Epobiocryn
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE EPОBІОКRІN (Epobiocrinum)
Composizione:
Principio attivo: Epoetin alfa;
1 ml di soluzione contiene eritropoietina ricombinante umana 1000 UI oppure 2000 UI oppure 4000 UI oppure 10 000 UI;
Eccipienti: albumina umana, citrato di sodio, cloruro di sodio, acido citrico monoidrato; acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile.
Principali caratteristiche fisico-chimiche: soluzione trasparente incolore.
Gruppo farmacoterapeutico. Agenti antianemici. Eritropoietina.
Codice ATC B03XA01.
Proprietà farmacologiche.
Farmacodinamica.
L’eritropoietina ricombinante umana è biologicamente e immunologicamente identica all’eritropoietina umana, un ormone glicoproteico naturale che agisce come fattore stimolante la mitosi ed è l’ormone responsabile della stimolazione dell’eritropoiesi, il processo di formazione dei globuli rossi a partire dalle cellule staminali precursori. Nei soggetti sani, l’eritropoietina è normalmente sintetizzata per il 90% dai reni e per il 10% dalle cellule di Kupffer del fegato. Il livello di sintesi è determinato dalla saturazione del sangue con l’ossigeno. L’eritropoietina stimola la proliferazione e la differenziazione delle cellule eritroidi in globuli rossi maturi. Il suo effetto agisce nelle fasi iniziali dell’eritropoiesi, a livello delle unità formanti colonie eritroidi (CFU-E) e delle unità formanti aggregati eritroidi (BFU-E), e successivamente a livello del proeritroblasto, eritroblasto e reticolocita (la sensibilità di queste cellule all’eritropoietina è proporzionale al loro grado di maturazione). L’eritropoietina normalizza i livelli di emoglobina ed ematocrito e allevia i sintomi associati all’anemia.
La massa molecolare dell’epoetina alfa è di circa 30.600 Dalton. La parte proteica rappresenta circa il 60% della massa molecolare ed è costituita da 165 aminoacidi. Quattro catene carboidratiche sono legate alla proteina tramite tre legami N-glicosidici e un legame O-glicosidico.
Farmacocinetica.
Somministrazione endovenosa. Dopo somministrazione endovenosa del medicinale, l’emivita di eliminazione in soggetti con normale funzionalità renale è di circa 4 ore; nei pazienti con compromissione della funzionalità renale è di circa 5 ore. L’emivita nei bambini è di circa 6 ore.
Somministrazione sottocutanea. Le concentrazioni plasmatiche dopo somministrazione sottocutanea sono significativamente più basse rispetto a quelle ottenute con somministrazione endovenosa. Dopo somministrazione sottocutanea, la concentrazione del farmaco nel sangue aumenta lentamente, raggiungendo il picco dopo 12 – 18 ore dall’iniezione. La concentrazione massima nel plasma dopo somministrazione sottocutanea è inferiore a quella ottenuta con somministrazione endovenosa (circa 1/20 del valore).
Non si osserva effetto cumulativo, ovvero le concentrazioni misurate di eritropoietina nel siero rimangono costanti, indipendentemente dal fatto che la misurazione venga effettuata 24 ore dopo la prima iniezione o 24 ore dopo l’ultima.
Non sono disponibili dati riguardo alla capacità dell’eritropoietina ricombinante umana di attraversare la barriera placentaria o di essere escreta nel latte materno; tuttavia, questa sostanza non attraversa la barriera ematoencefalica.
L’emivita dopo somministrazione sottocutanea è di circa 24 ore.
La biodisponibilità del farmaco dopo somministrazione sottocutanea è significativamente inferiore rispetto a quella endovenosa e corrisponde a circa il 20%.
Caratteristiche cliniche.
Indicazioni.
Trattamento dell'anemia sintomatica associata all'insufficienza renale cronica:
- trattamento dell'anemia associata all'insufficienza renale cronica in adulti e bambini in emodialisi e in adulti in dialisi peritoneale;
- trattamento dell'anemia grave di origine renale accompagnata da sintomi clinici in adulti con insufficienza renale non ancora sottoposti ad emodialisi.
Trattamento dell'anemia e riduzione del volume delle emazie necessarie in adulti sottoposti a chemioterapia per neoplasia non mieloma, linfoma maligno o mieloma multiplo, e nei quali il rischio di trasfusione è considerato elevato in base allo stato generale del paziente (compreso lo stato cardiovascolare e l'anemia preesistente prima dell'inizio della chemioterapia).
Epobiocryn può essere utilizzato nell'ambito di programmi di predeposito in pazienti adulti con anemia lieve o moderata (livello di emoglobina 10 – 13 g/dl (6,2 – 8,1 mmol/l), assenza di carenza di ferro) prima di interventi chirurgici maggiori, al fine di facilitare il prelievo di sangue autologo e ridurre il rischio associato all'uso di emazie allogene, qualora il fabbisogno previsto di sangue per trasfusione superi la quantità ottenibile mediante prelievo autologo senza l'uso di epoetina alfa.
Epobiocryn è indicato negli adulti con anemia lieve o moderata (emoglobina tra 10 – 13 g/dl in assenza di carenza di ferro) prima di interventi ortopedici maggiori con perdita ematica media prevista (900–1800 ml di sangue), per ridurre la necessità di emazie allogene e facilitare il recupero del sistema eritropoietico.
Controindicazioni.
Ipersensibilità a uno qualsiasi dei componenti del medicinale.
Sviluppo di aplasia eritroide vera in seguito al trattamento con epoetina alfa (vedere sezione «Informazioni particolari di impiego»).
Ipertensione non controllata.
Controindicazioni relative al programma di prelievo di sangue autologo in pazienti in trattamento con epoetina alfa.
Gravi malattie coronariche, malattie arteriosclerotiche periferiche, malattie carotidee o cerebrovascolari, nonché recente infarto miocardico o ictus in pazienti sottoposti a chirurgia ortopedica elettiva che non hanno partecipato a un programma di prelievo autologo. Impossibilità di applicare una profilassi antitrombotica adeguata nei pazienti chirurgici.
Interazioni con altri medicinali ed altre forme di interazione.
Non esistono dati che indichino che il trattamento con epoetina alfa influenzi il metabolismo di altri medicinali.
Farmaci che rallentano l'eritropoiesi possono ridurre la risposta al trattamento con epoetina alfa.
Poiché il ciclosporina si lega agli eritrociti, esiste la possibilità di un'interazione farmacologica. Quando Epobiocryn viene somministrato contemporaneamente al ciclosporina, si raccomanda di monitorare i livelli ematici di quest'ultimo e, se necessario, di aggiustarne la dose.
Non esistono evidenze di interazioni tra epoetina alfa e G-CSF (fattore stimolante le colonie di granulociti) o GM-CSF (fattore stimolante le colonie di granulociti e macrofagi) riguardo alla differenziazione ematologica o alla proliferazione cellulare tumorale in campioni di biopsia in vitro.
In pazienti con carcinoma mammario metastatico, la somministrazione sottocutanea di epoetina alfa alla dose di 40000 UI/ml contemporaneamente a trastuzumab alla dose di 6 mg/kg non ha influenzato la farmacocinetica di trastuzumab.
Caratteristiche particolari di impiego.
La pressione arteriosa deve essere costantemente monitorata in tutti i pazienti durante il trattamento con Epobiocryn. Il medicinale deve essere utilizzato con cautela nei pazienti con ipertensione non trattata o con controllo inadeguato dell'ipertensione. Durante il trattamento con Epobiocryn può rendersi necessario iniziare o intensificare una terapia antipertensiva. Se la pressione non può essere controllata, l'uso dell'epoetina alfa deve essere interrotto.
Sono stati osservati casi di crisi ipertensiva con encefalopatia e convulsioni, che hanno richiesto un immediato intervento medico e terapia intensiva, anche in pazienti con pressione arteriosa normale o bassa all'inizio del trattamento. Si deve prestare particolare attenzione all'insorgenza di un improvviso mal di testa pulsante e lancinante, che può essere un segnale d'allarme (vedere la sezione «Effetti indesiderati»).
L'epoetina alfa deve essere utilizzata con cautela nei pazienti con epilessia, storia di convulsioni o condizioni mediche che rappresentano fattori di rischio per lo sviluppo di convulsioni, come infezioni del sistema nervoso centrale o metastasi cerebrali.
L'epoetina alfa deve essere utilizzata con cautela nei pazienti con insufficienza epatica cronica. La sicurezza dell'uso dell'epoetina alfa in questa categoria di pazienti non è stata stabilita.
Nei pazienti che ricevono medicinali stimolanti l'eritropoiesi si osserva un aumento del rischio di malattie vascolari con complicanze trombotiche (vedere la sezione «Effetti indesiderati»), inclusi trombosi e embolia venosa ed arteriosa (anche con esito fatale), come trombosi venosa profonda, embolia polmonare, trombosi della vena retinica e infarto miocardico. Sono stati inoltre riportati casi di ictus (inclusi ictus ischemico, emorragico e attacchi ischemici transitori).
Prima di iniziare il trattamento con epoetina alfa, si devono attentamente valutare i rischi di malattie vascolari con complicanze trombotiche, specialmente nei pazienti con fattori di rischio concomitanti, inclusi obesità e storia di malattie vascolari (ad esempio trombosi venosa profonda, embolia polmonare e ictus).
Si deve monitorare attentamente il livello di emoglobina in tutti i pazienti a causa del potenziale aumento del rischio di complicanze tromboemboliche e di esito letale nel caso di utilizzo del medicinale con livelli di emoglobina superiori a quelli raccomandati per le indicazioni d'uso.
Durante il trattamento può verificarsi un lieve aumento dose-dipendente del numero di piastrine entro i limiti normali. Questo parametro si riduce durante il proseguimento del trattamento. Sono stati inoltre riportati casi di trombocitosi. Si raccomanda di monitorare regolarmente il numero di piastrine durante le prime 8 settimane di trattamento.
Tutte le altre cause di anemia (carenza di ferro, acido folico, vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi o fibrosi del midollo osseo di qualsiasi origine) devono essere identificate e trattate prima di iniziare la terapia con epoetina alfa e prima di decidere di aumentare il dosaggio. Nella maggior parte dei casi, i valori di ferritina nel siero diminuiscono contemporaneamente all'aumento dell'ematocrito. Per garantire una risposta ottimale al trattamento con epoetina alfa, è necessario assicurare un adeguato apporto di ferro (vedere la sezione «Modalità di somministrazione e dosi»):
- ai pazienti con insufficienza renale cronica si raccomanda l'assunzione di ferro (200–300 mg/giorno per adulti e 100–200 mg/giorno per bambini per via orale, calcolati come ferro elementare), se il livello di ferritina nel siero è inferiore a 100 ng/ml;
- ai pazienti con neoplasie si raccomanda l'assunzione di ferro (200–300 mg/giorno per via orale, calcolati come ferro elementare), se la saturazione della transferrina è inferiore al 20%;
- ai pazienti che partecipano a programmi di raccolta autologa di sangue si raccomanda l'assunzione di ferro (200 mg/giorno per via orale, calcolati come ferro elementare) alcune settimane prima dell'inizio della raccolta autologa di sangue, al fine di raggiungere significative riserve di ferro prima dell'inizio della terapia e durante il corso del trattamento con epoetina alfa;
- ai pazienti prima di interventi ortopedici elettivi estesi si raccomanda l'assunzione di ferro (200 mg/giorno per via orale, calcolati come ferro elementare) durante il corso del trattamento con epoetina alfa. Ove possibile, l'assunzione di ferro dovrebbe iniziare prima dell'inizio della terapia con epoetina alfa per raggiungere significative riserve di ferro.
Molto raramente sono stati riportati casi di sviluppo o peggioramento di porfiria preesistente in pazienti trattati con epoetina alfa. L'epoetina alfa deve essere utilizzata con cautela nei pazienti con porfiria.
Durante un trattamento prolungato con epoetina alfa, può svilupparsi gravi reazioni avverse cutanee, inclusi la sindrome di Stevens-Johnson e la necrolisi epidermica tossica, che possono essere potenzialmente letali o avere esito fatale (vedere la sezione «Effetti indesiderati»).
I pazienti devono essere informati della possibilità di reazioni avverse cutanee. In caso di comparsa di segni e sintomi di reazioni avverse cutanee, il trattamento con epoetina alfa deve essere immediatamente interrotto e si devono considerare metodi alternativi di trattamento.
Le informazioni sul nome commerciale dei medicinali stimolanti l'eritropoiesi precedentemente utilizzati devono essere chiaramente indicate nella cartella clinica del paziente. La conversione da un agente stimolante l'eritropoiesi a un altro può avvenire solo sotto la supervisione di un medico.
Pura aplasia eritroide (PRCA).
Sono stati riportati casi di sviluppo di pura aplasia eritroide (PRCA) mediata da anticorpi dopo mesi o anni di somministrazione sottocutanea di epoetina, prevalentemente in pazienti con insufficienza renale cronica. Sono stati inoltre riportati casi di pura aplasia eritroide in pazienti con epatite C trattati con interferone e ribavirina contemporaneamente a agenti stimolanti l'eritropoiesi. L'epoetina alfa non è indicata per il trattamento dell'anemia associata all'epatite C.
I pazienti che mostrano una perdita improvvisa di efficacia terapeutica (evidenziata da una riduzione del livello di emoglobina di 1–2 g/dl al mese) con aumento della necessità di trasfusioni devono essere indirizzati per esami del numero di reticolociti e per l'identificazione delle cause tipiche di ridotta risposta clinica (carenza di ferro, acido folico, vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi o fibrosi del midollo osseo di qualsiasi origine).
In caso di riduzione paradossale dell'emoglobina e sviluppo di grave anemia associata a basso numero di reticolociti, il trattamento con Epobiocryn deve essere interrotto e si deve verificare la presenza di anticorpi contro l'eritropoietina, nonché eseguire un esame del midollo osseo per confermare la diagnosi di pura aplasia eritroide.
Non si deve iniziare il trattamento con altri agenti stimolanti l'eritropoiesi, poiché esiste la possibilità di reazione crociata.
Trattamento dell'anemia sintomatica in adulti e bambini con insufficienza renale cronica.
Nei pazienti con insufficienza renale cronica trattati con epoetina alfa, il livello di emoglobina deve essere monitorato regolarmente fino al raggiungimento di un livello stabile, quindi periodicamente. L'aumento dell'emoglobina dovrebbe essere di circa 1 g/dl (0,62 mmol/l) al mese e non deve superare 2 g/dl (1,25 mmol/l) al mese, al fine di minimizzare il rischio di sviluppo di ipertensione arteriosa.
Nei pazienti con insufficienza renale cronica, il livello di emoglobina raggiunto non deve superare il limite superiore della concentrazione desiderata (vedere la sezione «Modalità di somministrazione e dosi»). Negli studi clinici è stato osservato un aumento del rischio di esiti letali e di gravi effetti indesiderati cardiovascolari con l'uso di agenti stimolanti l'eritropoiesi per raggiungere concentrazioni di emoglobina superiori a 12 g/dl (7,5 mmol/l).
Studi clinici controllati non hanno dimostrato vantaggi significativi nell'uso degli epoetini con concentrazioni di emoglobina superiori al livello necessario per controllare i sintomi dell'anemia e prevenire trasfusioni.
Si deve aumentare con cautela la dose di Epobiocryn nei pazienti con insufficienza renale cronica, poiché alte dosi cumulative di eritropoietina possono essere associate a un aumento del rischio di mortalità e di gravi eventi cardiovascolari e cerebrovascolari. Nei pazienti con risposta inadeguata al trattamento con epoetini, si devono considerare altre opzioni per affrontare la scarsa risposta (vedere la sezione «Modalità di somministrazione e dosi»).
Lo stato dei pazienti con insufficienza renale cronica trattati con Epobiocryn per via sottocutanea deve essere monitorato regolarmente per la perdita di efficacia del trattamento, definita come riduzione o perdita della risposta al trattamento con epoetina alfa in pazienti che precedentemente avevano risposto alla terapia. La perdita di efficacia è caratterizzata da una riduzione persistente del livello di emoglobina indipendentemente dall'aumento della dose di epoetina alfa (vedere la sezione «Effetti indesiderati»).
Con schemi di trattamento con intervalli di dosaggio prolungati (somministrazione di epoetina alfa meno di una volta alla settimana), in alcuni pazienti il livello di emoglobina può diminuire; in questi pazienti potrebbe essere necessario un aumento della dose. Il livello di emoglobina deve essere monitorato regolarmente.
Nei pazienti in emodialisi sono state osservate trombosi dello shunt, specialmente in quelli predisposti all'ipotensione o con complicanze delle fistole artero-venose (ad esempio stenosi, aneurismi, ecc.). A questi pazienti si raccomanda il controllo dello shunt e la prevenzione della trombosi, ad esempio con acido acetilsalicilico.
In singoli casi è stata osservata iperkaliemia, sebbene non sia stato stabilito un nesso causale. Nei pazienti con insufficienza renale cronica è necessario monitorare i livelli di elettroliti nel siero. In caso di aumento del livello di potassio nel sangue, oltre al trattamento specifico per l'iperkaliemia, si deve considerare la possibilità di sospendere temporaneamente Epobiocryn fino alla completa correzione dello stato di iperkaliemia.
A causa dell'aumento del livello di ematocrito, i pazienti in emodialisi che ricevono Epobiocryn spesso necessitano di un aumento della dose di eparina durante la dialisi. In caso di insufficiente eparinizzazione, può svilupparsi occlusione del sistema di dialisi.
Secondo le informazioni attualmente disponibili, l'uso di Epobiocryn nei pazienti pre-dialisi non accelera il progresso dell'insufficienza renale.
Trattamento di pazienti con anemia indotta da chemioterapia.
Nei pazienti con neoplasie in trattamento con epoetina alfa, il livello di emoglobina deve essere monitorato regolarmente fino al raggiungimento di un livello stabile, quindi periodicamente.
Gli epoetini sono fattori di crescita che stimolano principalmente la produzione di eritrociti. I recettori dell'eritropoietina sono stati riscontrati anche sulla superficie di diverse cellule tumorali. Come con altri fattori di crescita, non si può escludere la possibilità che gli epoetini stimolino la crescita di alcuni tipi di tumori.
Non si può escludere l'effetto dei farmaci stimolanti l'eritropoiesi sul progresso del tumore o sulla riduzione della sopravvivenza libera da progressione della malattia. In studi clinici controllati, l'uso di Epobiocryn e di altri agenti stimolanti l'eritropoiesi è stato associato a una riduzione del controllo locoregionale del tumore o della sopravvivenza globale:
- riduzione del controllo locoregionale in pazienti con cancro alla testa e al collo in progressione, in trattamento con radioterapia, quando utilizzato per aumentare il livello di emoglobina oltre 14 g/dl (8,7 mmol/l);
- riduzione della sopravvivenza globale e aumento dei casi letali dovuti alla progressione della malattia entro 4 mesi in pazienti con cancro al seno metastatico in trattamento con chemioterapia, quando utilizzato per aumentare il livello di emoglobina a 12-14 g/dl (7,5-8,7 mmol/l);
- aumento del rischio di morte quando utilizzato per aumentare il livello di emoglobina a 12 g/dl (7,5 mmol/l) in pazienti con malattia maligna attiva che non ricevono né chemioterapia né radioterapia. I farmaci stimolanti l'eritropoiesi sono controindicati in questo gruppo di pazienti;
- aumento del 9% del rischio di progressione della malattia o morte nel gruppo di pazienti che ricevevano epoetina alfa e trattamento standard, e un aumento del 15% statisticamente non escludibile, in pazienti con cancro al seno metastatico in trattamento con chemioterapia, quando utilizzato per aumentare il livello di emoglobina a 10-12 g/dl (6,2-7,5 mmol/l).
Alla luce di quanto sopra, in alcune situazioni cliniche può essere preferibile la trasfusione di sangue per il trattamento dell'anemia nei pazienti oncologici. La decisione sull'uso di eritropoietine ricombinanti deve basarsi su una valutazione rischio-beneficio per il singolo paziente, tenendo conto del contesto clinico specifico. I fattori da considerare in tale valutazione devono includere il tipo e lo stadio del tumore; il grado di anemia; la sopravvivenza prevista; le condizioni di trattamento del paziente e le preferenze dello stesso.
Nei pazienti con neoplasie in trattamento con chemioterapia, di solito si osserva un ritardo di 2-3 settimane nella risposta (dal momento della somministrazione dell'eritropoietina all'apparizione dei globuli rossi indotti). Questa caratteristica deve essere considerata nella valutazione dell'efficacia del trattamento (specialmente nei pazienti con necessità di trasfusioni).
Pazienti sottoposti a intervento chirurgico e partecipanti a programmi di raccolta autologa di sangue.
Si devono osservare tutte le precauzioni specifiche relative ai programmi di raccolta autologa di sangue, specialmente le procedure di ripristino del volume circolante.
Pazienti prima di un intervento ortopedico elettivo esteso.
Si devono sempre seguire le pratiche appropriate di trasfusione ematica nel periodo pre- e post-operatorio.
I pazienti prima di un intervento ortopedico elettivo esteso devono ricevere misure di prevenzione antitrombotica adeguata, poiché dopo gli interventi chirurgici in questi pazienti possono verificarsi complicanze trombotiche e vascolari, specialmente in presenza di malattie cardiovascolari concomitanti. Particolare cautela deve essere esercitata nel trattamento di pazienti predisposti alla trombosi venosa profonda. Inoltre, nei pazienti con livello iniziale di emoglobina > 13 g/dl, il rischio di complicanze trombotiche o vascolari post-operatorie associate al trattamento con epoetina alfa è significativamente più elevato. Pertanto, l'uso di epoetina alfa in pazienti con livello iniziale di emoglobina > 13 g/dl non è raccomandato.
Pazienti di età avanzata.
La sicurezza dell'uso di epoetina alfa in questa categoria di pazienti non è stata stabilita.
Uso durante la gravidanza o l'allattamento.
Gravidanza.
Attualmente non sono disponibili risultati di studi controllati sull'uso del medicinale Epobiocryn in donne in gravidanza. Studi sugli animali hanno mostrato tossicità riproduttiva. Pertanto, Epobiocryn deve essere utilizzato in donne in gravidanza solo se il beneficio terapeutico potenziale supera il rischio potenziale per il feto. L'uso di epoetina alfa in donne in gravidanza che partecipano a programmi di raccolta autologa di sangue non è raccomandato.
Allattamento.
Non è noto se l'epoetina alfa esogena venga escreta nel latte materno. L'epoetina alfa deve essere utilizzata con cautela in donne che allattano. La decisione di continuare o interrompere l'allattamento o di continuare o interrompere il trattamento con epoetina alfa deve essere presa considerando il beneficio dell'allattamento al seno per il bambino e il beneficio del trattamento con epoetina alfa per la donna.
L'uso di epoetina alfa in pazienti che partecipano a programmi di raccolta autologa di sangue durante l'allattamento non è raccomandato.
Fertilità.
Non sono stati condotti studi sull'effetto dell'epoetina alfa sulla fertilità negli uomini o nelle donne.
Capacità di influire sulla velocità di reazione nella guida di autoveicoli o di altri macchinari.
Non sono stati condotti studi sull'effetto sulla velocità di reazione nella guida di autoveicoli o di altri macchinari.
Modalità e dosi di somministrazione.
L'epoetina alfa può essere somministrata per via sottocutanea e per via endovenosa.
Come per tutti i medicinali parenterali, il medicinale Epobiocryn deve essere ispezionato prima dell'uso per verificare l'assenza di particelle visibili e di cambiamenti nel colore della soluzione.
Tutte le altre cause di anemia (carenza di ferro, acido folico, vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi o fibrosi del midollo osseo di qualsiasi eziologia) devono essere identificate e trattate prima di iniziare la terapia con epoetina alfa e prima di decidere di aumentare il dosaggio. Per ottenere una risposta ottimale al trattamento con epoetina alfa, è necessario garantire un adeguato apporto di ferro e, se necessario, somministrare ulteriori preparati di ferro (vedere la sezione «Speciali avvertenze e precauzioni per l'uso»).
Somministrazione endovenosa.
L'epoetina alfa viene somministrata mediante iniezione della durata da 1 a 5 minuti, a seconda della dose del medicinale. Nei pazienti sottoposti a emodialisi, l'iniezione in bolo può essere somministrata direttamente durante la procedura attraverso un apposito porto venoso nella linea di dialisi. Il medicinale può anche essere somministrato al termine della procedura di emodialisi attraverso la fistola o il catetere, seguito dall'infusione di 10 ml di soluzione fisiologica di sodio cloruro per il lavaggio del circuito e una corretta distribuzione del medicinale nella circolazione sanguigna.
La somministrazione lenta è preferita principalmente nei pazienti che manifestano sintomi di tipo influenzale.
L'epoetina alfa non deve essere somministrata per infusione endovenosa né miscelata con altri medicinali.
Somministrazione sottocutanea.
Il volume massimo di somministrazione sottocutanea del medicinale in un'unica sede è di 1 ml. Se necessario somministrare volumi maggiori, l'iniezione sottocutanea deve essere effettuata in più sedi.
Il medicinale deve essere iniettato sottocutaneamente negli arti o nella parete anteriore dell'addome.
Se, a giudizio del medico, il paziente o un caregiver possono somministrare in modo sicuro ed efficace Epobiocryn per via sottocutanea, devono essere istruiti correttamente sul dosaggio e sull'uso del medicinale.
Trattamento dell'anemia sintomatica negli adulti e nei bambini con insufficienza renale cronica.
Nei pazienti con insufficienza renale cronica, quando è possibile utilizzare la via endovenosa (pazienti in emodialisi), questa è considerata la via preferita. Quando la somministrazione endovenosa risulta problematica (pazienti non ancora sottoposti a emodialisi o pazienti in dialisi peritoneale), l'epoetina alfa può essere somministrata per via sottocutanea.
I sintomi dell'anemia e le complicanze possono variare in base all'età, al sesso e alle condizioni indotte dalla malattia; pertanto è necessaria una valutazione clinica individuale da parte del medico.
Epobiocryn deve essere utilizzato per aumentare i livelli di emoglobina fino a un massimo di 12 g/dl (7,5 mmol/l). È necessario evitare un aumento dei livelli di emoglobina superiore a 2 g/dl (1,25 mmol/l) in un periodo di quattro settimane. In tal caso, la dose deve essere ridotta come indicato più avanti.
A causa della variabilità individuale, i valori periodici dell'emoglobina in ciascun paziente possono essere superiori o inferiori al livello desiderato.
Il livello di emoglobina deve essere controllato mediante aggiustamento della dose, mantenendolo compreso tra 10 g/dl (6,2 mmol/l) e 12 g/dl (7,5 mmol/l). Nei bambini, il livello raccomandato di emoglobina è compreso tra 9,5 e 11 g/dl (5,9–6,8 mmol/l).
È necessario evitare un livello costante di emoglobina superiore a 12 g/dl (7,5 mmol/l). Se la concentrazione di emoglobina aumenta di almeno 2 g/dl (1,25 mmol/l) al mese o se il livello costante di emoglobina supera i 12 g/dl (7,5 mmol/l), la dose di epoetina deve essere ridotta del 25%. Se il livello di emoglobina supera i 13 g/dl (8,1 mmol/l), il trattamento deve essere interrotto fino a quando l'emoglobina non scende a 12 g/dl (7,5 mmol/l), dopodiché il trattamento con epoetina alfa deve essere ripreso con una dose ridotta del 25% rispetto alla dose precedente.
I pazienti devono essere attentamente monitorati per garantire che la dose minima efficace dei farmaci stimolanti l'eritropoiesi assicuri un controllo adeguato dei sintomi dell'anemia.
Il livello di ferritina (o la concentrazione di ferro nel siero) deve essere determinato in tutti i pazienti prima dell'inizio e durante il trattamento con Epobiocryn. Se necessario, devono essere somministrati ulteriori preparati di ferro. Altre forme di anemia (come carenza di vitamina B12 o di acido folico) devono essere escluse prima di iniziare la terapia con Epobiocryn. L'assenza di risposta clinica al trattamento con Epobiocryn richiede l'individuazione di fattori concomitanti, come carenza di ferro, acido folico o vitamina B12, intossicazione da alluminio, infezioni intercorrenti, processi infiammatori o traumi, emolisi, fibrosi del midollo osseo di qualsiasi eziologia.
Pazienti adulti in emodialisi.
Nei pazienti in emodialisi, il medicinale viene somministrato per via endovenosa.
Il trattamento è suddiviso in due fasi.
Fase di correzione.
50 UI/kg 3 volte a settimana.
Se necessario, la dose può essere aumentata in modo graduale (non più di una volta ogni 4 settimane) di 25 UI/kg 3 volte a settimana, fino al raggiungimento della concentrazione ottimale di emoglobina (10–12 g/dl, 6,2–7,5 mmol/l).
Fase di mantenimento.
Regolazione della dose per mantenere il livello desiderato di emoglobina (Hb) tra 10 e 12 g/dl (6,2–7,5 mmol/l).
La dose settimanale raccomandata è compresa tra 75 e 300 UI/kg.
I pazienti con livelli iniziali di emoglobina molto bassi (< 6 g/dl, o < 3,75 mmol/l) possono richiedere dosi più elevate per mantenere la concentrazione rispetto ai pazienti con anemia meno grave (emoglobina > 8 g/dl, o > 5 mmol/l).
Bambini in emodialisi.
Nei bambini in emodialisi, il medicinale viene somministrato per via endovenosa.
Il trattamento è suddiviso in due fasi.
Fase di correzione.
50 UI/kg 3 volte a settimana.
Se necessario, la dose può essere aumentata in modo graduale (non più di una volta ogni 4 settimane) di 25 UI/kg 3 volte a settimana, fino al raggiungimento della concentrazione ottimale di emoglobina (9,5–11 g/dl, 5,9–6,8 mmol/l).
Fase di mantenimento.
Regolazione della dose per mantenere il livello desiderato di emoglobina (Hb) tra 9,5 e 11 g/dl (5,9–6,8 mmol/l).
I bambini con peso corporeo inferiore a 30 kg richiedono una dose di mantenimento maggiore rispetto agli adulti e ai bambini con peso corporeo superiore a 30 kg.
Dosi di mantenimento di epoetina alfa:
| Dosaggio (UI/kg per 3 settimane) |
||
| Peso (kg) |
Dosaggio medio |
Dosaggio di mantenimento abituale |
| < 10 |
100 |
75–150 |
| 10–30 |
75 |
60–150 |
| > 30 |
33 |
30–100 |
I pazienti con livelli iniziali di emoglobina molto bassi (< 6,8 g/dl, o < 4,25 mmol/l) possono richiedere dosi più elevate per il mantenimento della concentrazione rispetto ai pazienti con anemia meno grave (emoglobina > 6,8 g/dl, o > 4,25 mmol/l).
Pazienti adulti con insufficienza renale nel periodo pre-dialitico.
Nei pazienti con insufficienza renale in periodo pre-dialitico, in assenza di accesso per somministrazione endovenosa, il medicinale può essere somministrato per via sottocutanea.
Il trattamento è suddiviso in due fasi.
Fase correttiva.
50 UI/kg, 3 volte alla settimana.
Se necessario, la dose può essere aggiustata aumentando di 25 UI/kg 3 volte alla settimana, con intervalli tra gli aumenti di almeno 4 settimane, fino al raggiungimento di un livello di emoglobina compreso tra 10–12 g/dl (6,2–7,5 mmol/l).
Fase di mantenimento.
Durante la fase di mantenimento, Epobiocryn può essere somministrato 3 volte alla settimana oppure, in caso di somministrazione sottocutanea, 1 volta alla settimana o 1 volta ogni 2 settimane. Le dosi e gli intervalli tra le somministrazioni devono essere aggiustati al fine di mantenere il livello desiderato di emoglobina (Hb) tra 10 e 12 g/dl (6,2–7,5 mmol/l). L’allungamento degli intervalli tra le somministrazioni può richiedere un aumento della dose. La dose massima non deve superare 150 UI/kg 3 volte alla settimana, 240 UI/kg (massimo fino a 20000 UI) una volta alla settimana oppure 480 UI/kg (massimo fino a 40000 UI) una volta ogni 2 settimane.
Pazienti adulti in dialisi peritoneale.
Nei pazienti in dialisi peritoneale, in assenza di accesso per somministrazione endovenosa, il medicinale può essere somministrato per via sottocutanea.
Il trattamento è suddiviso in due fasi.
Fase correttiva.
50 UI/kg, 2 volte alla settimana.
Fase di mantenimento.
Generalmente, la dose necessaria per mantenere il livello desiderato di emoglobina (Hb) tra 10 e 12 g/dl (6,2–7,5 mmol/l) è compresa tra 25 e 50 UI/kg 2 volte alla settimana, mediante somministrazione di due iniezioni equivalenti.
Trattamento dei pazienti con anemia indotta da chemioterapia.
Nei pazienti con anemia (ad esempio, concentrazione di emoglobina ≤ 10 g/dl (6,2 mmol/l)), Epobiocryn deve essere somministrato per via sottocutanea. I sintomi e le complicanze dell’anemia dipendono dall’età, dal sesso e dalle condizioni cliniche del paziente legate alla malattia; pertanto è necessaria una valutazione individuale da parte del medico riguardo al decorso clinico e allo stato del paziente.
A causa della variabilità individuale, i valori periodici dell’emoglobina in ciascun paziente possono risultare superiori o inferiori al livello desiderato. Il livello di emoglobina deve essere controllato mediante aggiustamento della dose, tenendo presente che tale livello deve rimanere compreso tra 10 g/dl (6,2 mmol/l) e 12 g/dl (7,5 mmol/l). Si deve evitare un livello costante di emoglobina superiore a 12 g/dl (7,5 mmol/l). Le indicazioni per l’aggiustamento della dose in caso di livelli di emoglobina superiori a 12 g/dl (7,5 mmol/l) sono descritte di seguito.
La terapia con epoetina alfa deve proseguire per un mese dopo l’interruzione della chemioterapia.
La dose iniziale per il trattamento dell’anemia in questo gruppo di pazienti è di 150 UI/kg 3 volte alla settimana. L’epoetina alfa può essere somministrata, in alternativa, alla dose iniziale di 450 UI/kg per via sottocutanea una volta alla settimana.
Se dopo 4 settimane di trattamento con la dose iniziale il livello di emoglobina è aumentato di almeno 1 g/dl (0,6 mmol/l) (oppure il livello di reticolociti è aumentato a ≥ 40000 cellule/ml), la dose deve rimanere a 150 UI/kg 3 volte alla settimana oppure a 450 UI/kg per via sottocutanea una volta alla settimana. Se dopo 4 settimane di trattamento con la dose iniziale il livello di emoglobina è aumentato di 1 g/dl (0,62 mmol/l) oppure il livello di reticolociti è aumentato a < 40000 cellule/ml, la dose deve essere aumentata a 300 UI/kg 3 volte alla settimana oppure a 40000 UI una volta alla settimana.
Se dopo 4 settimane di trattamento con la dose aumentata di 300 UI/kg 3 volte alla settimana il livello di emoglobina è aumentato di ≥ 1 g/dl (≥ 0,62 mmol/l) oppure il livello di reticolociti è aumentato a ≥ 40000 cellule/ml, la dose non viene modificata. Tuttavia, se il livello di emoglobina è aumentato di < 1 g/dl (≥ 0,62 mmol/l) oppure il livello di reticolociti è aumentato di < 40000 cellule/ml, la risposta clinica è considerata negativa e il trattamento deve essere interrotto.
Schema del regime posologico raccomandato:
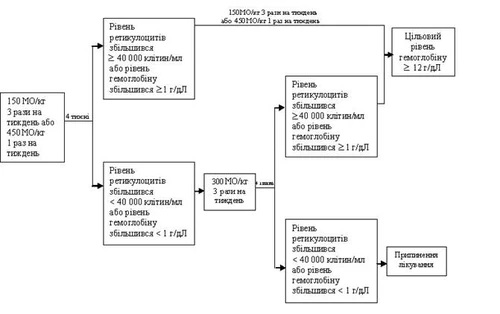
I pazienti devono essere attentamente monitorati per garantire che la dose più bassa approvata dei farmaci stimolanti l’eritropoiesi assicuri un adeguato controllo dei sintomi dell’anemia.
Aggiustamento della dose per il mantenimento del livello obiettivo di emoglobina tra 10 e 12 g/dl.
Se l’incremento del livello di emoglobina supera i 2 g/dl (1,25 mmol/l) al mese e il livello totale di emoglobina si avvicina a 12 g/dl (7,5 mmol/l), la dose di Epobiocryn deve essere ridotta del 25–50%, a seconda della velocità di incremento dell’emoglobina. Se il livello di emoglobina supera 13 g/dl (8,1 mmol/l), la terapia deve essere temporaneamente interrotta fino al ritorno del livello a 12 g/dl (7,5 mmol/l), dopodiché il trattamento deve essere ripreso con una dose del 25% inferiore rispetto alla precedente.
Pazienti adulti che partecipano a un programma di predeposito di sangue autologo prima di interventi chirurgici.
Si raccomanda la somministrazione endovenosa.
L’epoetina alfa deve essere somministrata dopo ogni procedura di prelievo di sangue.
Nei pazienti con anemia moderata (ematocrito compreso tra 33–39%), che necessitano di ≥ 4 unità di sangue, il trattamento con epoetina alfa deve essere effettuato alla dose di 600 UI/kg 2 volte alla settimana per 3 settimane precedenti l’intervento chirurgico.
Tutti i pazienti trattati con epoetina alfa devono ricevere un adeguato apporto di ferro (200 mg al giorno per via orale) per tutta la durata del trattamento. Per garantire un livello sufficiente di ferro nell’organismo, la somministrazione di farmaci a base di ferro deve essere iniziata il prima possibile, anche diverse settimane prima dell’inizio del programma di predeposito di sangue autologo.
Pazienti adulti sottoposti a chirurgia ortopedica elettiva.
Si raccomanda la somministrazione per via sottocutanea.
Il regime posologico raccomandato è di 600 UI/kg alla settimana per 3 settimane precedenti l’intervento (21°, 14° e 7° giorno prima dell’intervento) e nel giorno dell’intervento.
Se per motivi clinici è necessario ridurre il periodo preoperatorio a meno di 3 settimane, Epobiocryn deve essere somministrato giornalmente alla dose di 300 UI/kg per 10 giorni consecutivi prima dell’intervento, nel giorno dell’intervento e per 4 giorni dopo l’intervento. Se durante i controlli ematologici preoperatori il livello di emoglobina raggiunge o supera 15 g/dl, l’uso di epoetina alfa deve essere completamente interrotto.
Tutti i pazienti trattati con epoetina alfa devono ricevere un adeguato apporto di ferro (200 mg al giorno per via orale) per tutta la durata del trattamento. La somministrazione di farmaci a base di ferro deve essere iniziata il prima possibile, anche diverse settimane prima dell’inizio del programma di predeposito di sangue autologo.
Bambini.
L’epoetina alfa è indicata per il trattamento dell’anemia associata all’insufficienza renale cronica nei bambini di età compresa tra 1 mese e 18 anni sottoposti a dialisi. La sicurezza e l’efficacia del medicinale nei bambini di età inferiore a 1 mese non sono state stabilite.
Sovradosaggio.
Il medicinale ha un ampio indice terapeutico. In caso di sovradosaggio di epoetina alfa si verificano effetti che riflettono il massimo grado di espressione dell’azione farmacologica dell’ormone. In presenza di livelli estremamente elevati di emoglobina, può essere necessaria una flebotomia. Se necessario, si applica una terapia sintomatica.
Effetti indesiderati.
L'effetto indesiderato più comune durante il trattamento con epoetina alfa in pazienti oncologici e in pazienti con insufficienza renale cronica è l'aumento dose-dipendente della pressione arteriosa o il peggioramento di una preesistente ipertensione. Il controllo della pressione arteriosa deve essere effettuato fin dall'inizio del trattamento. Altri effetti indesiderati comuni includono trombosi venosa profonda, embolia polmonare, convulsioni, diarrea, nausea, cefalea, stato simil-influenzale, piressia, eruzioni cutanee e vomito.
All'inizio del trattamento possono manifestarsi sintomi simili a quelli del raffreddore, come cefalea, dolori muscolari e articolari e brividi. La frequenza può variare a seconda delle indicazioni.
Può inoltre verificarsi un peggioramento della pervietà delle vie respiratorie, inclusa la congestione nasale e la nasofaringite.
Tra gli effetti indesiderati gravi vi sono trombosi venosa e arteriosa, embolia (inclusi casi con esito fatale), trombosi venosa profonda, embolia polmonare, trombosi arteriosa (inclusi infarto miocardico e ischemia miocardica), trombosi della retina e trombosi dello shunt (inclusa l'occlusione del sistema di dialisi). Possono inoltre verificarsi complicanze cerebrovascolari (inclusi ictus ed emorragie cerebrali) e attacchi ischemici transitori, aneurismi e reazioni di ipersensibilità, inclusi eruzioni cutanee, orticaria, reazioni anafilattiche e angioedema.
Inoltre, durante il trattamento con epoetina alfa, in pazienti con pressione inizialmente normale o ridotta, può manifestarsi una crisi ipertensiva con encefalopatia e convulsioni, che richiede un immediato intervento medico e un trattamento intensivo. Particolare attenzione va posta in caso di comparsa improvvisa di cefalea acuta di tipo emicranico, che potrebbe rappresentare un segnale premonitorio.
Molto raramente può verificarsi una vera aplasia eritroide mediata da anticorpi dopo mesi o anni di trattamento.
Frequenza degli effetti indesiderati: molto frequente (≥1/10); frequente (≥1/100 fino a <1/10); non frequente (≥1/1000 fino a <1/100); raro (≥1/10000 fino a <1/1000); molto raro (<1/10000); frequenza sconosciuta.
Dal punto di vista del sangue e del sistema linfatico.
Non frequente – trombocitemia (pazienti affetti da cancro).
Frequenza sconosciuta – trombocitemia (pazienti con insufficienza renale cronica).
Molto raro – vera aplasia eritroide dipendente da anticorpi.
Dal punto di vista del sistema immunitario.
Frequenza sconosciuta – reazioni anafilattiche, reazioni di ipersensibilità.
Reazioni anafilattiche si verificano raramente: complicanze potenzialmente gravi associate a disturbi respiratori o a ipotensione; reazioni immunitarie (ha una minima capacità di indurre la formazione di anticorpi).
Dal punto di vista del sistema nervoso.
Molto frequente – cefalea (pazienti affetti da cancro).
Frequente – convulsioni (pazienti con insufficienza renale cronica), cefalea (pazienti con insufficienza renale cronica), ictus.
Non frequente – emorragie cerebrali, convulsioni (pazienti affetti da cancro).
Frequenza sconosciuta – ictus cerebrovascolare, encefalopatia ipertensiva, attacco ischemico transitorio, vertigini, sonnolenza.
Dal punto di vista degli organi della vista.
Frequenza sconosciuta – trombosi retinica.
Dal punto di vista del cuore.
Frequenza sconosciuta – infarto del miocardio.
Dal punto di vista del sistema vascolare.
Frequente – trombosi venosa profonda (pazienti affetti da cancro), ipertensione arteriosa.
Frequenza sconosciuta – trombosi venosa profonda (pazienti con insufficienza renale cronica), trombosi arteriosa, crisi ipertensiva.
Dal punto di vista del sistema respiratorio.
Frequente – embolia polmonare (pazienti affetti da cancro), tosse.
Non frequente – peggioramento della pervietà delle vie respiratorie.
Frequenza sconosciuta – embolia polmonare (pazienti con insufficienza renale cronica).
Dal punto di vista del tratto gastrointestinale.
Molto frequente – nausea.
Frequente – diarrea (pazienti affetti da cancro), vomito.
Non frequente – diarrea (pazienti con insufficienza renale cronica).
Dal punto di vista della cute.
Frequente – eruzioni cutanee, eczema.
Frequenza sconosciuta – angioedema, orticaria, prurito, edema di Quincke, sindrome di Stevens-Johnson, necrolisi epidermica tossica (che possono essere potenzialmente letali o causare esito fatale).
Dal punto di vista del sistema muscoloscheletrico e dei tessuti connettivi.
Molto frequente – artralgia (pazienti con insufficienza renale cronica), dolore osseo, dolore agli arti.
Frequente – artralgia (pazienti affetti da cancro).
Non frequente – mialgia (pazienti affetti da cancro).
Frequenza sconosciuta – mialgia (pazienti con insufficienza renale cronica).
Malattie congenite, ereditarie/genetiche.
Frequenza sconosciuta – porfiria.
Disturbi generali e disturbi nel sito di somministrazione.
Molto frequente – piressia (pazienti affetti da cancro), stato simil-influenzale (pazienti con insufficienza renale cronica), reazioni nel sito di iniezione, edema periferico.
Frequente – stato simil-influenzale (pazienti affetti da cancro).
Frequenza sconosciuta – brividi, mancata risposta al trattamento.
Esami di laboratorio.
Frequenza sconosciuta – presenza di anticorpi contro l'eritropoietina, iperkaliemia, iperfosfatemia, aumento della concentrazione di urea, creatinina e acido urico nel plasma (in pazienti con insufficienza renale cronica).
Lesioni, avvelenamenti e complicanze da procedure.
Frequente – trombosi dello shunt, inclusa l'apparecchiatura per dialisi (pazienti con insufficienza renale cronica).
Pazienti con insufficienza renale cronica.
Nei pazienti con insufficienza renale cronica, un livello di emoglobina superiore a 12 g/dl può essere associato a un aumentato rischio di complicanze cardiovascolari, inclusi casi con esito fatale.
In pazienti sottoposti ad emodialisi, specialmente in caso di predisposizione all'ipotensione o presenza di complicanze a livello della fistola artero-venosa (stenoosi, aneurismi, ecc.), sono stati descritti casi di trombosi dello shunt.
Pazienti con patologie oncologiche.
Lo sviluppo di complicanze trombotiche è possibile in pazienti trattati con farmaci stimolanti l'eritropoiesi, inclusa l'epoetina alfa (vedi sezione «Informazioni importanti sull'uso»).
Pazienti chirurgici adulti.
Non può essere esclusa la possibilità che il trattamento con epoetina alfa in pazienti con livello di emoglobina stabile >13 g/dl possa essere associato a un aumentato rischio di complicanze trombotiche/vascolari post-operatorie.
Descrizione di singoli effetti indesiderati.
Sono state riportate reazioni di ipersensibilità, compresi casi di eruzioni cutanee (inclusa orticaria), reazioni anafilattiche e angioedema (vedi sezione «Informazioni importanti sull'uso»).
Sono stati osservati casi di crisi ipertensiva con encefalopatia e convulsioni, che richiedevano immediata visita medica e terapia intensiva, in pazienti con pressione arteriosa normale o bassa all'inizio del trattamento. È necessario prestare particolare attenzione alla comparsa di improvvisa cefalea acuta di tipo emicranico, che potrebbe rappresentare un segnale di allarme (vedi sezione «Informazioni importanti sull'uso»).
Molto raramente (< 1 su 10000 pazienti-anno) sono stati riportati casi di vera aplasia eritroide mediata da anticorpi (PRCA) in pazienti trattati con farmaci contenenti eritropoietina per mesi o anni (vedi sezione «Informazioni importanti sull'uso»).
Bambini con insufficienza renale cronica in emodialisi.
L'esperienza sull'uso di eritropoietina nei bambini con insufficienza renale cronica in emodialisi, durante studi clinici e nel periodo post-marketing, è limitata. Non sono stati osservati effetti indesiderati specifici per l'età pediatrica non riportati in tabella, né effetti indesiderati non correlati alla patologia sottostante.
Segnalazione di effetti indesiderati sospetti.
La segnalazione degli effetti indesiderati dopo l'autorizzazione del medicinale è di grande importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale.
I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, possono segnalare tutti i casi sospetti di effetti indesiderati o di mancata efficacia dei medicinali prodotti da «FZ «Biopharma» SRL» in qualsiasi modo comodo (per posta, e-mail o telefono), in forma cartacea o elettronica. Le segnalazioni riguardanti effetti indesiderati o mancata efficacia dei medicinali prodotti da «FZ «Biopharma» SRL» devono essere inviate al reparto di farmacovigilanza all'indirizzo: via M. Amosova, 12, Kiev, 03680, telefono +38 044 459 4600, indirizzo e-mail: [email protected]. In caso di effetti indesiderati o domande sulla sicurezza d'uso dei medicinali prodotti da «FZ «Biopharma» SRL», si prega di rivolgersi al reparto di farmacovigilanza.
Inoltre, le segnalazioni di effetti indesiderati possono essere inviate tramite il Sistema Informatizzato Automatizzato di Farmacovigilanza al seguente link: https://aisf.dec.gov.ua
Periodo di validità. 2 anni.
Condizioni di conservazione. Conservare nella confezione originale al riparo dalla luce, a una temperatura compresa tra 2 e 8 °C. Non congelare. Non agitare.
Conservare fuori dalla portata dei bambini.
Incompatibilità. Poiché non sono disponibili studi di compatibilità, Epobiocryn non deve essere somministrato contemporaneamente ad altri medicinali.
Confezione. 1000 UI, 2000 UI, 4000 UI, 10 000 UI in siringhe preriempite o fiale. 5 siringhe preriempite o 5 fiale per blister. 1 blister per confezione.
Categoria di prescrizione. Su prescrizione medica.
Produttore.
FZ «STADA» SRL, Ucraina.
Sede del produttore e indirizzo del luogo di esercizio dell'attività.
Ucraina, 09100, Oblast' di Kiev, città di Bila Tserkva, via Kyivska, 37.