Epobiokryn
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU Epobiokryn (Epobiocrinum)
Skład:
substancja czynna: Epoetin alfa;
1 ml roztworu zawiera 1000 MI, lub 2000 MI, lub 4000 MI, lub 10 000 MI rekombinowanego erytropoetyny ludzkiej;
substancje pomocnicze: albumina ludzka, cytrynian sodu, chlorek sodu, kwas cytrynowy jednowodny; woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny roztwór.
Grupa farmakoterapeutyczna. Leki przeciwanemiczne. Erytropoetyna.
Kod ATC B03XA01.
Właściwości farmakodynamiczne.
Farmakodynamika.
Rekombinowany erytropoetyna ludzka pod względem aktywności biologicznej i immunologicznej odpowiada naturalnej erytropoetynie ludzkiej – glikoproteinowemu hormonowi, który pełni rolę czynnika stymulującego mitozę i jest hormonem stymulującym erytropoezę, czyli proces powstawania czerwonych krwinek z komórek macierzystych. U zdrowej osoby erytropoetyna jest wytwarzana w nerkach (90 %) oraz w komórkach Kupffera wątroby (10 %). Poziom jej syntezy zależy od nasycenia krwi tlenem. Erytropoetyna stymuluje proliferację i różnicowanie komórek erytropojetycznych do dojrzałych erytrocytów. Działa na wczesnych etapach erytropoezy na poziomie jednostek tworzących kolonie erytrocytów i jednostek tworzących skupiska erytrocytów, a następnie na poziomie proerytroblastu, erytroblastu i retikulocytu (wrażliwość tych komórek na erytropoetynę jest proporcjonalna do ich dojrzałości). Erytropoetyna normalizuje poziom hemoglobiny i hematokrytu oraz likwiduje objawy związane z anemią.
Masa cząsteczkowa epoetyny alfa wynosi około 30 600 daltonów. Część białkowa stanowi około 60 % masy cząsteczkowej i zawiera 165 aminokwasów. Cztery łańcuchy węglowodanowe są przyłączone do białka za pomocą trzech wiązań N-glikozydowych i jednego wiązania O-glikozydowego.
Farmakokinetyka.
Podanie dożylne. Po dożylnej administracji leku okres półtrwania u osób z prawidłową funkcją nerek wynosi około 4 godziny; u pacjentów z zaburzeniami funkcji nerek – około 5 godzin. Okres półtrwania u dzieci wynosi około 6 godzin.
Podanie podskórne. Stężenia w osoczu po podaniu podskórnym są znacznie niższe niż po podaniu dożylnym. Po podaniu podskórnym stężenie leku we krwi wzrasta powoli i osiąga maksimum po 12–18 godzinach od wstrzyknięcia. Maksymalne stężenie w osoczu po podaniu podskórnym jest niższe niż po dożylnej aplikacji (około 1/20 wartości).
Nie występuje efekt kumulacji, tj. mierzone stężenia erytropoetyny w surowicy pozostają na tym samym poziomie, niezależnie od tego, czy oznaczenie stężenia leku przeprowadzono po 24 godzinach od pierwszej iniekcji, czy po 24 godzinach od ostatniej.
Brak danych na temat możliwości przenikania rekombinowanej ludzkiej erytropoetyny przez barierę łożyskową lub do mleka matki; jednakże przez barierę krew-mózg ta substancja nie przenika.
Okres półtrwania po podaniu podskórnym wynosi około 24 godziny.
Biodostępność leku po podaniu podskórnym jest znacznie niższa niż po dożylnej aplikacji i wynosi około 20 %.
Charakterystyka kliniczna.
Wskazania.
Leczenie anemii symptomatycznej związanej z przewlekłą niewydolnością nerek:
- leczenie anemii związanej z przewlekłą niewydolnością nerek u dzieci i dorosłych poddawanych hemodializie oraz u dorosłych pacjentów poddawanych dializie otrzewnowej;
- leczenie ciężkiej anemii o podłożu nerkowym towarzyszącej objawom klinicznym u dorosłych pacjentów z niewydolnością nerek, którzy jeszcze nie poddawani byli hemodializie.
Leczenie anemii oraz zmniejszenie objętości wymaganych transfuzji krwi u dorosłych pacjentów poddawanych chemioterapii z powodu nowotworu niemielomatycznego, złośliwej chłoniaki lub szpiczaka mnogiego, u których ryzyko transfuzji oceniane jest na podstawie ogólnego stanu pacjenta (w tym stanu sercowo-naczyniowego, istniejącej anemii przed rozpoczęciem chemioterapii).
Epobiokryn można stosować w ramach programu przedoperacyjnego u pacjentów przed dużymi zabiegami chirurgicznymi z umiarkowanymi objawami anemii (poziom hemoglobiny 10 – 13 g/dl (6,2 – 8,1 mmol/l), brak niedoboru żelaza) w celu ułatwienia poboru krwi autologicznej i zmniejszenia ryzyka związanego z użyciem transfuzji allogennych, jeśli oczekiwane zapotrzebowanie na krew do przetaczania przekracza ilość, jaką można uzyskać metodą poboru autologicznego bez zastosowania alfa epoetyny.
Epobiokryn stosuje się dorosłym pacjentom z łagodnym i umiarkowanym stopniem anemii (hemoglobina w zakresie 10 – 13 g/dl przy braku niedoboru żelaza) przed przeprowadzeniem rozległych zabiegów ortopedycznych z oczekiwanym umiarkowanym stopniem utraty krwi (900–1800 ml krwi) w celu zmniejszenia potrzeby transfuzji allogennych oraz wspomagania regeneracji układu erytropoetycznego.
Przeciwwskazania.
Podwyższona wrażliwość na którykolwiek składnik leku.
Rozwój prawdziwej aplazji czerwonych komórek krwi w wyniku leczenia alfa epoetyną (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Niekontrolowana nadciśnienie tętnicze.
Przeciwwskazania związane z programem poboru krwi autologicznej u pacjentów otrzymujących alfa epoetynę.
Ciężkie choroby wieńcowe, obwodowe choroby tętnicze, choroby tętnic szyjnych lub mózgowo-naczyniowe, a także niedawno przebyty zawał mięśnia sercowego lub udar u pacjentów kwalifikowanych do planowej chirurgii ortopedycznej, którzy nie uczestniczyli w programie poboru krwi autologicznej. Niemożność zastosowania odpowiedniej profilaktyki przeciwzakrzepowej u pacjentów chirurgicznych.
Interakcje z innymi lekami oraz inne rodzaje interakcji.
Nie ma danych wskazujących, że leczenie alfa epoetyną wpływa na metabolizm innych leków.
Leki hamujące erytropoetynę mogą obniżać odpowiedź na leczenie alfa epoetyną.
Ponieważ cyklosporyna wiąże się z erytrocytami, istnieje możliwość interakcji lekowej. W przypadku jednoczesnego stosowania Epobiokrynu i cyklosporyny należy monitorować poziom tego ostatniego we krwi i w razie potrzeby korygować dawkę.
Nie stwierdzono potwierdzonych interakcji między alfa epoetyną a G-CSF (czynnikiem stymulującym kolonie granulocytów) ani GM-CSF (czynnikiem stymulującym kolonie granulocytów i makrofagów) w odniesieniu do różnicowania hematologicznego lub proliferacji komórek nowotworowych w próbce biopsji in vitro.
U pacjentek z przerzutowym rakiem piersi podskórne stosowanie alfa epoetyny w dawce 40000 IU/ml jednocześnie z trastuzumabem w dawce 6 mg/kg nie wpływało na farmakokinetykę trastuzumabu.
Szczególne środki ostrożności.
Ciśnienie tętnicze należy stale monitorować u wszystkich pacjentów podczas leczenia lekiem Epobiokryn. Lek należy stosować z ostrożnością u pacjentów z nieleczoną nadciśnieniem tętniczym lub niedostatecznie kontrolowanym nadciśnieniem tętniczym. W trakcie leczenia Epobiokrynem może zaistnieć potrzeba rozpoczęcia lub wzmocnienia terapii przeciwnadciśnieniowej. Jeśli nie uda się kontrolować ciśnienia, należy przerwać stosowanie epoetyny alfa.
Przypadki hipertensyjnego kryzysu z encefalopatią i drgawkami, wymagającego natychmiastowego badania lekarskiego i intensywnej terapii, obserwowano również u pacjentów z normalnym lub niskim ciśnieniem tętniczym na początku leczenia. Należy zwrócić szczególną uwagę na nagły, migrenopodobny, pulsujący ból głowy, który może być sygnałem ostrzegawczym (patrz sekcja „Efekty uboczne”).
Epoetynę alfa należy stosować z ostrożnością u pacjentów z padaczką, z przeszłością drgawek lub stanami medycznymi stanowiącymi czynniki ryzyka wystąpienia drgawek, takimi jak infekcje układu nerwowego lub przerzuty do mózgu.
Epoetynę alfa należy stosować z ostrożnością u pacjentów z przewlekłą niewydolnością wątroby. Bezpieczeństwo stosowania epoetyny alfa u tej grupy pacjentów nie zostało ustalone.
U pacjentów otrzymujących leki stymulujące erytropoetynę obserwuje się zwiększony ryzyko chorób naczyniowych z powikłaniami zakrzepowymi (patrz sekcja „Efekty uboczne”), w tym zakrzepicy żył głębokich, zakrzepicy tętniczej i żylnych, zakrzepicy płucnej, zakrzepicy żył siatkówki i zawału mięśnia sercowego. Zgłaszano również przypadki udarów mózgu (w tym udarów niedokrwnych, udarów krwotocznych i przejściowych ataków niedokrwiennych).
Przed rozpoczęciem leczenia epoetyną alfa należy dokładnie ocenić ryzyko chorób naczyniowych z powikłaniami zakrzepowymi, szczególnie u pacjentów z istniejącymi czynnikami ryzyka, w tym nadmierną masą ciała i chorobami naczyniowymi w wywiadzie (np. zakrzepicą żył głębokich, zakrzepicą płucną i udarem mózgu).
Należy dokładnie monitorować poziom hemoglobiny u wszystkich pacjentów ze względu na potencjalnie zwiększone ryzyko powikłań zakrzepowo-zatorowych i śmiertelnych w przypadku stosowania leku przy poziomie hemoglobiny wyższym niż docelowy w wskazaniach do stosowania.
Podczas leczenia może wystąpić umiarkowane, zależne od dawki zwiększenie liczby płytek krwi w granicach normy. Ten parametr zmniejsza się w dalszym ciągu leczenia. Zgłaszano również przypadki trombocytozy. Zaleca się regularne monitorowanie liczby płytek krwi w pierwszych 8 tygodniach leczenia.
Wszystkie inne przyczyny anemii (niedobór żelaza, kwasu foliowego, witaminy B12, zatrucie glinem, infekcja lub stan zapalny, utrata krwi, hemoliza lub włóknienie szpiku kostnego o dowolnym pochodzeniu) należy ustalić i wyleczyć przed rozpoczęciem terapii epoetyną alfa oraz przed podjęciem decyzji o zwiększeniu dawki. W większości przypadków wartości ferrytyny w surowicy krwi obniżały się równocześnie ze wzrostem hematokrytu. Aby zagwarantować optymalną odpowiedź na leczenie epoetyną alfa, należy zapewnić odpowiednie dostarczanie żelaza (patrz sekcja „Sposób stosowania i dawki”):
- pacjentom z przewlekłą niewydolnością nerek zaleca się przyjmowanie żelaza (200–
300 mg/dobę dla dorosłych i 100–200 mg/dobę dla dzieci doustnie, przeliczając na żelazo pierwiastkowe), jeśli poziom ferrytyny w surowicy krwi jest niższy niż 100 ng/ml; - pacjentom z chorobami nowotworowymi zaleca się przyjmowanie żelaza (200–300 mg/dobę doustnie, przeliczając na żelazo pierwiastkowe), jeśli nasycenie transferyny jest niższe niż 20%;
- pacjentom biorącym udział w programie poboru krwi autologicznej zaleca się przyjmowanie żelaza (200 mg/dobę doustnie, przeliczając na żelazo pierwiastkowe) kilka tygodni przed rozpoczęciem poboru krwi autologicznej w celu osiągnięcia znacznych zapasów żelaza w organizmie przed rozpoczęciem terapii oraz w trakcie leczenia epoetyną alfa;
- pacjentom przed planowanymi dużymi zabiegami ortopedycznymi zaleca się przyjmowanie żelaza (200 mg/dobę doustnie, przeliczając na żelazo pierwiastkowe) w trakcie leczenia epoetyną alfa. Jeśli to możliwe, należy rozpocząć przyjmowanie żelaza przed rozpoczęciem terapii epoetyną alfa w celu osiągnięcia znacznych zapasów żelaza w organizmie.
Bardzo rzadko zgłaszano rozwój lub pogorszenie istniejącej porfirii u pacjentów leczonych epoetyną alfa. Pacjentom z porfirią epoetynę alfa należy stosować z ostrożnością.
Przy długotrwałym leczeniu epoetyną alfa może wystąpić rozwój ciężkich efektów ubocznych ze strony skóry, w tym zespół Stevensa-Johnsona i toksyczny epidermalny nekroliz, które mogą zagrażać życiu lub mieć śmiertelny skutek (patrz sekcja „Efekty uboczne”).
Pacjenci powinni być poinformowani o możliwych efektach ubocznych ze strony skóry. W przypadku wystąpienia objawów i objawów rozwoju skórnych efektów ubocznych należy natychmiast przerwać leczenie epoetyną alfa i rozważyć alternatywne metody leczenia.
Informacje o handlowej nazwie leków stymulujących erytropoetynę, które były stosowane w leczeniu, należy wyraźnie wskazać w karcie medycznej pacjenta. Przejście pacjenta z jednego środka stymulującego erytropoetynę na inny jest możliwe tylko pod nadzorem lekarza.
Prawdziwa aplazja czerwonych krwinki (PRCA).
Istnieją doniesienia o przypadkach rozwoju PRCA po wielomiesięcznym lub wieloletnim podskórnej aplikacji epoetyny, głównie u pacjentów z przewlekłą niewydolnością nerek. Zgłaszano również przypadki prawdziwej aplazji czerwonych krwinek u pacjentów z wirusem zapalenia wątroby typu C, którzy jednocześnie otrzymywali interferon i rybawirynę z lekami stymulującymi erytropoetynę. Epoetyna alfa nie jest wskazana w leczeniu anemii związanej z wirusem zapalenia wątroby typu C.
Pacjentów, u których występuje nagła utrata skuteczności terapii (objawiająca się spadkiem poziomu hemoglobiny o 1–2 g/dl na miesiąc) z koniecznością zwiększenia transfuzji, należy skierować na badanie liczby retikulocytów krwi i wykrycie typowych przyczyn obniżenia odpowiedzi klinicznej (niedobór żelaza, kwasu foliowego, witaminy B12, zatrucie glinem, infekcja lub stan zapalny, utrata krwi, hemoliza lub włóknienie szpiku kostnego o dowolnym pochodzeniu).
W przypadku paradoksalnego spadku hemoglobiny i rozwoju ciężkiej anemii związanej z niską liczbą retikulocytów należy przerwać leczenie Epobiokrynem i określić obecność przeciwciał przeciwko erytropoetynie, a także przeprowadzić badanie szpiku kostnego w celu potwierdzenia rozpoznania prawdziwej aplazji czerwonych krwinek.
Pacjentom nie należy przepisywać leczenia innymi lekami stymulującymi erytropoetynę, ponieważ istnieje możliwość reakcji krzyżowej.
Leczenie anemii symptomatycznej u dorosłych pacjentów i dzieci z przewlekłą niewydolnością nerek.
U pacjentów z przewlekłą niewydolnością nerek otrzymujących epoetynę alfa należy regularnie monitorować poziom hemoglobiny do osiągnięcia stabilnego poziomu, a następnie okresowo. Tempo wzrostu poziomu hemoglobiny powinno wynosić około 1 g/dl (0,62 mmol/l) miesięcznie i nie powinno przekraczać 2 g/dl (1,25 mmol/l) miesięcznie w celu minimalizacji ryzyka rozwoju nadciśnienia tętniczego.
U pacjentów z przewlekłą niewydolnością nerek osiągnięty poziom hemoglobiny nie powinien przekraczać górnej granicy pożądanego stężenia hemoglobiny we krwi (patrz sekcja „Sposób stosowania i dawki”). W badaniach klinicznych obserwowano zwiększone ryzyko śmiertelności i poważnych niepożądanych reakcji ze strony układu sercowo-naczyniowego przy stosowaniu leków stymulujących erytropoetynę w celu osiągnięcia stężenia hemoglobiny powyżej 12 g/dl (7,5 mmol/l).
Kontrolowane badania kliniczne nie wykazały istotnych korzyści z zastosowania epoetyn przy stężeniu hemoglobiny wyższym niż poziom konieczny do zapewnienia kontroli objawów anemii i zapobiegania przetaczaniu krwi.
Należy ostrożnie zwiększać dawkę Epobiokrynu u pacjentów z przewlekłą niewydolnością nerek, ponieważ wysokie dawki skumulowane epoetyny mogą być związane ze zwiększonym ryzykiem śmiertelności, poważnych zaburzeń sercowo-naczyniowych i mózgowo-naczyniowych. U pacjentów z niewystarczającą odpowiedzią na leczenie epoetynami warto rozważyć inne sposoby przezwyciężenia niewystarczającej odpowiedzi (patrz sekcja „Sposób stosowania i dawki”).
Stan pacjentów z przewlekłą niewydolnością nerek, którym stosuje się Epobiokryn podskórnie, należy regularnie monitorować pod kątem utraty skuteczności leczenia, zdefiniowanej jako spadek lub utrata odpowiedzi na leczenie epoetyną alfa u pacjentów, u których wcześniej obserwowano odpowiedź na terapię. Utrata skuteczności charakteryzuje się trwałym spadkiem poziomu hemoglobiny niezależnie od zwiększenia dawki epoetyny alfa (patrz sekcja „Efekty uboczne”).
W trybie leczenia z wydłużonymi interwałami dawkowania (podawanie epoetyny alfa rzadziej niż raz w tygodniu) u niektórych pacjentów może dojść do spadku poziomu hemoglobiny, u których może być konieczne zwiększenie dawki. Należy regularnie monitorować poziom hemoglobiny.
U pacjentów poddawanych hemodializie obserwowano zakrzepicę szuntów, szczególnie u osób skłonnych do hipotensji lub powikłań przetok tętniczo-żylnych (np. zwężenie, tętniaki itp.). Takim pacjentom zaleca się kontrolę szuntu i zapobieganie zakrzepicy poprzez stosowanie np. kwasu acetylosalicylowego.
W pojedynczych przypadkach obserwowano hiperkaliemię, choć nie stwierdzono związku przyczynowego. U pacjentów z przewlekłą niewydolnością nerek należy monitorować poziom elektrolitów w surowicy krwi. W przypadku wzrostu poziomu potasu we krwi oprócz odpowiedniego leczenia hiperkaliemii należy rozważyć możliwość tymczasowego odstawienia Epobiokrynu do pełnej korekty stanu hiperkaliemii.
W wyniku wzrostu poziomu hematokrytu pacjenci poddawani hemodializie i otrzymujący Epobiokryn często wymagają zwiększenia dawki heparyny podczas dializy. W przypadku niedostatecznej heparynizacji możliwe jest rozwinięcie się okluzyjnego uszkodzenia układu dializacyjnego.
Zgodnie z obecną wiedzą, stosowanie Epobiokrynu pacjentom przeddializacyjnym nie przyspiesza postępu niewydolności nerek.
Leczenie pacjentów z anemią spowodowaną chemioterapią.
U pacjentów z chorobami nowotworowymi leczonych epoetyną alfa należy regularnie monitorować poziom hemoglobiny do osiągnięcia stabilnego poziomu, a następnie okresowo.
Epoetyny są czynnikami wzrostu, które głównie stymulują produkcję erytrocytów. Receptory erytropoetyny wykryto również na powierzchni różnych komórek nowotworowych. Tak jak w przypadku innych czynników wzrostu, nie można wykluczyć możliwości stymulacji przez epoetyny wzrostu niektórych typów nowotworów.
Nie można wykluczyć wpływu leków stymulujących erytropoetynę na postęp nowotworu lub zmniejszenie przeżycia bez postępu choroby. W kontrolowanych badaniach klinicznych zastosowanie Epobiokrynu i innych leków stymulujących erytropoetynę było związane ze zmniejszeniem lokalnego kontroli nowotworu lub ogólnego przeżycia:
- zmniejszenie lokalnego kontroli u pacjentów z postępującym rakiem głowy i szyi, którzy otrzymywali radioterapię, przy zastosowaniu w celu podniesienia poziomu hemoglobiny powyżej 14 g/dl (8,7 mmol/l);
- skrócenie ogólnego przeżycia i zwiększenie liczby śmiertelnych przypadków w wyniku postępu choroby w ciągu 4 miesięcy u pacjentek z przerzutowym rakiem piersi, które otrzymywały chemioterapię, przy zastosowaniu w celu podniesienia poziomu hemoglobiny do 12-14 g/dl (7,5-8,7 mmol/l);
- zwiększone ryzyko śmiertelności przy zastosowaniu w celu podniesienia poziomu hemoglobiny do 12 g/dl (7,5 mmol/l) u pacjentów z aktywną chorobą nowotworową, którzy nie otrzymują ani chemioterapii, ani radioterapii. Leki stymulujące erytropoetynę są przeciwwskazane tej grupie pacjentów;
- zwiększenie ryzyka śmiertelności o 9% w grupie pacjentów, którzy otrzymywali epoetynę alfa i standardowe leczenie, oraz 15% zwiększone ryzyko, którego nie można wykluczyć statystycznie, u pacjentek z przerzutowym rakiem piersi, które otrzymywały chemioterapię, przy zastosowaniu w celu podniesienia poziomu hemoglobiny do 10-12 g/dl (6,2-7,5 mmol/l).
Ze względu na powyższe, w niektórych sytuacjach klinicznych warto rozważyć przetaczanie krwi w leczeniu anemii u pacjentów z nowotworem. Decyzja o zastosowaniu rekombinowanego erytropoetyny powinna opierać się na ocenie korzyści i ryzyka dla konkretnego pacjenta, z uwzględnieniem specyficznego kontekstu klinicznego. Czynniki, które należy wziąć pod uwagę przy tej ocenie, powinny obejmować typ i stadium nowotworu; stopień anemii; przewidywany czas życia; warunki, w jakich leczony jest pacjent, oraz życzenia samego pacjenta.
U pacjentów z chorobami nowotworowymi, którzy otrzymują chemioterapię, zazwyczaj występuje opóźnienie odpowiedzi o 2–3 tygodnie (od momentu przepisania erytropoetyny do pojawienia się wywołanych przez nią czerwonych krwinek). Należy wziąć pod uwagę tę cechę przy ocenie skuteczności terapii (szczególnie w odniesieniu do pacjentów wymagających transfuzji).
Pacjenci poddawani zabiegom chirurgicznym i biorący udział w programie poboru krwi autologicznej.
Należy przestrzegać wszystkich szczególnych środków ostrożności związanych z programem poboru krwi autologicznej, szczególnie procedur przywracania objętości krwi krążącej.
Pacjenci przed dużym planowanym zabiegiem ortopedycznym.
Należy zawsze przestrzegać odpowiednich zasad transfuzjologii w okresie przed- i pooperacyjnym.
Pacjenci przed dużym planowanym zabiegiem ortopedycznym powinni otrzymywać środki w celu odpowiedniej profilaktyki przeciwzakrzepowej, ponieważ po zabiegach chirurgicznych u tych pacjentów mogą wystąpić powikłania zakrzepowe i naczyniowe, szczególnie na tle współistniejących chorób sercowo-naczyniowych. Szczególną ostrożność należy zachować w terapii pacjentów skłonnych do rozwoju zakrzepicy żył głębokich. Co więcej, u pacjentów z początkowym poziomem hemoglobiny > 13 g/dl ryzyko rozwoju powikłań zakrzepowych lub naczyniowych pooperacyjnych związanych z terapią epoetyną alfa jest znacznie wyższe. W związku z tym stosowanie epoetyny alfa pacjentom z początkowym poziomem hemoglobiny > 13 g/dl nie jest zalecane.
Pacjenci w wieku starszym
Bezpieczeństwo stosowania epoetyny alfa u tej grupy pacjentów nie zostało ustalone.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża.
Obecnie nie ma wyników kontrolowanych badań dotyczących stosowania leku Epobiokryn u ciężarnych kobiet. Badania na zwierzętach wykazały toksyczność rozrodczą. Dlatego lek Epobiokryn należy stosować ciężarnym kobietom tylko wtedy, gdy potencjalna korzyść z terapii przewyższa możliwe ryzyko dla płodu. Stosowanie epoetyny alfa ciężarnym kobietom biorącym udział w programie poboru krwi autologicznej nie jest zalecane.
Karmienie piersią.
Nie wiadomo, czy egzogenna epoetyna alfa wydzielana jest w mleku matki. Należy ostrożnie stosować epoetynę alfa u kobiet karmiących piersią. Decyzję o kontynuowaniu lub przerwaniu karmienia piersią lub kontynuowaniu lub przerwaniu stosowania epoetyny alfa należy podjąć, biorąc pod uwagę korzyści karmienia piersią dla dziecka i korzyści z leczenia epoetyną alfa dla kobiety.
Stosowanie epoetyny alfa pacjentkom biorącym udział w programie poboru krwi autologicznej w czasie karmienia piersią nie jest zalecane.
Plodność.
Nie przeprowadzono badań wpływu epoetyny alfa na płodność mężczyzn ani kobiet.
Wpływ na szybkość reakcji przy prowadzeniu pojazdów mechanicznych lub innych urządzeń.
Nie przeprowadzono badań wpływu na szybkość reakcji przy prowadzeniu pojazdów mechanicznych lub innych urządzeń.
Sposób stosowania i dawki.
Epoetyna alfa może być stosowana drogą podskórną oraz dożylną.
Tak jak w przypadku stosowania wszystkich leków podawanych pozajelitowo, lek Epobiokryn należy przed zastosowaniem sprawdzić pod kątem braku widocznych zanieczyszczeń oraz zmiany barwy roztworu.
Wszystkie inne przyczyny anemii (niedobór żelaza, kwasu foliowego, witaminy B12, zatrucie glinem, infekcja lub stan zapalny, utrata krwi, hemoliza lub fibroza szpiku kostnego o dowolnej etiologii) należy zdiagnozować i wyleczyć przed rozpoczęciem terapii epoetyną alfa oraz przed podjęciem decyzji o zwiększeniu dawki. W celu osiągnięcia optymalnej odpowiedzi na leczenie epoetyną alfa należy zapewnić odpowiedni poziom dopływu żelaza do organizmu oraz, w razie potrzeby, dodatkowo przepisać przyjmowanie leków zawierających żelazo (patrz sekcja „Szczególne wskazania”).
Podanie dożylne.
Epoetynę alfa stosuje się w formie iniekcji trwającej od 1 do 5 minut, w zależności od dawki leku. Pacjentom poddawanym hemodializie iniekcję bolusową można podać bezpośrednio w trakcie procedury przez odpowiedni port żylną w linii dializacyjnej. Lek można również podać po zakończeniu procedury hemodializy przez fystulę lub kaniulę z podłączeniem 10 ml izotonicznego roztworu natrii chloridum w celu przepłukania układu i odpowiedniego rozprowadzenia leku w krążeniu.
Podanie powolne stosuje się głównie u pacjentów z objawami przypominającymi przeziębienie.
Epoetyny alfa nie wolno stosować w formie dożylnej infuzji ani mieszać z innymi lekami.
Podanie podskórne.
Maksymalna objętość leku podawanego podskórnie w jednym miejscu wynosi 1 ml. W przypadku konieczności podania większej objętości, podanie podskórne należy wykonać w kilku miejscach.
Lek należy podawać podskórnie w kończynach lub w przednią ścianę brzucha.
Jeśli lekarz uzna, że pacjent lub opiekun może bezpiecznie i skutecznie samodzielnie podawać Epobiokryn podskórnie, należy ich odpowiednio poinstruować w zakresie właściwego dawkowania i stosowania.
Leczenie anemii symptomatycznej u dorosłych pacjentów i dzieci z przewlekłą niewydolnością nerek.
U pacjentów z przewlekłą niewydolnością nerek, jeśli istnieje możliwość stosowania drogi dożylnej (pacjenci poddawani hemodializie), droga ta jest preferowana. W przypadku trudności z podawaniem leku drogą dożylną (pacjenci, u których hemodializa nie jest jeszcze wskazana, lub pacjenci poddawani dializie otnęciowej), epoetynę alfa można stosować podskórnie.
Objawy anemii oraz powikłania mogą się różnić w zależności od wieku, płci oraz stanów chorobowych; ocena indywidualnego przebiegu klinicznego i stanu pacjenta przez lekarza jest konieczna.
Epobiokryn należy stosować w celu podniesienia poziomu hemoglobiny nie wyższego niż 12 g/dl (7,5 mmol/l). Należy unikać wzrostu poziomu hemoglobiny o więcej niż 2 g/dl (1,25 mmol/l) w ciągu 4-tygodniowego okresu. W takim przypadku należy zmniejszyć dawkę, zgodnie z poniższymi wskazaniami.
Ze względu na indywidualną zmienność, okresowe wartości poziomu hemoglobiny u każdego pacjenta mogą być wyższe lub niższe niż pożądany poziom.
Poziom hemoglobiny należy kontrolować poprzez dobrać odpowiednią dawkę, biorąc pod uwagę, że jego wartość powinna utrzymywać się w zakresie od 10 g/dl (6,2 mmol/l) do 12 g/dl (7,5 mmol/l). U dzieci zalecany optymalny poziom hemoglobiny wynosi 9,5–11 g/dl (5,9–6,8 mmol/l).
Należy unikać trwałego poziomu hemoglobiny powyżej 12 g/dl (7,5 mmol/l). Jeśli stężenie hemoglobiny wzrośnie o co najmniej 2 g/dl (1,25 mmol/l) w ciągu miesiąca lub trwały poziom hemoglobiny przekracza 12 g/dl (7,5 mmol/l), dawkę epoetyny należy zmniejszyć o 25%. Jeśli poziom hemoglobiny przekracza 13 g/dl (8,1 mmol/l), leczenie należy wstrzymać do czasu obniżenia poziomu hemoglobiny do 12 g/dl (7,5 mmol/l), a następnie wznowić leczenie epoetyną alfa w dawce o 25% niższej niż poprzednia.
Stan pacjentów należy dokładnie monitorować w celu zapewnienia, że najniższa zatwierdzona dawka leków stymulujących erytropoetynę zapewnia adekwatną kontrolę objawów anemii.
Poziom ferrytyny (lub stężenie żelaza w surowicy krwi) należy oznaczyć u wszystkich pacjentów przed rozpoczęciem oraz w trakcie leczenia Epobiokrynem. W razie potrzeby należy dodatkowo stosować leki zawierające żelazo. Inne rodzaje anemii (takie jak niedobór witaminy B12 lub kwasu foliowego) należy wykluczyć przed rozpoczęciem terapii Epobiokrynem. Brak odpowiedzi klinicznej na leczenie Epobiokrynem wymaga poszukania czynników wpływających, takich jak: niedobór żelaza, kwasu foliowego lub witaminy B12, zatrucie glinem, infekcje współistniejące, procesy zapalne lub urazy, hemoliza, fibroza szpiku kostnego o dowolnej etiologii.
Pacjenci dorośli poddawani hemodializie.
Pacjentom poddawanym hemodializie lek podaje się dożylnie.
Leczenie dzieli się na dwa etapy.
Faza korygująca.
50 JМ/кг 3 razy w tygodniu.
W razie potrzeby dawkę można stopniowo zwiększać (nie częściej niż raz na 4 tygodnie) o 25 JМ/кг 3 razy w tygodniu, aż do osiągnięcia optymalnego stężenia hemoglobiny (10–12 g/dl, 6,2–7,5 mmol/l).
Faza utrzymania.
Dawkę należy dostosować w celu utrzymania pożądanego poziomu hemoglobiny (Hb) w zakresie od 10 do 12 g/dl (6,2–7,5 mmol/l).
Zalecana dawka tygodniowa wynosi od 75 do 300 JМ/кг.
Pacjenci, u których początkowy poziom hemoglobiny jest stosunkowo niski (< 6 g/dl lub < 3,75 mmol/l), mogą wymagać wyższych dawek utrzymania niż pacjenci z łagodniejszą anemią (hemoglobina > 8 g/dl lub > 5 mmol/l).
Dzieci poddawane hemodializie.
Pacjentom poddawanym hemodializie lek podaje się dożylnie.
Leczenie dzieli się na dwa etapy.
Faza korygująca.
50 JМ/кг 3 razy w tygodniu.
W razie potrzeby można stopniowo zwiększać dawkę (nie częściej niż raz na 4 tygodnie) o 25 JМ/кг 3 razy w tygodniu, aż do osiągnięcia optymalnego stężenia hemoglobiny 9,5–11 g/dl (5,9–6,8 mmol/l).
Faza utrzymania.
Dawkę należy dostosować w celu utrzymania pożądanego poziomu hemoglobiny (Hb) w zakresie od 9,5–11 g/dl (5,9–6,8 mmol/l).
Dzieci o masie ciała poniżej 30 kg wymagają wyższej dawki utrzymania niż dorośli oraz dzieci o masie ciała powyżej 30 kg.
Dawki utrzymania epoetyny alfa:
| Dawka (JMi/kg przez 3 tygodnie) |
||
| Masa ciała (kg) |
Średnia dawka |
Zwykła dawka utrzymująca |
| < 10 |
100 |
75–150 |
| 10–30 |
75 |
60–150 |
| > 30 |
33 |
30–100 |
Pacjenci, u których początkowy poziom hemoglobiny jest bardzo niski (< 6,8 g/dl lub < 4,25 mmol/l), mogą wymagać wyższych dawek w celu utrzymania stężenia w porównaniu z pacjentami z mniej nasilonym stopniem anemii (hemoglobina > 6,8 g/dl lub > 4,25 mmol/l).
Dorośli pacjenci z niewydolnością nerek w okresie przeddializacyjnym.
Pacjentom z niewydolnością nerek, którzy znajdują się w okresie przeddializacyjnym, w przypadku braku dostępu do drogi dożylnej lek można podawać podskórnie.
Leczenie dzieli się na dwa etapy.
Faza korekcyjna.
50 J/ kg 3 razy w tygodniu.
W razie potrzeby dawkę można korygować, zwiększając o 25 J/kg 3 razy w tygodniu, z odstępem między zwiększeniami nie mniejszym niż 4 tygodnie, aż do osiągnięcia poziomu hemoglobiny w zakresie 10–12 g/dl (6,2–7,5 mmol/l).
Faza utrzymania.
W trakcie fazy utrzymania Epobiokryn można stosować 3 razy w tygodniu lub, w przypadku podania podskórnego, 1 raz w tygodniu lub 1 raz na 2 tygodnie. Dawkę i odstępy między podaniami należy dostosować w celu utrzymania pożądanego poziomu hemoglobiny: (Hb) od 10 do 12 g/dl (6,2–7,5 mmol/l). Wydłużenie odstępów między podaniami może wymagać zwiększenia dawki. Maksymalna dawka nie powinna przekraczać 150 J/kg 3 razy w tygodniu, 240 J/kg (maksymalnie do 20000 J) 1 raz w tygodniu lub 480 J/kg (maksymalnie do 40000 J) 1 raz na 2 tygodnie.
Dorośli pacjenci poddawani dializie otnierzowej.
Pacjentom poddawanym dializie otnierzowej, w przypadku braku dostępu do drogi dożylnej, lek można stosować podskórnie.
Leczenie dzieli się na dwa etapy.
Faza korekcyjna.
50 J/kg 2 razy w tygodniu.
Faza utrzymania.
Zwykle dawka utrzymania pożądanego poziomu hemoglobiny (Hb) od 10 do 12 g/dl (6,2–7,5 mmol/l) wynosi od 25 do 50 J/kg 2 razy w tygodniu poprzez podanie dwóch równoważnych iniekcji.
Leczenie pacjentów z anemią wywołaną chemioterapią.
Pacjentom z anemią (np. stężenie hemoglobiny ≤ 10 g/dl (6,2 mmol/l)) Epobiokryn należy podawać podskórnie. Objawy anemii i powikłania zależą od wieku, płci i stanu pacjenta wywołanego chorobą; konieczna jest ocena indywidualnego przebiegu klinicznego i stanu pacjenta przez lekarza.
Ze względu na indywidualną zmienność okresowe wartości poziomu hemoglobiny u każdego pacjenta mogą być wyższe lub niższe niż pożądany poziom. Poziom hemoglobiny należy kontrolować poprzez dobór dawki, biorąc pod uwagę, że jego wartość powinna utrzymywać się w zakresie od 10 g/dl (6,2 mmol/l) do 12 g/dl (7,5 mmol/l). Należy unikać stałego poziomu hemoglobiny powyżej 12 g/dl (7,5 mmol/l). Wskazówki dotyczące korekty dawki przy poziomach hemoglobiny przekraczających 12 g/dl (7,5 mmol/l) opisano poniżej.
Leczenie epoetyną alfa należy kontynuować przez jeden miesiąc po zakończeniu chemioterapii.
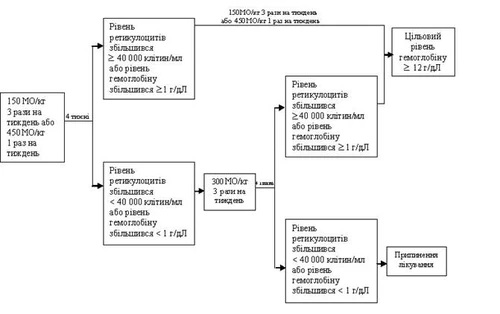
Początkowa dawka leczenia anemii u tej grupy pacjentów wynosi 150 J/kg 3 razy w tygodniu. Epoetynę alfa można stosować alternatywnie w dawce początkowej 450 J/kg podskórnie 1 raz w tygodniu.
Jeśli po 4 tygodniach stosowania dawki początkowej poziom hemoglobiny wzrósł co najmniej o 1 g/dl (0,6 mmol/l) (lub poziom retikulocytów wzrósł do ≥ 40000 komórek/ml), dawkę należy zachować na poziomie 150 J/kg 3 razy w tygodniu lub 450 J/kg podskórnie 1 raz w tygodniu. Jeśli po 4 tygodniach stosowania dawki początkowej poziom hemoglobiny wzrósł o 1 g/dl (0,62 mmol/l) lub poziom retikulocytów wzrósł do < 40000 komórek/ml, dawkę należy zwiększyć do 300 J/kg trzykrotnie w tygodniu lub 40000 J 1 raz w tygodniu.
Jeśli po 4 tygodniach leczenia zwiększoną dawką 300 J/kg 3 razy w tygodniu poziom hemoglobiny wzrósł o ≥ 1 g/dl (≥ 0,62 mmol/l) lub poziom retikulocytów wzrósł do ≥ 40000 komórek/ml, dawkę nie zmienia się. Jeśli jednak poziom hemoglobiny wzrósł o < 1 g/dl (≥ 0,62 mmol/l) lub poziom retikulocytów wzrósł o < 40000 komórek/ml, odpowiedź kliniczna jest ujemna i leczenie należy przerwać.
Schemat zalecanego trybu dawkowania:
Stan pacjentów należy dokładnie monitorować w celu zapewnienia, że najniższa zatwierdzona dawka leków stymulujących erytropoezę zapewnia odpowiednią kontrolę objawów anemii.
Dobór dawki w celu utrzymania docelowego poziomu hemoglobiny 10–12 g/dl.
Jeśli tempo wzrostu poziomu hemoglobiny przekracza 2 g/dl (1,25 mmol/l) w ciągu 1 miesiąca i ogólny poziom hemoglobiny zbliża się do 12 g/dl (7,5 mmol/l), należy zmniejszyć dawkę Epobiokrynu o 25–50% w zależności od tempa wzrostu poziomu hemoglobiny. Jeśli poziom hemoglobiny przekroczy 13 g/dl (8,1 mmol/l), leczenie należy tymczasowo przerwać do czasu spadku poziomu do 12 g/dl (7,5 mmol/l), a następnie wznowić leczenie dawką o 25% niższą niż poprzednia.
Dorośli pacjenci uczestniczący w programie poboru krwi autologicznej przed zabiegami chirurgicznymi.
Należy stosować podanie dożylne.
Epoetynę alfa należy podawać po zakończeniu każdej procedury poboru krwi.
Pacjentom ze średnim stopniem anemii (poziom hematokrytu 33–39%), którzy wymagają ≥ 4 jednostek krwi, należy leczyć epoetyną alfa w dawce 600 J/kg 2 razy w tygodniu przez 3 tygodnie przed zabiegiem chirurgicznym.
Wszyscy pacjenci leczeni epoetyną alfa powinni otrzymywać odpowiednie uzupełnienie żelaza (200 mg doustnie na dobę) przez cały okres terapii. W celu zapewnienia wystarczającego poziomu żelaza w organizmie leki żelaza należy przepisywać jak najwcześniej, nawet kilka tygodni przed rozpoczęciem programu poboru krwi autologicznej.
Dorośli pacjenci kwalifikowani do planowej chirurgii ortopedycznej.
Należy stosować podanie podskórne.
Zalecany tryb dawkowania leku to 600 J/kg na tydzień przez 3 tygodnie poprzedzające zabieg (21., 14. i 7. dzień przed operacją) oraz w dniu operacji.
Jeśli z powodów medycznych konieczne jest skrócenie okresu przedoperacyjnego do mniej niż 3 tygodnie, Epobiokryn należy podawać codziennie w dawce 300 J/kg przez 10 kolejnych dni przed operacją, w dniu operacji oraz przez 4 dni po operacji. Jeśli w wyniku badań hematologicznych w okresie przedoperacyjnym poziom hemoglobiny osiągnie 15 g/dl lub więcej, stosowanie epoetyny alfa należy całkowicie przerwać.
Wszyscy pacjenci leczeni epoetyną alfa powinni otrzymywać odpowiednie uzupełnienie żelaza (200 mg doustnie na dobę) przez cały okres terapii. Leki żelaza należy przepisywać jak najwcześniej, nawet kilka tygodni przed rozpoczęciem programu poboru krwi autologicznej.
Dzieci.
Epoetyna alfa jest wskazana w leczeniu anemii związanej z przewlekłą niewydolnością nerek u dzieci w wieku od 1 miesiąca do 18 lat poddawanych dializie. Bezpieczeństwo i skuteczność stosowania leku u dzieci poniżej 1 miesiąca życia nie zostały ustalone.
Przedawkowanie.
Lek wykazuje szerokie działanie terapeutyczne. W przypadku przedawkowania epoetyny alfa występują efekty odzwierciedlające najwyższy stopień działania farmakologicznego hormonu. Przy wyjątkowo wysokich poziomach hemoglobiny możliwe jest przeprowadzenie flebotomii. W razie potrzeby stosuje się leczenie objawowe.
Niepożądane działania.
Najczęstszym niepożądanym działaniem towarzyszącym leczeniu epoetyną alfa u pacjentów onkologicznych oraz u pacjentów z przewlekłą niewydolnością nerek jest zależne od dawki podwyższenie ciśnienia tętniczego lub nasilenie istniejącej nadciśnienia tętniczego. Kontrolę ciśnienia tętniczego należy prowadzić od początku leczenia. Innymi częstymi niepożądanymi działaniami są: zakrzepica żył głębokich, zatorowość płucna, drgawki, biegunka, nudności, ból głowy, stan grypowy, pyreksja, wysypka oraz wymioty.
Przeważnie na początku leczenia mogą występować objawy przeziębienia, takie jak ból głowy, ból mięśni i stawów, dreszcze. Częstość występowania może się różnić w zależności od wskazań.
Może również występować nasilenie niedrożności dróg oddechowych, w tym zatkanie nosa oraz zapalenie nosa i gardła.
Poważne niepożądane działania – zakrzepica żylna i tętnicza, zatorowość (w tym zakończona śmiercią), zakrzepica żył głębokich, zatorowość płucna, zakrzepica tętnicza (w tym zawód serca i niedokrwienie mięśnia sercowego), zakrzepica siatkówki oraz zakrzepica sztucznej przetoki (w tym okluzja układu dializacyjnego). Mogą również występować powikłania naczyniowo-mózgowe (w tym udar mózgu i krwotoki do mózgu) oraz przejściowe ataki niedokrwienne, aneurysmy oraz reakcje nadwrażliwości, w tym wysypka, pokrzywka, reakcje anafilaktyczne i obrzęk naczynioruchowy.
Ponadto, podczas leczenia epoetyną alfa u pacjentów z wcześniejszo normalnym lub obniżonym ciśnieniem może występować kryz nadciśnieniowy z encefalopatią i drgawkami, wymagający natychmiastowego skontaktowania się z lekarzem oraz intensywnej pomocy medycznej. Szczególną uwagę należy zwrócić na nagły, ostry ból głowy typu migrenowego jako możliwy sygnał ostrzegawczy.
Bardzo rzadko może występować opośredniona przeciwciałami prawdziwa aplazja czerwonych krwinek po wielomiesięcznym lub wieloletnim leczeniu.
Częstość występowania niepożądanych działań: bardzo często (≥1/10); często (≥1/100 do <1/10); rzadko (≥1/1000 do <1/100); nieczęsto (≥1/10000 do <1/1000); bardzo rzadko (<1/10000); częstość nieznana.
Ze strony układu krwiotwórczego i chłonnego.
Niezbyt często – trombocytoza (pacjenci z nowotworami).
Częstość nieznana – trombocytoza (pacjenci z przewlekłą niewydolnością nerek).
Bardzo rzadko – przeciwciałozależna prawdziwa aplazja czerwonych krwinek.
Ze strony układu immunologicznego.
Częstość nieznana – reakcje anafilaktyczne, reakcje nadwrażliwości.
Rzadko występują reakcje anafilaktyczne: potencjalnie poważne powikłania związane z zaburzeniami oddychania lub obniżeniem ciśnienia tętniczego, reakcje immunologiczne (ma minimalną zdolność indukowania tworzenia przeciwciał).
Ze strony układu nerwowego.
Bardzo często – ból głowy (pacjenci z nowotworami).
Często – drgawki (pacjenci z przewlekłą niewydolnością nerek), ból głowy (pacjenci z przewlekłą niewydolnością nerek), udar mózgu.
Niezbyt często – krwotoki do mózgu, drgawki (pacjenci z nowotworami).
Częstość nieznana – udar mózgu, encefalopatia nadciśnieniowa, przejściowy atak niedokrwienny, zawroty głowy, senność.
Ze strony narządów wzroku.
Częstość nieznana – zakrzepica siatkówki.
Ze strony serca.
Częstość nieznana – zawał mięśnia sercowego.
Ze strony układu naczyniowego.
Często – zakrzepica żył głębokich (pacjenci z nowotworami), nadciśnienie tętnicze.
Częstość nieznana – zakrzepica żył głębokich (pacjenci z przewlekłą niewydolnością nerek), zakrzepica tętnicza, kryz nadciśnieniowy.
Ze strony układu oddechowego.
Często – zatorowość płucna (pacjenci z nowotworami), kaszel.
Niezbyt często – nasilenie niedrożności dróg oddechowych.
Częstość nieznana – zatorowość płucna (pacjenci z przewlekłą niewydolnością nerek).
Ze strony przewodu pokarmowego.
Bardzo często – nudności.
Często – biegunka (pacjenci z nowotworami), wymioty.
Niezbyt często – biegunka (pacjenci z przewlekłą niewydolnością nerek).
Ze strony skóry.
Często – wysypka, egzema.
Częstość nieznana – obrzęk naczynioruchowy, pokrzywka, swędzenie, obrzęk Quinckego, zespół Stevensa–Johnsona, toksyczny epidermalny nekrolioza (mogą zagrażać życiu lub mieć skutek śmiertelny).
Ze strony mięśni, tkanki łącznej i kości.
Bardzo często – artralgia (pacjenci z przewlekłą niewydolnością nerek), ból kości, ból kończyn.
Często – artralgia (pacjenci z nowotworami).
Niezbyt często – mialgia (pacjenci z nowotworami).
Częstość nieznana – mialgia (pacjenci z przewlekłą niewydolnością nerek).
Wady wrodzone, dziedziczne i zaburzenia genetyczne.
Częstość nieznana – porfiria.
Zaburzenia ogólne i zaburzenia w miejscu podania.
Bardzo często – pyreksja (pacjenci z nowotworami), stan grypowy (pacjenci z przewlekłą niewydolnością nerek), reakcje w miejscu wstrzyknięcia, obrzęk obwodowy.
Często – stan grypowy (pacjenci z nowotworami).
Częstość nieznana – dreszcze, brak odpowiedzi na leczenie.
Badania.
Częstość nieznana – obecność przeciwciał przeciw erytropoetynie, hiperkaliemia, hiperfosfatemia, podwyższenie stężenia mocznika, kreatyniny, kwasu moczowego we krwi (u pacjentów z przewlekłą niewydolnością nerek).
Urazy, zatrucia i powikłania proceduralne.
Często – zakrzepica sztucznej przetoki, w tym sprzętu dializacyjnego (pacjenci z przewlekłą niewydolnością nerek).
Pacjenci z przewlekłą niewydolnością nerek.
U pacjentów z przewlekłą niewydolnością nerek poziom hemoglobiny powyżej 12 g/dl może być związany ze zwiększonym ryzykiem powikłań sercowo-naczyniowych, w tym śmiertelnych.
U pacjentów poddawanych hemodializie, szczególnie w przypadku skłonności do hipotensji lub występowania powikłań ze strony przetoki tętniczo-żylnej (zwężenia, aneurysmy itp.), opisywano przypadki zakrzepicy przetoki.
Pacjenci z chorobami nowotworowymi.
Rozwój powikłań zakrzepowych jest możliwy u pacjentów stosujących leczenie lekami stymulującymi erytropoezę, w tym epoetyną alfa (patrz rozdział „Szczególne wskazania”).
Chirurgiczni pacjenci dorośli.
Nie można wykluczyć możliwości, że leczenie epoetyną alfa u pacjentów z ustalonym poziomem hemoglobiny >13 g/dl może wiązać się ze zwiększonym ryzykiem powikłań zakrzepowo-naczyniowych po zabiegu chirurgicznym.
Opis poszczególnych niepożądanych działań.
Opisywano reakcje nadwrażliwości, w tym przypadki wysypki (w tym pokrzywki), reakcje anafilaktyczne oraz obrzęk naczynioruchowy (patrz rozdział „Szczególne wskazania”).
Zgłaszano przypadki kryzu nadciśnieniowego z encefalopatią i drgawkami, wymagające natychmiastowego badania przez lekarza i intensywnej terapii, u pacjentów z normalnym lub niskim ciśnieniem tętniczym na początku leczenia. Należy zwracać szczególną uwagę na nagły, strzelający ból głowy typu migrenowego, który może być sygnałem ostrzegawczym (patrz rozdział „Szczególne wskazania”).
Bardzo rzadko (<1/10000 przypadków na pacjenta-rok) zgłaszano przypadki opośrednionej przeciwciałami prawdziwej aplazji czerwonych krwinek (PRCA) u pacjentów leczonych lekami na bazie erytropoetyny przez miesiące lub lata (patrz rozdział „Szczególne wskazania”).
Dzieci z przewlekłą niewydolnością nerek poddawane hemodializie.
Doświadczenie z zastosowaniem erytropoetyny u dzieci z przewlekłą niewydolnością nerek poddawanych hemodializie w trakcie badań klinicznych i w okresie postmarketingowym jest ograniczone. Nie wykryto żadnych niepożądanych działań specyficznych dla dzieci, które nie zostały wymienione w tabeli; nie stwierdzono również niepożądanych działań, które nie odpowiadałyby istniejącej chorobie.
Zgłaszanie podejrzewanych niepożądanych działań.
Zgłaszanie niepożądanych działań po rejestracji leku ma duże znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka stosowania tego leku.
Lekarze, farmaceuci, a także pacjenci lub ich ustawowi przedstawiciele mogą zgłaszać wszelkie przypadki podejrzewanych niepożądanych działań lub braku skuteczności leków produkowanych przez spółkę z o.o. „FZ «Biopharma»” w dowolny wygodny dla nich sposób (listem, e-mailem lub telefonicznie) w formie papierowej lub elektronicznej. O przypadkach niepożądanych działań lub braku skuteczności leków produkowanych przez spółkę z o.o. „FZ «Biopharma»” należy informować dział farmakologii klinicznej pod adresem: ul. M. Amosova 12, miasto Kijów, 03680, tel. +38 044 459 4600, adres e-mail: [email protected]. W przypadku wystąpienia działań niepożądanych lub pytań dotyczących bezpieczeństwa stosowania leków produkowanych przez spółkę z o.o. „FZ «Biopharma»” prosimy o kontakt z działem farmakologii klinicznej.
Dodatkowo zgłoszenia dotyczące niepożądanych działań mogą być przesyłane poprzez Automatyczny System Informacyjny Farmakologii Klinicznej pod adresem: https://aisf.dec.gov.ua
Okres ważności. 2 lata.
Warunki przechowywania. Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem, w temperaturze od 2 do 8 °C. Nie zamrażać. Nie wstrząsać.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność. Ponieważ brak badań dotyczących niezgodności, Epobiokryn nie może być stosowany jednocześnie z innymi lekami.
Opakowanie. 1000 J, 2000 J, 4000 J, 10 000 J w strzykawkach wstępnie napełnionych lub ampułkach. Po 5 strzykawek wstępnie napełnionych lub 5 ampułek w blisterze. Po 1 blisterze w opakowaniu.
Kategoria wydania. Na receptę.
Producent.
Spółka z o.o. „FZ «STADA», Ukraina.
Adres siedziby producenta i miejsce prowadzenia działalności.
Ukraina, 09100, obwód kijowski, miasto Biała Cerkiew, ul. Kijewska 37.