31 z 59 wcześniej nieleczonych pacjentów bez inhibitorów obserwowano tymczasowe zmniejszenie przyrostowego odbudowania (IR) czynnika VIII po podaniu Esperoct. U 17 wcześniej nieleczonych pacjentów stwierdzono kolejne pomiary obniżonego IR; u wszystkich tych pacjentów stwierdzono obecność przeciwciał IgG przeciwko PEG. Nie można wykluczyć związku między przeciwciałami przeciwko PEG a niskim IR.
Skuteczność kliniczna leku Esperoct w leczeniu epizodów krwawień oraz w trybie na żądanie
Skuteczność kliniczną leku Esperoct w leczeniu epizodów krwawień wykazano u wszystkich wcześniej leczonych pacjentów we wszystkich grupach wiekowych.
W większości przypadków krwawienia leczone za pomocą leku Esperoct miały lekki lub umiarkowany stopień ciężkości.
Ogólny wskaźnik skutecznego leczenia hemostatycznego krwawień u wcześniej leczonych pacjentów wyniósł 84,4%.
Wskaźniki skutecznego leczenia hemostatycznego w poszczególnych grupach wiekowych wcześniej leczonych pacjentów wynosiły odpowiednio: 89,4% (0–5 lat), 82,6% (6–11 lat), 78,9% (12–17 lat) i 84,9% (≥ 18 lat); 94,2% krwawień zostało skutecznie wyleczonych, a krwawienia ustały po podaniu 1–2 iniekcji.
Skuteczność leku Esperoct w leczeniu epizodów krwawień wykazano również u wcześniej nieleczonych pacjentów w wieku < 6 lat. Ogólny wskaźnik skutecznego leczenia hemostatycznego wyniósł 91,9%; 93,3% krwawień zostało skutecznie wyleczonych, a krwawienia ustały po podaniu 1–2 iniekcji.
W badaniu podstawowym 12 pacjentów w wieku od 18 lat zdecydowało się pozostać na leczeniu na żądanie. U tych pacjentów leczono 1270 krwawień przy średniej dawce 37,5 j.m./kg (zakres: 20–75 j.m./kg). Po podaniu 1–2 iniekcji leku Esperoct zatrzymano 97% wszystkich krwawień.
Skuteczność kliniczna leku Esperoct w zastosowaniu podczas zabiegów chirurgicznych
Efekt hemostatyczny leku Esperoct w zabiegach chirurgicznych oceniano w czterech badaniach, w tym w jednym badaniu specjalnym przeprowadzonym podczas interwencji chirurgicznych.
W specjalnym badaniu chirurgicznym przeanalizowano 49 dużych zabiegów chirurgicznych u 35 wcześniej leczonych pacjentów w wieku dorosłym i młodzieńczym. W dniu operacji pacjenci otrzymali średnią dawkę przedoperacyjną 55,7 j.m./kg (zakres: 27,2–86,2 j.m./kg), a średnia dawka pozapowikacyjna wyniosła 30,7 j.m./kg (zakres: 10,1–58,8 j.m./kg). Ogólny wskaźnik skutecznego leczenia hemostatycznego lekiem Esperoct podczas dużych zabiegów chirurgicznych wyniósł 95,9%, przy czym efektywność hemostazy oceniono jako doskonałą lub dobrą w 47 z 49 dużych zabiegów.
W dwóch badaniach z udziałem wcześniej leczonych dzieci (w wieku do 12 lat) 24 pacjentów przeszło łącznie 46 zabiegów chirurgicznych, z których tylko jeden był dużym zabiegiem z udaną odpowiedzią hemostatyczną. Małe operacje u tych pacjentów nie powodowały żadnych powikłań, choć skuteczność hemostazy i poziom czynnika VIII nie były kontrolowane podczas tych zabiegów. W badaniu z udziałem wcześniej nieleczonych pacjentów u 26 dzieci (w wieku do 6 lat) skuteczny efekt hemostatyczny odnotowano we wszystkich 4 dużych zabiegach chirurgicznych oraz w 25 z 30 małych operacji. Esperoct stosowano zgodnie z zaleceniami dotyczącymi dawkowania.
Farmakokinetyka.
Ogółem oceniono 129 profili farmakokinetycznych (PK), uzyskanych po pojedynczym podaniu leku Esperoct u 86 pacjentów (w tym 24 pacjentów pediatrycznych w wieku od 0 do 12 lat).
Wszystkie badania farmakokinetyki leku Esperoct przeprowadzono u pacjentów z ciężką hemofilią typu A (poziom czynnika VIII < 1%), którzy wcześniej byli leczeni. Pacjenci otrzymali pojedynczą dawkę 50 j.m./kg, a pobieranie próbek krwi do analizy przeprowadzono przed podaniem leku oraz wielokrotnie w ciągu 96 godzin po jego podaniu.
U dorosłych pacjentów okres półtrwania leku Esperoct był 1,6 raza dłuższy niż przy stosowaniu leków niemodyfikowanego czynnika VIII.
Parametry farmakokinetyczne
Ogółem oceniono 108 profili farmakokinetycznych, uzyskanych po pojedynczym podaniu dawki leku Esperoct 50 j.m./kg u 69 pacjentów. Parametry farmakokinetyczne leku po pojedynczym podaniu były porównywalne u młodszych dzieci (0–6 lat) i dzieci w średnim wieku (6–12 lat), jak również u starszych dzieci (12–17 lat) i dorosłych (≥ 18 lat).
Zgodnie z oczekiwaniami, u dzieci w wieku do 12 lat w porównaniu z dorosłymi i dziećmi w wieku od 12 lat stwierdzono niższe przyrostowe odbudowanie (IR) oraz wyższy przeliczony na masę ciała klirens. Ogólnie obserwowano tendencję do wzrostu przyrostowego odbudowania (IR) i spadku klirensu (ml/godz/kg) wraz z wiekiem. Jest to zgodne z faktem, że objętość dystrybucji leku na kilogram masy ciała u dzieci w wieku do 12 lat jest wyższa niż u dorosłych (tabela 1).
Parametry farmakokinetyczne leku Esperoct po pojedynczym podaniu, określone po 28 tygodniach leczenia profilaktycznego, odpowiadały początkowym parametrom farmakokinetycznym.
W tabeli 3 przedstawiono parametry farmakokinetyczne leku Esperoct po jego pojedynczym podaniu.
Tabela 3. Parametry farmakokinetyczne leku Esperoct 50 j.m./kg po jego pojedynczym podaniu u wcześniej leczonych pacjentów, wyznaczone metodą chromogenną [średnia geometryczna (CV% – współczynnik wariacji)]
| Parametr PK, |
Pacjenci w wieku od 0 do 6 lat (N = 13) |
Pacjenci w wieku od 6 do 12 lat (N = 11) |
Pacjenci w wieku od 12 do 18 lat (N = 3) |
Pacjenci w wieku od 18 lat (N = 42) |
| Liczba profili PK |
13 |
11 |
5 |
79 |
| IR (j.m./dL)/(j.m./kg)a |
1,80 (29) |
1,99 (25) |
2,79 (12) |
2,63 (22) |
| Maksymalna aktywność czynnika VIII (j.m./dL)a |
101,2 (28) |
119,6 (25) |
133,2 (9) |
134,4 (23) |
| t1/2 (godziny) |
13,6 (20) |
14,2 (26) |
15,8 (43) |
19,9 (34) |
| AUCinf (j.m.*godz/dL) |
2 147 (47) |
2 503 (42) |
3 100 (44) |
3 686 (35) |
| CL (ml/godz/kg) |
2,6 (45) |
2,4 (40) |
1,5 (43) |
1,4 (32) |
| Vss (ml/kg) |
44,2 (34) |
41,2 (25) |
33,4 (10) |
37,7 (27) |
| MRT (godziny) |
17,0 (22) |
17,3 (31) |
21,7 (45) |
25,2 (29)b |
Skróty: AUC – pole pod krzywą farmakokinetyczną opisującą dynamikę czasową aktywności czynnika VIII; t1/2 – czas półtrwania eliminacji; MRT – średni czas retencji leku w organizmie; CL – klarans; Vss – objętość rozkładu w stanie równowagi; IR – przyrostowe odtworzenie.
a Przyrost odtworzenia (IR) i aktywność czynnika VIII oznaczano 30 minut po podaniu leku u pacjentów w wieku od 12 lat oraz 60 minut po podaniu (pierwsza pobrana próbka do analizy) u dzieci poniżej 12. roku życia.
b Obliczenia oparte na danych z 67 profili.
W badaniu z udziałem wcześniej nieleczonych dzieci ocenę przyrostu odtworzenia (IR) czynnika VIII przeprowadzono u 46 pacjentów w wieku do 6 lat po pierwszym podaniu leku, średnia wartość geometryczna (CV %) wyniosła 1,76 (34) [IU/dl]/[IU/kg]. U 17 z 59 wcześniej nieleczonych pacjentów bez inhibitorów stwierdzono kolejne pomiarowane tymczasowe obniżenie przyrostu odtworzenia (IR) czynnika VIII w zakresie od 5 do 10 dawek skutecznych (szczegółowe informacje zawarte w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Średnie wartości minimalnej aktywności czynnika VIII u pacjentów wcześniej leczonych i wcześniej nieleczonych według grup wiekowych zestawiono w tabeli 4.
Tabela 4. Średnie obliczone wartości minimalnej aktywności czynnika VIII u pacjentów wcześniej leczonych i wcześniej nieleczonych według grup wiekowych
| Minimalna aktywność czynnika VIII |
Wcześniej leczeni pacjenci, 60 JM/kg leku Esperoct na profilaktykę dwa razy w tygodniu |
Wcześniej leczeni pacjenci, 50 JM/kg leku Esperoct na profilaktykę raz na 4 dni |
Wcześniej nieleczani pacjenci, 60 JM/kg leku Esperoct na profilaktykę dwa razy w tygodniu |
||
| Grupy wiekowe w momencie włączenia do badania |
0–5 lat |
6–11 lat |
12–17 lat |
≥ 18 lat |
0–5 lat |
| Liczba pacjentów włączonych do analizy |
31 |
34 |
23 |
143 |
81 |
| Liczba minimalnych wartości włączonych do analizy |
144 |
161 |
112 |
722 |
355 |
| Liczba minimalnych wartości poniżej LLOQ |
62 |
43 |
16 |
107 |
128a |
| Wyniki modelu mieszanegob: |
|||||
| 1,2 |
2,0 |
2,7 |
3,0 |
1,5 |
|
| 0,8; 1,6 |
1,5; 2,7 |
1,8; 4,0 |
2,6; 3,5 |
1,1; 1,9 |
|
LLOQ – dolna granica oznaczalności
a Wartości stężeń aktywności czynnika VIII w osoczu poniżej dolnej granicy oznaczalności (LLOQ), która wynosi 0,009 MI/ml, ustawiono na poziomie połowy LLOQ (0,0045 MI/ml).
b Model mieszany logarytmicznie przekształconych wartości aktywności czynnika VIII w osoczu, w którym grupa wiekowa była efektem stałym, a pacjent – efektem losowym. Modelowanie oddzielne przeprowadzono dla każdej profilaktycznej terapii (czyli dla każdej częstotliwości dawkowania). Minimalny poziom został przekształcony z powrotem do skali naturalnej.
Do analizy włączono wyłącznie pomiary przed podaniem leku, uzyskane w stanie równowagi dla tej terapii profilaktycznej.
Dane przedkliniczne dotyczące bezpieczeństwa
Dane z badań przedklinicznych wskazują na brak szczególnego ryzyka dla człowieka, biorąc pod uwagę wyniki standardowych badań bezpieczeństwa i toksyczności przy wielokrotnym stosowaniu.
Właściwości kliniczne.
Wskazania.
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (dziedzicznym niedoborem czynnika VIII).
Esperoct może być stosowany we wszystkich grupach wiekowych pacjentów.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na inne substancje pomocnicze leku (patrz rozdział „Skład”).
Znana reakcja alergiczna na białka chomika.
Interakcje z innymi lekami i inne rodzaje oddziaływań.
Nie ma doniesień o przypadkach interakcji czynnika krzepnięcia krwi VIII ludzkiego (rDNA) z innymi lekami.
Szczególne wskazania dotyczące stosowania.
Śledzenie
W celu poprawy śledzenia produktów biologicznych nazwa i numer serii podawanego leku powinny być wyraźnie wskazane.
Nadwrażliwość
Podczas stosowania leku Esperoct mogą występować reakcje nadwrażliwościowe o charakterze alergicznym. Środki te zawierają śladowe ilości białek chomika, które mogą wywoływać reakcje alergiczne u niektórych pacjentów. Pacjentom należy zalecić natychmiastowe zaprzestanie stosowania tego leku i skontaktowanie się z lekarzem w przypadku wystąpienia objawów nadwrażliwości. Pacjentów należy poinformować o wczesnych objawach nadwrażliwości, w tym o wysypce alergicznej, pokrzywce uogólnionej, uczuciu ściskania w klatce piersiowej, świstach w klatce piersiowej, hipotensji i anafilaksji.
W przypadku wystąpienia wstrząsu należy zastosować standardowe leczenie w przypadku wstrząsu.
Inhibitory
Znanym powikłaniem leczenia osób z hemofilią typu A jest powstawanie neutralizujących przeciwciał (inhibitorów) czynnika VIII. Inhibitory te są zazwyczaj immunoglobulinami klasy IgG, których działanie skierowane jest przeciwko aktywności krzepnej czynnika VIII, co ilościowo określa się w jednostkach Bethesda (JB) na 1 ml osocza krwi, stosując zmodyfikowaną metodę analizy ilościowej. Ryzyko powstawania inhibitorów zależy od ciężkości choroby oraz od poziomu ekspozycji na czynnik VIII, przy czym ryzyko to jest największe w ciągu pierwszych 50 dni ekspozycji, jednakże utrzymuje się przez całe życie, choć pojawia się rzadko.
Kliniczne znaczenie powstawania inhibitorów zależy od ich miana, przy czym niższe miano inhibitorów oznacza mniejsze ryzyko niewystarczającej odpowiedzi klinicznej niż wyższe miano. Ogólnie należy dokładnie monitorować wszystkich pacjentów stosujących leki zawierające czynnik krzepnięcia krwi VIII, aby wykryć powstawanie inhibitorów poprzez odpowiednią obserwację kliniczną i badania laboratoryjne. Jeśli oczekiwane poziomy aktywności czynnika VIII we krwi nie zostaną osiągnięte lub jeśli krwawienia nie da się zatrzymać stosując odpowiednią dawkę, należy sprawdzić, czy nie pojawiły się inhibitory czynnika VIII. U pacjentów z wysokim poziomem inhibitorów terapia z wykorzystaniem czynnika VIII może okazać się nieskuteczna, dlatego należy rozważyć inne opcje leczenia.
Leczeniem takich pacjentów powinni zajmować się lekarze z doświadczeniem w leczeniu hemofilii i przypadków powstawania inhibitorów czynnika VIII.
Obniżenie aktywności czynnika VIII u pacjentów wcześniej leczonych
W doniesieniach po wprowadzeniu na rynek zgłaszano przypadki obniżenia aktywności czynnika VIII w przypadku braku wykrytych inhibitorów czynnika VIII u pacjentów wcześniej leczonych. Obniżenie aktywności czynnika VIII obserwowano podczas przejścia na Esperoct i w niektórych przypadkach mogło być związane z przeciwciałami przeciwko PEG. Należy rozważyć możliwość odpowiedniego oznaczenia aktywności czynnika VIII po przejściu. Dodatkowe informacje zawiera sekcja „Działania niepożądane”.
Zjawiska sercowo-naczyniowe
U pacjentów z istniejącymi czynnikami ryzyka sercowo-naczyniowego stosowanie terapii zastępczej czynnikiem VIII może prowadzić do zwiększenia ryzyka sercowo-naczyniowego.
Powikłania związane z użyciem kaniuli
Jeśli istnieje potrzeba użycia urządzenia do dostępu do żyły centralnej (CVC), należy wziąć pod uwagę możliwość wystąpienia powikłań związanych z CVC, w tym infekcji miejscowych, bakteriemii i zakrzepicy w miejscu wprowadzenia kaniuli.
Dzieci
Wymienione ostrzeżenia i środki ostrożności mają zastosowanie zarówno w leczeniu dorosłych, jak i dzieci.
Obniżenie przyrostu inkrementalnego (IR) poziomu czynnika krzepnięcia krwi VIII u pacjentów wcześniej nieleczonych
W badaniach klinicznych u 31 z 59 pacjentów wcześniej nieleczonych obserwowano obniżenie przyrostu inkrementalnego (IR) poziomu czynnika VIII przy braku widocznych inhibitorów czynnika VIII. U 14 z nich zarejestrowano tylko jedno pomiarowe stwierdzenie niskiego przyrostu inkrementalnego (IR), podczas gdy u 17 pacjentów zanotowano 2 lub więcej kolejnych pomiarów niskiego przyrostu inkrementalnego (IR) w przedziale 5–10 skutecznych dawek. Obniżenie przyrostu inkrementalnego (IR) miało charakter tymczasowy i w przedziale 15–70 skutecznych dawek przyrost inkrementalny (IR) powrócił do wartości > 0,6 (MO/dl)/(MO/kg). Obniżenie przyrostu inkrementalnego (IR) występowało przy wzroście miana przeciwciał przeciwko polietylenglikolowi (anti-PEG IgG) u pacjentów wcześniej nieleczonych, bez powstawania inhibitorów czynnika VIII. Kolejne niskie wartości przyrostu inkrementalnego (IR) mogą potencjalnie wiązać się ze zmniejszoną skutecznością leku w tym okresie. Zaleca się obserwację pacjentów w wieku dziecięcym, w tym monitorowanie aktywności czynnika VIII po podaniu leku. Jeśli przy stosowaniu zalecanej dawki leku Esperoct krwawienie nie jest kontrolowane i/lub oczekiwane poziomy aktywności czynnika VIII nie są osiągnięte przy braku inhibitorów czynnika VIII, należy rozważyć możliwość dostosowania dawki lub częstotliwości stosowania leku lub zaprzestania jego stosowania.
Substancje pomocnicze
Ten lek zawiera 30,5 mg sodu w fiolce z odtworzonym lekiem, co odpowiada 1,5% rekomendowanego przez WHO maksymalnego dziennego spożycia sodu – 2,0 g – dla dorosłego człowieka.
Stosowanie w czasie ciąży lub karmienia piersią.
Badania dotyczące rozrodczości u zwierząt po zastosowaniu czynnika VIII nie były prowadzone. Hemofilia typu A u kobiet występuje rzadko, dlatego doświadczenie stosowania czynnika VIII w czasie ciąży i laktacji jest ograniczone. Z tego powodu czynnik VIII należy stosować w czasie ciąży i laktacji wyłącznie w przypadkach, gdy wskazanie do takiego zastosowania jest wyraźne.
Wpływ na zdolność prowadzenia pojazdów lub obsługi urządzeń.
Esperoct nie wpływa lub ma nieznaczny wpływ na zdolność prowadzenia pojazdów lub obsługi urządzeń.
Sposób stosowania i dawki
Leczenie należy rozpoczynać pod nadzorem lekarza z doświadczeniem w leczeniu hemofilii.
Monitorowanie w trakcie leczenia
W trakcie leczenia zaleca się odpowiednie oznaczanie poziomów aktywności czynnika VIII w celu umożliwienia dostosowania schematu dawkowania leku Esperoct w razie potrzeby. Odpowiedź na czynnik VIII może różnić się u poszczególnych pacjentów, co objawia się różnymi wartościami okresu półwylucania i wskaźnika przyrostowego odbudowy (IR). U pacjentów z niedożywieniem lub nadmierną masą ciała może być konieczna korekta dawki w zależności od masy ciała. W przypadku dużych zabiegów chirurgicznych należy monitorować proces terapii zastępczej czynnikiem VIII poprzez oznaczanie aktywności czynnika VIII we krwi.
Aktywność czynnika VIII w składzie leku Esperoct można oznaczać standardowymi metodami ilościowego oznaczania czynnika VIII – metodą chromogenną i jednoetapową metodą oznaczania. W przypadku stosowania jednoetapowej metody oznaczania krzepnięcia krwi opartej na oznaczaniu czasu tromboplastynowego częściowego aktywowanego (APTT – aktywowany częściowy czas tromboplastynowy) in vitro do oznaczania aktywności czynnika VIII w próbkach krwi pacjentów, wyniki aktywności czynnika VIII we krwi mogą znacząco zależeć zarówno od typu odczynnika APTT, jak i od zastosowanego wzorca odniesienia podczas oznaczania.
W przypadku stosowania jednoetapowej metody oznaczania krzepnięcia krwi należy unikać stosowania niektórych odczynników na bazie krzemionki, ponieważ mogą one prowadzić do uzyskania zaniżonych wyników. Ponadto mogą występować istotne rozbieżności wyników oznaczeń uzyskanych metodą jednoetapową opartą na oznaczaniu APTT oraz metodą chromogenną zgodnie z Europejską Farmakopeą. Ma to szczególne znaczenie przy zmianie laboratorium analitycznego i/lub odczynników stosowanych do przeprowadzenia analizy.
Dawkowanie
Dawka, odstępy między dawkami oraz trwałość terapii zastępczej zależą od stopnia ciężkości niedoboru czynnika VIII, lokalizacji i nasilenia krwawienia, pożądanego poziomu aktywności czynnika VIII oraz stanu klinicznego pacjenta. Ilość podanych jednostek czynnika VIII wyraża się w jednostkach międzynarodowych (J.M.), które odnoszą się do obowiązującego standardu WHO dla koncentratów leków zawierających czynnik VIII. Aktywność czynnika VIII we krwi wyraża się albo w procentach (w stosunku do normalnego poziomu we krwi człowieka), albo w jednostkach międzynarodowych na jeden decylitr (w stosunku do obowiązującego międzynarodowego standardu dla czynnika VIII we krwi).
Jedna jednostka międzynarodowa (J.M.) aktywności czynnika VIII odpowiada ilości czynnika VIII w 1 ml osocza krwi człowieka.
Leczenie na żądanie i leczenie epizodów krwawienia
Obliczenie wymaganej dawki czynnika VIII opiera się na empirycznie uzyskanych wynikach, z których wynika, że jedna jednostka międzynarodowa (J.M.) czynnika VIII na jeden kilogram masy ciała prowadzi do wzrostu aktywności czynnika VIII we krwi o 2 J.M./dl.
Wymaganą dawkę wyznacza się według poniższego wzoru:
Wymagana liczba jednostek (J.M.) = masa ciała (kg) × pożądane zwiększenie poziomu czynnika VIII (%) (J.M./dl) × 0,5 (J.M./kg na J.M./dl).
Wymaganą liczbę i częstotliwość stosowania należy zawsze dostosować do skuteczności klinicznej w każdym przypadku i u każdego pacjenta indywidualnie.
Zalecenia dotyczące dawkowania leku Esperoct w leczeniu na żądanie oraz w leczeniu epizodów krwawienia przedstawiono w tabeli 5. Poziomy aktywności czynnika VIII należy utrzymywać na poziomie równym lub wyższym niż wskazane poziomy we krwi (w J.M. na jeden decylitr lub w % normy). W leczeniu krwawienia można stosować maksymalną pojedynczą dawkę leku 75 J.M./kg oraz maksymalną dawkę sumaryczną 200 J.M./kg/24 godziny.
Tabela 5. Zalecenia dotyczące leczenia epizodów krwawienia lekiem Esperoct
| Stopień ciężkości krwawienia |
Wymagany poziom czynnika VIII (JE/dl lub % normy)a |
Częstotliwość dawkowania (godziny) |
Trwanie terapii |
| Lekki Wczesny hemartroz, lekkie krwawienie do mięśni lub jamy ustnej |
20–40 |
12–24 |
Do ustania krwawienia |
| Umiarkowany Bardziej zaawansowany hemartroz, krwawienie do mięśni, siniak |
30–60 |
12–24 |
Do ustania krwawienia |
| Silne lub zagrożliwe dla życia krwawienia |
60–100 |
8–24 |
Do usunięcia zagrożenia |
a Wymaganą dawkę ustala się według następującego wzoru:
Wymagana liczba jednostek (JE) = masa ciała (kg) × pożądane zwiększenie poziomu czynnika VIII (%) (JE/dl) × 0,5 (JE/kg na JE/dl).
Zastosowanie w okresie okołoperacyjnym
Dawka oraz odstępy między dawkami podczas zabiegu chirurgicznego zależą od zastosowanej procedury chirurgicznej oraz lokalnej praktyki klinicznej. Można zastosować maksymalną dawkę pojedynczą preparatu Esperoct 75 JE/kg oraz maksymalną dawkę dobową 200 JE/kg/24 godziny.
Częstotliwość dawkowania i czas trwania terapii należy zawsze dobrać indywidualnie, w zależności od odpowiedzi klinicznej u każdego pacjenta.
W tabeli 6 przedstawiono ogólne zalecenia dotyczące dawkowania preparatu Esperoct w okresie okołoperacyjnym. Należy zwrócić uwagę na utrzymanie aktywności czynnika VIII na poziomie równym lub wyższym od zakresu docelowego.
Tabela 6. Zalecenia dotyczące dawkowania preparatu Esperoct w okresie okołoperacyjnym
| Typ zabiegu chirurgicznego |
Wymagany poziom czynnika VIII (jednostek/dl (%) (jednostek/dl)a |
Częstotliwość dawkowania (godziny) |
Trwanie terapii |
| Lekki zabieg chirurgiczny (w tym usunięcie zęba) |
30–60 |
W ciągu jednej godziny przed zabiegiem chirurgicznym. W razie potrzeby powtórzyć po 24 godzinach. |
Podanie pojedyncze lub powtarzane co 24 godziny przez co najmniej jeden dzień, aż do uzyskania wyleczenia. |
| Poważny zabieg chirurgiczny |
80–100 (przed i po zabiegu chirurgicznym) |
W ciągu jednej godziny przed zabiegiem chirurgicznym w celu osiągnięcia poziomu aktywności czynnika VIII w zakresie docelowym. Powtarzać co 8–24 godziny w celu utrzymania aktywności czynnika VIII w zakresie docelowym. |
Powtarzać wstrzyknięcie co 8–24 godziny, zgodnie z potrzebą, w celu odpowiedniego stopnia gojenia rany. Należy rozważyć kontynuację terapii przez kolejne 7 dni w celu utrzymania aktywności czynnika VIII na poziomie od 30% do 60% (jednostek/dl). |
a Potrzebną dawkę ustala się według następującego wzoru:
Potrzebna ilość jednostek (JE) = masa ciała (kg) × pożądane zwiększenie stężenia czynnika VIII (%) (JE/dl) × 0,5 (JE/kg na JE/dl).
Profilaktyka
Zalecana dawka leku Esperoct u dorosłych to 50 JE na kilogram masy ciała co 4 dni.
Można rozważyć dostosowanie dawki i częstotliwości podawania w zależności od osiągniętych stężeń czynnika VIII oraz indywidualnej tendencji do krwawień u każdego pacjenta.
Dzieci
Zalecana dawka dla dzieci (powyżej 12 roku życia) jest taka sama jak u dorosłych.
Zalecana dawka profilaktyczna dla dzieci do 12 roku życia to 65 JE na 1 kg masy ciała (50–75 JE/kg) dwa razy w tygodniu. Dostosowanie dawki i częstotliwości podawania leku zależy od osiągniętego stężenia czynnika VIII oraz indywidualnej skłonności do krwawień.
Szczegółowe informacje dotyczące pacjentów w wieku dziecięcym zawarte są w sekcjach „Szczególne wytyczne dotyczące stosowania”, „Farmakodynamika” oraz „Farmakokinetyka”.
Sposób podania
Lek Esperoct przeznaczony jest do wstrzykiwania dożylnego.
Lek Esperoct należy podawać w formie wstrzyknięcia dożylnego (przez około 2 minuty) po odtworzeniu proszku za pomocą 4 ml rozpuszczalnika dostarczonego wraz z lekiem (roztwór do wstrzykiwań chlorku sodu, 9 mg/ml (0,9 %)).
Instrukcje dotyczące odtwarzania leku przed jego zastosowaniem znajdują się poniżej w podsekcji „Zalecenia dotyczące postępowania z lekiem i jego utylizacji”.
Zalecenia dotyczące postępowania z lekiem i jego utylizacji
Esperoct należy podawać dożylnie po odtworzeniu proszku za pomocą rozpuszczalnika znajdującego się w strzykawce. Po odtworzeniu roztwór powinien być klarowny, bezbarwny i nie powinien zawierać widocznych cząstek. Przed podaniem odtworzony lek należy wizualnie sprawdzić pod kątem obecności cząstek stałych oraz zmiany barwy. Roztwór musi być klarowny i bezbarwny. Nie należy stosować roztworu, który jest mętny lub zawiera osad.
Instrukcje dotyczące odtwarzania leku przed zastosowaniem znajdują się w podsekcji „Instrukcja obsługi leku Esperoct”.
Szybkość podania należy dostosować do poziomu komfortu pacjenta, przez około 2 minuty.
Będzie również potrzebny zestaw do infuzji (igła motylkowa z rurką), sterylne alkoholowe waciki, gaziki i plaster. Urządzenia te nie są zawarte w zestawie Esperoct.
Należy zawsze stosować technikę bezpieczną.
Utylizacja
Po wstrzyknięciu należy bezpiecznie zutylizować strzykawkę, zestaw do infuzji oraz fiolkę z przejściówką do fiolki.
Nie wykorzystany lek lub odpady należy zutylizować zgodnie z lokalnymi przepisami.
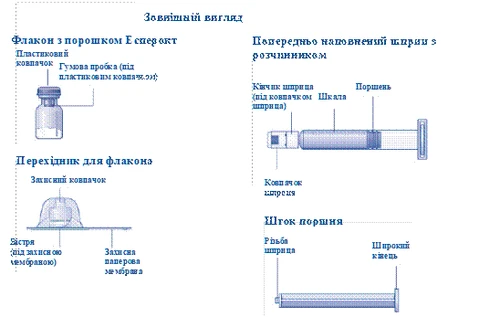
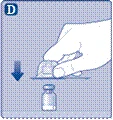
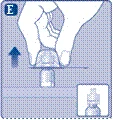
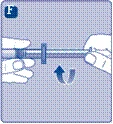
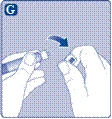
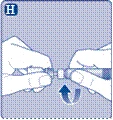
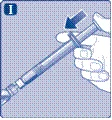
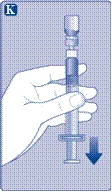
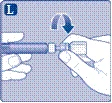
| Instrukcja obsługi leku Esperoct Uważnie przeczytaj niniejszą instrukcję przed zastosowaniem leku Esperoct. Lek Esperoct dostarczany jest w postaci proszku. Przed wstrzyknięciem należy go rozpuścić za pomocą roztworu znajdującego się w strzykawce. Roztwór ten to roztwór do wstrzykiwań chlorku sodu 9 mg/ml (0,9%). Odtworzony lek należy podawać dożylnie (wstrzyknięcie i.v.). Sprzęt dostarczony w tym opakowaniu przeznaczony jest do odtworzenia i podania leku Esperoct. Będziesz również potrzebować:
Te przedmioty nie są zawarte w opakowaniu z lekiem Esperoct. Nie stosuj sprzętu, jeśli nie otrzymałeś odpowiedniego szkolenia od lekarza lub pielęgniarki. Zawsze myj ręce i zapewnij czystą przestrzeń wokół siebie. Podczas przygotowania i podania leku bezpośrednio do żyły ważne jest stosowanie metod zapewniających czystość i sterylność (metody aseptyczne). Nieprawidłowe stosowanie tych metod może prowadzić do wprowadzenia drobnoustrojów i zakażenia krwi. Nie otwieraj sprzętu, dopóki nie będziesz gotowy go użyć. Nie używaj sprzętu, jeśli upadł lub jest uszkodzony. W takim przypadku użyj nowego opakowania. Nie używaj sprzętu po upływie daty ważności. W takim przypadku użyj nowego opakowania. Data ważności jest wydrukowana na tekturowym opakowaniu, na fiolce, na łączniku do fiolki i na wstępnie wypełnionej strzykawce. Nie używaj sprzętu, jeśli podejrzewasz jego zanieczyszczenie. W takim przypadku użyj nowego opakowania. Nie wyrzucaj żadnych przedmiotów, dopóki nie wykonasz wstrzyknięcia odtworzonego roztworu leku. Ten sprzęt przeznaczony jest wyłącznie do jednorazowego użytku. |
|
| Zawartość Opakowanie zawiera:
|
|
|
|
|
Nie stosuj innych sposobów podgrzewania fiolki i wstępnie wypełnionej strzykawki. |
|
Nie dotykaj gumowej septy palcami, ponieważ może to prowadzić do wprowadzenia drobnoustrojów. |
|
Jeśli ochronna papierowa membrana nie jest całkowicie przylepiona lub jest rozerwana, nie stosuj tego łącznika do fiolki. Nie wyjmuj łącznika do fiolki z ochronnego kapturka palcami. Jeśli dotkniesz igły łącznika do fiolki, może to prowadzić do wprowadzenia drobnoustrojów z Twoich palców. |
|
Gdy łącznik jest już podłączony do fiolki, nie odłączaj go z fiolki. |
|
Nie odłączaj łącznika od fiolki, gdy zdejmujesz ochronny kapturzek. |
|
|
|
Nie dotykaj końcówki strzykawki znajdującej się pod kapturkiem. Jeśli dotkniesz końcówki strzykawki, może to prowadzić do wprowadzenia drobnoustrojów z Twoich palców. Jeśli plastikowy kapturzek jest poluzowany lub brakuje go, nie używaj tej wstępnie wypełnionej strzykawki. |
|
|
|
|
|
Nie wstrząsaj fiolką, ponieważ może to prowadzić do pienienia się roztworu.
|
|
| Esperoct zaleca się zastosować natychmiast po odtworzeniu. Jeśli nie możesz zastosować odtworzonego roztworu leku Esperoct natychmiast, należy go zastosować:
Odtworzony roztwór należy przechowywać w fiolce. Nie mroź odtworzonego roztworu i nie przechowuj go w strzykawkach. Przechowuj odtworzony roztwór w miejscu chronionym przed bezpośrednim światłem słonecznym. Jeśli do uzyskania potrzebnej dawki konieczne jest użycie więcej niż jednej fiolki leku, powtórz kroki od A do J z dodatkowymi fiolkami, łącznikami do fiolki i wstępnie wypełnionymi strzykawkami, aż uzyskasz wymaganą ilość dawki. |
|
|
|
Nie dotykaj końcówki strzykawki. Jeśli dotkniesz końcówki strzykawki, może to prowadzić do wprowadzenia drobnoustrojów z Twoich palców. |
|
Obecnie lek Esperoct jest gotowy do wstrzyknięcia dożylnego.
Nie mieszać leku Esperoct z żadnymi innymi wlewami dożylnymi ani lekami. Podawanie leku Esperoct przez bezigłowe konektory do cewników dożylnych (i.v.). Uwaga! Wstępnie wypełniona strzykawka wykonana jest ze szkła i przeznaczona jest do użycia ze standardowym połączeniem Luer-lock. Niektóre bezigłowe konektory z wewnętrznym kolcem są niezgodne z tą wstępnie wypełnioną strzykawką. Taka niezgodność może utrudnić podanie leku i spowodować uszkodzenie bezigłowego konektora. Podawanie roztworu przez urządzenie do dostępu do żyły centralnej (UDŻC), takie jak cewnik do żyły centralnej lub port podskórny:
Jeśli system UDŻC należy przemyć przed wstrzyknięciem leku Esperoct lub po nim, użyj roztworu do wstrzykiwań chlorku sodu o stężeniu 9 mg/ml (0,9%). |
|
| Unieszkodliwienie.
Nie wyrzucaj tych materiałów ze zwykłymi odpadami komunalnymi. |
|
| Nie rozłączaj sprzętu na części przed jego unieszkodliwieniem. Nie używaj tego sprzętu ponownie. |
|


Dzieci
Leczenie i profilaktyka krwawień u pacjentów z hemofilią typu A (dziedziczny deficyt czynnika VIII).
Esperoct może być stosowany u wszystkich grup wiekowych.
Szczegółowe informacje dotyczące pacjentów w wieku dziecięcym podano w sekcjach „Właściwości farmakologiczne” (podsekcje „Farmakodynamika” i „Farmakokinetyka”), „Szczególne wskazania”, „Sposób podania i dawka”.
Przedawkowanie.
Nie odnotowano doniesień o objawach przedawkowania rekombinowanym czynnikiem krzepnięcia krwi VIII.
Efekty uboczne.
Podsumowanie profilu bezpieczeństwa
Nadwrażliwość lub reakcje alergiczne (w tym obrzęk naczynioruchowy, uczucie pieczenia i mrowienia w miejscu infuzji, dreszcze, zawroty głowy, uogólnione pokrzywienie, ból głowy, wysypka alergiczna, hipotensja, senność, nudności, pobudzenie, tachykardia, uczucie ucisku w klatce piersiowej, mrowienie, wymioty, świsty w oddychaniu) obserwowano rzadko i w niektórych przypadkach mogły one postępować do ciężkiej anafilaksji (w tym wstrząs).
Bardzo rzadko obserwowano powstawanie przeciwciał przeciwko białkom chomika z odpowiednimi reakcjami nadwrażliwości.
U pacjentów z hemofilią A leczonych czynnikiem VIII, w tym lekiem Esperoct, mogą powstawać przeciwciała neutralizujące (inhibitory). Jeśli takie inhibitory powstają, objawia się to niewystarczającą odpowiedzią kliniczną na leczenie. W takich przypadkach zaleca się skonsultowanie się ze specjalistycznym ośrodkiem hemofilii.
Lista efektów ubocznych
Dane dotyczące częstości występowania efektów ubocznych pochodzące z sześciu badań klinicznych z udziałem 270 pacjentów wcześniej leczonych oraz 81 pacjentów wcześniej nieleczonych z ciężką hemofilią A (< 1% aktywności endogennego czynnika VIII) i bez wcześniejszych doniesień o inhibitorach czynnika VIII przedstawiono w tabeli 7. Poniżej wymienione kategorie zarejestrowanych efektów ubocznych odpowiadają klasyfikacji układów i narządów MedDRA (według klas układów organizmu i preferowanych terminów) podanych w tabeli 7.
Częstość występowania efektów ubocznych szacowano według następujących kategorii: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), nieczęsto (od ≥ 1/1000 do < 1/100), rzadko (od ≥ 1/10000 do < 1/1000), bardzo rzadko (< 1/10000); nieznane (niemożliwe do oszacowania na podstawie dostępnych danych). Poniżej wymieniono efekty uboczne występujące u pacjentów wcześniej leczonych.
Tabela 7. Częstość efektów ubocznych zarejestrowanych w trakcie badań klinicznych
| Układów narządów zgodnie z klasyfikacją |
Terminy występowania działań niepożądanych |
Częstość występowania (wcześniej leczeni pacjenci) |
Częstość występowania (wcześniej nieleczieni pacjenci) |
| Zaburzenia układu krwi i chłonnego |
Inhibicja czynnika VIII * |
Niekorzystne |
Bardzo często** |
| Zaburzenia skóry i tkanek podskórnych |
Świerdzenie |
Często |
- |
| Zapalenie |
Często |
Często |
|
| Wysypka |
Często |
Często |
|
| Zaburzenia ogólne i reakcje w miejscu podania |
Reakcje w miejscu wstrzyknięcia*** |
Często |
Często |
| Zaburzenia układu odpornościowego |
Nadwrażliwość na lek |
- |
Często |
| Nadwrażliwość |
Niekorzystne |
- |
|
| Badania |
Obniżony poziom czynnika krzepnięcia krwi VIII |
Nieznane**** |
- |
* Pacjenta z potwierdzonym inhibitorem czynnika VIII zdefiniowano jako wynik testu inhibitora początkowego ≥ 0,6 jednostek Bethesda (JB), potwierdzony drugą próbką pobraną nie później niż w ciągu 2 tygodni.
** W tym pacjenci z potwierdzonym inhibitorem czynnika VIII wśród pacjentów z grupy ryzyka (co najmniej 10 dni stosowania leku).
*** Najczęstsze działania niepożądane obejmowały reakcje w miejscu wstrzyknięcia: reakcja w miejscu wstrzyknięcia, siniak w miejscu nakłucia naczynia, reakcja w miejscu infuzji, zaczerwienienie w miejscu wstrzyknięcia, wysypka w miejscu wstrzyknięcia, ból w miejscu nakłucia naczynia i obrzęk w miejscu wstrzyknięcia.
**** Na podstawie doniesień z okresu po wprowadzeniu na rynek.
Opis wybranych działań niepożądanych
Wytwarzanie inhibitorów czynnika VIII
Zarejestrowano jeden potwierdzony przypadek powstawania inhibitora czynnika VIII podczas profilaktycznego stosowania leku Esperoct u 18-letniego pacjenta, który wcześniej był leczony. Pacjent miał inwersję intronu 22 w genie czynnika VIII i wysokie ryzyko powstawania inhibitorów czynnika VIII.
Brak danych sugerujących zwiększone ryzyko powstawania inhibitorów czynnika VIII podczas leczenia lekiem Esperoct w porównaniu z innymi lekami czynnika VIII.
Wytwarzanie przeciwciał przeciw lekowi
Zarejestrowano jeden przypadek trwałego wytwarzania przeciwciał przeciw lekowi, który pokrywał się czasowo z potwierdzonym przypadkiem powstawania inhibitorów czynnika VIII (patrz sekcja dotycząca powstawania inhibitorów czynnika VIII). U trzech pacjentów po podaniu leku Esperoct wynik testu na przeciwciała był tymczasowo pozytywny, jednak nie stwierdzono korelacji z działaniami niepożądanymi.
Przeciwciała przeciwko PEG
W trakcie programu badań klinicznych u 37 pacjentów stwierdzono obecność przeciwciał przeciwko PEG przed podaniem leku Esperoct. Test na przeciwciała przeciwko PEG był negatywny u 20 z 37 pacjentów po podaniu leku. U 17 pacjentów zaobserwowano tymczasowe niskie miana przeciwciał przeciwko PEG. Nie stwierdzono korelacji z działaniami niepożądanymi.
Zgodnie z doniesieniami z okresu po wprowadzeniu na rynek obserwowano również powstawanie przeciwciał przeciwko PEG podczas przejścia na Esperoct. U niektórych pacjentów przeciwciała przeciwko PEG mogły być związane z niższym niż oczekiwany poziomem aktywności FVIII.
Dzieci
Nie stwierdzono różnic w profilu bezpieczeństwa u dzieci wcześniej leczonych i dorosłych pacjentów.
Tymczasowe zmniejszenie przyrostu odzyskania (IR) poziomu czynnika VIII obserwowano u niektórych pacjentów wcześniej nieleczonych, bez obecności inhibitorów czynnika VIII (szczegółowe informacje zawarte są w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Zgłaszanie podejrzewanych działań niepożądanych i braku skuteczności leku
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Osoby pracujące w zawodach medycznych i farmaceutycznych, a także pacjenci lub ich przedstawiciele prawni powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych i braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
Nieotwierany fiolka (przed rozcieńczeniem):
36 miesięcy w temperaturze lodówki (2–8 °C).
W trakcie okresu ważności lek może być przechowywany:
- w temperaturze pokojowej (≤ 30 °C) od momentu produkcji do rozpoczęcia rozcieńczania nie dłużej niż 12 miesięcy
lub
- w temperaturze powyżej temperatury pokojowej (od > 30 °C do 40 °C) od momentu produkcji do rozpoczęcia rozcieńczania nie dłużej niż 3 miesiące.
Po przechowywaniu leku poza lodówką nie można go ponownie umieszczać w lodówce.
Należy zanotować początek przechowywania poza lodówką oraz temperaturę przechowywania w odpowiednim miejscu na tece kartonowej.
Po rozcieńczeniu:
Stabilność chemiczna i fizyczna podczas użytkowania została wykazana:
- przez 24 godziny w temperaturze lodówki (2–8 °C) lub
- przez 4 godziny w temperaturze ≤ 30 °C lub
- przez 1 godzinę w temperaturze od > 30 °C do 40 °C, pod warunkiem, że przed rozcieńczeniem lek był przechowywany w temperaturze powyżej temperatury pokojowej (od > 30 °C do 40 °C) nie dłużej niż 3 miesiące.
Z mikrobiologicznego punktu widzenia lek należy stosować natychmiast po rozcieńczeniu. Jeżeli rozcieńczony lek nie jest stosowany natychmiast, odpowiedzialność za czas i warunki przechowywania podczas użytkowania spoczywa na użytkowniku; zaleca się nie przechowywać leku dłużej niż wskazano powyżej, z wyjątkiem przypadków, gdy rozcieńczenie przeprowadzono w kontrolowanych i walidowanych warunkach zapewniających sterylność.
Rozcieńczony roztwór należy przechowywać w fiolce.
Warunki przechowywania.
Przechowywać w lodówce (2–8 °C). Nie zamrażać. Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem.
Informacje dotyczące przechowywania w temperaturze pokojowej (≤ 30 °C) lub w temperaturze do 40 °C oraz warunki przechowywania po rozcieńczeniu leku znajdują się w sekcji „Okres ważności”. Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Ponieważ nie przeprowadzono badań zgodności, tego leku nie wolno mieszać z innymi lekami ani rozcieńczać innymi roztworami do wstrzykiwań niż dołączony do opakowania leku rozcieńczalnik – chlorku sodu.
Rozcieńczonego leku nie wolno podawać jednocześnie z innymi lekami przez te same rurki do wstrzykiwań ani przez ten sam pojemnik, który jest już używany do podawania innych leków.
Opakowanie.
Każde kartonowe opakowanie leku Esperoct zawiera:
- 1 fiolkę szklaną (szkło typu I) z proszkiem, zamkniętą butylową septą, aluminiową pokrywką i plastikowym kapturkiem do odlamania;
- 1 sterylny adapter do fiolki do rozcieńczania;
- 1 wstępnie napełniony strzykawka o pojemności 4 ml z rozcieńczalnikiem (0,9 % roztwór chlorku sodu) z zaworem zwrotnym (polipropylen), tłokiem gumowym (bromobutyl) i kapturkiem gumowym (bromobutyl);
- 1 tłok strzykawki (polipropylen).
Kategoria wydania. Na receptę.
Właściciel zezwolenia na dopuszczenie do obrotu/Wytwórca.
A/S Novo Nordisk.
Siedziba właściciela zezwolenia na dopuszczenie do obrotu/wytwórcy oraz adres miejsca prowadzenia działalności.
Novo Allé,
Bagsværd, 2880,
Dania.