Emoklot
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku Emoklot
Skład:
substancja czynna: ludzki czynnik krzepnięcia krwi VIII;
1 fiolka z proszkiem zawiera 500 JM lub 1000 JM ludzkiego czynnika krzepnięcia krwi VIII;
substancje pomocnicze: sodu chloridum, sodu cytrynian, glicyna, wapnia chloridum;
fiolka z rozpuszczalnikiem zawiera wodę do wstrzykiwań 10 ml.
Odtworzony roztwór zawiera 50 JM/ml (500 JM/10 ml) lub 100 JM/ml (1000 JM/10 ml) ludzkiego czynnika krzepnięcia krwi VIII.
Specyficzna aktywność leku Emoklot wynosi około 80 JM/mg białka. Aktywność (JM) oznacza się metodą chromogeniczną zgodnie z Europejską Farmakopeą.
Preparat wytwarzany jest z osocza ludzkiego.
Preparat zawiera ludzki czynnik von Willebranda.
Substancje pomocnicze o znanym działaniu: lek zawiera do 41 mg sodu na fiolkę (10 ml).
Postać farmaceutyczna. Proszek i rozpuszczalnik do sporządzenia roztworu do wlewu.
Główne właściwości fizykochemiczne: proszek: biały lub bladoróżowy higroskopijny proszek lub krucha masa; rozpuszczalnik: przezroczysta, bezbarwna ciecz.
Grupa farmakoterapeutyczna. Środki hemostatyczne. Czynniki krzepnięcia krwi VIII. Czynnik krzepnięcia VIII. Kod ATC B02BD02.
Właściwości farmakodynamiczne.
Farmakodynamika.
Złożony preparat czynnika VIII/czynnika von Willebranda składa się z dwóch cząsteczek (czynnik VIII i czynnik von Willebranda) o różnych funkcjach fizjologicznych.
Po podaniu pacjentowi z hemofilią czynnik VIII wiąże się z obecnym we krwi pacjenta czynnikiem von Willebranda.
Aktywowany czynnik VIII działa jako kofaktor dla aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w formę aktywną. Aktywowany czynnik X przekształca protrombinę w trombinę, która z kolei przekształca fibrynogen w fibrynę, umożliwiając powstanie skrzepliny.
Hemofilia A to uwarunkowane genetycznie, związane z płcią zaburzenie krzepnięcia krwi, spowodowane obniżonym poziomem czynnika VIII:C, prowadzące do obfitych krwawień do stawów, mięśni lub narządów wewnętrznych, które występują spontanicznie lub jako wynik przypadkowych urazów czy zabiegów chirurgicznych. Terapia zastępcza zwiększa stężenie czynnika VIII w osoczu, tym samym umożliwiając tymczasową korektę jego niedoboru i zmniejszenie skłonności do krwawień.
Należy zaznaczyć, że średnioroczna częstość krwawień (ŚCK) różni się w zależności od zastosowanego koncentratu czynnika VIII oraz od poszczególnych badań klinicznych.
Czynnik von Willebranda pełni rolę stabilizatora czynnika VIII, pośredniczy w adhezji płytek krwi do uszkodzonych odcinków naczyń oraz bierze udział w agregacji płytek krwi.
Dziesięciu pacjentów z ciężką hemofilią A (mediana wieku 15 lat, zakres 5–51) z wysokim mianem inhibitorów, włączonych do rejestru PROFIT prowadzonego przez Włoskie Stowarzyszenie Ośrodków Hemofilii, otrzymywało leczenie lekiem Emoklot w celu wyeliminowania inhibitorów poprzez terapię metodą indukcji tolerancji immunologicznej (ITI). Ośmiu z tych dziesięciu pacjentów otrzymywało ITI jako terapię pierwszej linii, a dwóch pacjentów poddano pilnej terapii ITI po wcześniejszej nieudanej próbie z użyciem innego koncentratu czynnika VIII. Pięciu pacjentów otrzymywało codziennie leczenie w dawkach średnich/wysokich (100/200 J/mkg/dzień), a pięciu otrzymywało leczenie różnymi dawkami co drugi dzień lub trzy razy w tygodniu (50–150 J/mkg). Pełna lub częściowa odpowiedź, utrzymująca się po średnim okresie obserwacji wynoszącym 9 lat, została osiągnięta w 50% przypadków. U czterech pacjentów, u których osiągnięto pełny sukces, średni czas wyeliminowania inhibitora wyniósł 26 miesięcy.
Dodatkowo w literaturze opisano doświadczenie z 11 pacjentami (mediana wieku 17 lat) z wysokim mianem inhibitorów, którzy poddano terapii metodą ITI lekiem Emoklot; ogólnie ITI okazało się skuteczne u 9 z 11 pacjentów (82%), z pełnym wyeliminowaniem inhibitorów u 4 (36%) i częściowym sukcesem u 5 (45%) pacjentów.
Dzieci
125 dzieci w wieku do 6 lat bez inhibitorów, które wcześniej nie otrzymywały lub otrzymywały minimalnego leczenia czynnikiem krzepnięcia krwi ludzkiej VIII, otrzymywało lek Emoklot w ramach kontrolowanego, randomizowanego badania (SIPPET), którego celem była ocena częstości występowania inhibitorów u pacjentów leczonych czynnikiem pochodzącym z osocza lub rekombinowanym czynnikiem krzepnięcia krwi ludzkiej VIII. 61 z 125 pacjentów otrzymywało Emoklot w dawkowaniu ustalonym dla leczenia na żądanie lub w celu profilaktyki. 34 z 61 pacjentów otrzymywało leczenie na żądanie, 5 pacjentów – standardowe dawki profilaktyczne (3 infuzje/tydzień), 15 pacjentów – zmodyfikowaną profilaktykę (2 infuzje/tydzień) i 7 pacjentów otrzymywało różne kombinacje schematów leczenia.
W wyniku analizy post-hoc, której celem była ocena średniorocznej częstości krwawień (ŚCK) u pacjentów leczonych Emoklotem, otrzymano wartości ŚCK wynoszące 4,2 (342 epizody) u pacjentów leczonych schematem na żądanie, 7,5 (25 epizodów) u pacjentów leczonych standardową profilaktyką (z 25 epizodów krwawień zarejestrowanych w tej grupie, 24 epizody wystąpiły u jednego pacjenta; po wyłączeniu tego pacjenta wartość ŚCK spadła do 0,24), 5,8 (92 epizody) u pacjentów leczonych zmodyfikowaną profilaktyką oraz 5,9 (60 epizodów) u pacjentów leczonych różnymi kombinacjami schematów leczenia.
Farmakokinetyka.
Po podaniu leku około 2/3–3/4 czynnika VIII pozostaje w krążeniu.
Poziom aktywności czynnika VIII osiągany w osoczu wynosi 80–120% od obliczonej aktywności czynnika VIII w osoczu.
Aktywność czynnika VIII w osoczu maleje zgodnie z dwufazową krzywą eksponencjalną.
W wstępnej fazie rozkład między przestrzenią wewnątrznaczyniową a innymi płynami ustrojowymi odbywa się z okresem półtrwania z osocza wynoszącym 3–6 godzin.
W kolejnej, wolniejszej fazie (która najprawdopodobniej odzwierciedla wychwyt czynnika VIII) okres półtrwania waha się od 8 do 20 godzin i średnio wynosi 12 godzin, co odzwierciedla rzeczywisty biologiczny okres półtrwania.
Właściwości farmakokinetyczne leku Emoklot zostały przebadane w badaniu klinicznym „Badanie farmakokinetyki i skuteczności klinicznej koncentratu Czynnika VIII Emoklot u pacjentów z hemofilią A” (kod badania KB030), w którym wzięło udział 15 pacjentów z ciężką hemofilią A (z poziomem czynnika VIII < 1). Parametry farmakokinetyczne określano po dwóch oddzielnych infuzjach (dawka 25 J/mkg), przeprowadzonych w odstępie 3–6 miesięcy. W okresie między dwiema infuzjami pacjenci otrzymywali leczenie lekiem Emoklot zgodnie ze swoim standardowym schematem terapeutycznym (w celu leczenia lub profilaktyki).
Średnie wartości parametrów farmakokinetycznych leku Emoklot, ustalone podczas badania, przedstawiono w tabeli 1.
Tabela 1
| Wskaźnik |
Pierwsza infuzja |
Druga infuzja |
||
| Bez odjęcia wartości początkowych |
Z odjęciem wartości początkowych |
Bez odjęcia wartości początkowych |
Z odjęciem wartości początkowych |
|
| AUC0-t (MO·ml-1·h) |
10,94 |
9,96 |
10,75 |
8,95 |
| AUC0-∞ (MO·ml-1·h) |
13,08 |
11,22 |
12,07 |
9,89 |
| Cltot (ml·h-1·kg-1) |
2,63 |
2,89 |
2,51 |
2,99 |
| Stopniowe przywrócenie (%) |
2,688 |
2,671 |
||
| t1/2α (h) |
0,543 |
0,768 |
||
| t1/2β (h) |
12,05 |
15,16 |
||
Dzieci
Chociaż brak specyficznych danych dotyczących stosowania u dzieci, pojedyncze opublikowane dane farmakokinetyczne nie wykazały istotnych różnic między dorosłymi a dziećmi z tym samym schorzeniem.
Dane przedkliniczne dotyczące bezpieczeństwa.
Stężony czynnik krzepnięcia krwi człowieka VIII jest naturalnym składnikiem ludzkiej osocza i działa podobnie jak endogenny czynnik VIII.
Badania toksyczności pojedynczej dawki nie są istotne, ponieważ wysokie dawki powodują hipervolemię.
Badania toksyczności przy wielokrotnym (powtarzanym) podawaniu u zwierząt nie są możliwe ze względu na interferencję przeciwciał, które powstają przeciwko białku heterologicznemu.
Nawet dawki znacznie przekraczające zalecane dla człowieka na 1 kg masy ciała nie wykazują żadnego działania toksycznego u zwierząt badanych.
Ponieważ doświadczenie kliniczne stosowania czynnika krzepnięcia krwi człowieka VIII nie potwierdza jego działania kancerogennego ani mutagennego, przeprowadzenie badań eksperymentalnych, w szczególności z udziałem gatunków heterologicznych, nie jest uważane za konieczne.
Dane kliniczne.
Wskazania.
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (wrodzony niedobór czynnika krzepnięcia krwi człowieka VIII).
Leczenie nabytego niedoboru czynnika krzepnięcia krwi człowieka VIII.
Leczenie chorych na hemofilię z obecnością przeciwciał przeciwko czynnikowi krzepnięcia krwi człowieka VIII (inhibitory: patrz także „Szczególne ostrzeżenia i środki ostrożności”).
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych preparatu.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie zgłaszano interakcji leków zawierających czynnik krzepnięcia krwi człowieka VIII z innymi lekami.
Dzieci
Brak danych specyficznych dotyczących dzieci.
Szczególne wytyczne dotyczące stosowania.
Śledzenie
W celu poprawy śledzenia leków biologicznych zaleca się zapisywanie nazwy i numeru serii preparatu Emoklot za każdym razem, gdy jest on podawany pacjentowi.
Zwiększona wrażliwość
Podczas stosowania preparatu Emoklot możliwe są reakcje alergiczne związane ze zwiększona wrażliwością.
Preparat zawiera śladowe ilości białek ludzkich innych niż czynnik VIII. W przypadku wystąpienia objawów zwiększonej wrażliwości pacjentom należy zalecić natychmiastowe zaprzestanie stosowania preparatu i skonsultowanie się z lekarzem. Pacjentów należy poinformować o wczesnych objawach reakcji nadwrażliwości, w tym wysypce, pokrzywce uogólnionej, ucisku w klatce piersiowej, świstach podczas oddychania, hipotensji oraz reakcji anafilaktycznej.
W przypadku wystąpienia wstrząsu należy postępować zgodnie z obowiązującymi standardami medycznymi leczenia wstrząsu.
Ważne informacje dotyczące substancji pomocniczych w preparacie Emoklot
Ten lek zawiera do 41 mg sodu w fiolce (10 ml), co stanowi 2,05% zalecanej przez WHO maksymalnej dziennej dawki sodu dla dorosłych, wynoszącej 2 g.
Inhibitory
Tworzenie się przeciwciał neutralizujących (inhibitorów) przeciwko ludzkiemu czynnikowi VIII krzepnięcia krwi jest znanym powikłaniem leczenia pacjentów z hemofilią typu A. Inhibitory te zazwyczaj stanowią immunoglobuliny klasy IgG skierowane przeciwko działaniu prokrzepnemu czynnika VIII, których stężenie określa się w jednostkach Bethesda (JB) na 1 ml osocza metodą zmodyfikowanego badania. Ryzyko powstawania inhibitorów jest powiązane z ciężkością choroby oraz ekspozycją na czynnik VIII i jest najwyższe w ciągu pierwszych 50 dni leczenia. Choć ryzyko to występuje rzadko, istnieje przez całe życie.
Kliniczne znaczenie tworzenia się inhibitorów zależy od ich miana. Przy niskim mianie inhibitorów ryzyko niewystarczającej odpowiedzi klinicznej jest niższe niż przy wysokim mianie inhibitorów.
Ogólnie należy dokładnie monitorować wszystkich pacjentów otrzymujących leczenie preparatami czynnika VIII krzepnięcia krwi pod kątem powstawania inhibitorów, poprzez odpowiednią obserwację kliniczną i badania laboratoryjne. Jeśli po podaniu odpowiedniej dawki nie osiąga się oczekiwanego poziomu aktywności czynnika VIII w osoczu lub jeśli krwawienie nie ustępuje po podaniu odpowiedniej dawki, należy przeprowadzić badanie w celu wykrycia obecności inhibitorów czynnika VIII. U pacjentów z wysokimi poziomami inhibitorów terapia czynnikiem VIII może okazać się nieskuteczna, w takim przypadku należy rozważyć inne opcje leczenia. Leczenie takich pacjentów powinno być prowadzone pod nadzorem lekarza posiadającego doświadczenie w leczeniu pacjentów z hemofilią i inhibitorami czynnika VIII.
Powikłania sercowo-naczyniowe
U pacjentów z istniejącymi czynnikami ryzyka chorób układu sercowo-naczyniowego terapia zastępcza czynnikiem VIII może zwiększyć to ryzyko.
Powikłania związane z użyciem kaniuli
W przypadku potrzeby dostępu do żyły centralnej należy wziąć pod uwagę ryzyko powikłań związanych z użyciem urządzenia, w tym infekcji lokalnych, bakteriemii oraz zakrzepicy w miejscu wprowadzenia kaniuli.
Bezpieczeństwo wirusowe
Standardowe środki ostrożności mające na celu zapobieganie infekcjom wynikającym ze stosowania leków pochodzących z ludzkiej krwi lub osocza obejmują selekcję dawców, badanie indywidualnych partii i puli osocza na obecność specyficznych markerów infekcji oraz skuteczne procedury produkcyjne mające na celu inaktywację/eliminację wirusów.
Pomimo to, podczas podawania leków pochodzących z ludzkiej krwi lub osocza, nie można całkowicie wykluczyć możliwości przeniesienia patogenów, w tym nieznanych lub nowych wirusów i innych patogenów.
Środki, które są stosowane, uważane są za skuteczne wobec wirusów otoczkowych, takich jak wirus HIV (ludzkiego wirusa niedoboru odporności), wirus zapalenia wątroby typu B (HBW) i wirus zapalenia wątroby typu C (HCV), a także wobec wirusów nieotoczkowych, takich jak wirus zapalenia wątroby typu A (HAV). Środki te mogą mieć ograniczoną skuteczność wobec wirusów nieotoczkowych, takich jak parwowirus B19. Zakażenie parwowirusem B19 może mieć ciężki przebieg u kobiet w ciąży (zakażenie płodu) oraz u pacjentów z niedoborem odporności lub nasilonym erytropoezą (np. w anemii hemolitycznej).
Należy rozważyć możliwość przeprowadzenia odpowiedniej szczepionki (przeciwko wirusom HAV i HBW) u pacjentów, którym wielokrotnie lub powtarzalnie stosuje się preparaty czynnika VIII pochodzące z ludzkiego osocza.
W celu zachowania powiązania między pacjentem a serią preparatu, zaleca się pilnie rejestrować nazwę i numer serii preparatu Emoklot za każdym razem, gdy jest on podawany.
Dzieci
Brak danych specyficznych dotyczących dzieci.
Stosowanie w okresie ciąży lub karmienia piersią.
Badania wpływu czynnika VIII na funkcję rozrodczą u zwierząt nie były prowadzone. Ponieważ częstość występowania hemofilii typu A u kobiet jest niska, brak jest doświadczenia w stosowaniu czynnika VIII u kobiet w ciąży i karmiących piersią. Dlatego czynnik VIII należy stosować w okresie ciąży i karmienia piersią tylko w przypadku wyraźnych wskazań.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Emoklot nie wpływa na zdolność prowadzenia pojazdów i obsługiwanie innych urządzeń mechanicznych.
Sposób stosowania i dawki.
Leczenie należy rozpoczynać pod kontrolą lekarza posiadającego doświadczenie w leczeniu hemofilii.
Kontrola leczenia
Podczas leczenia zaleca się odpowiednie oznaczanie poziomów czynnika VIII w celu ustalenia odpowiedniej dawki oraz częstotliwości powtórnego podawania. Odpowiedź na czynnik VIII może różnić się u poszczególnych pacjentów, co objawia się różnymi okresami półtrwania i czasem wydzielania z organizmu. U pacjentów z niedożywieniem lub nadwagą może być konieczna korekta dawki obliczanej na podstawie masy ciała.
W przypadku dużych zabiegów operacyjnych konieczne jest szczególnie dokładne monitorowanie terapii zastępczej poprzez przeprowadzanie analiz krzepnięcia krwi (aktywność czynnika VIII w osoczu krwi).
W przypadku stosowania jednostopniowego testu krzepnięcia in vitro opartego na czasie tromboplastynowym częściowym (APTT) do oznaczania aktywności czynnika VIII w próbkach krwi pacjentów, wyniki aktywności czynnika VIII mogą znacznie zależeć zarówno od rodzaju użytego odczynnika APTT, jak i od zastosowanego standardu porównawczego. Mogą również występować istotne różnice między wynikami analizy uzyskanymi za pomocą jednostopniowego testu krzepnięcia opartego na APTT a analizą chromogenną zgodnie z Europejską Farmakopeą. Jest to szczególnie istotne przy zmianie laboratorium i/lub odczynników stosowanych do analizy.
Dozowanie
Dawkowanie i trwanie terapii zastępczej zależy od stopnia nasilenia niedoboru czynnika VIII, lokalizacji i nasilenia krwawienia, jak również od stanu klinicznego pacjenta.
Ilość podawanych jednostek czynnika VIII wyrażana jest w jednostkach międzynarodowych (MI), odpowiadających aktualnemu standardowi WHO dla preparatów czynnika VIII. Aktywność czynnika VIII w osoczu wyrażana jest w procentach (w stosunku do osocza normalnego człowieka) lub w jednostkach międzynarodowych (w stosunku do międzynarodowego standardu dla czynnika VIII w osoczu).
Aktywność jednej jednostki międzynarodowej (MI) czynnika VIII odpowiada ilości czynnika VIII zawartego w 1 ml osocza normalnego człowieka.
Leczenie na żądanie
Obliczenie wymaganej dawki czynnika VIII opiera się na danych empirycznych. 1 jednostka międzynarodowa (MI) czynnika VIII na 1 kg masy ciała zwiększa aktywność czynnika VIII w osoczu o 1,5–2% aktywności normalnej.
Wymaganą dawkę oblicza się według następującego wzoru:
Wymagana liczba jednostek = masa ciała (kg) × pożądane zwiększenie czynnika VIII (%) (MI/dl) × 0,4
Ilość podawanego leku oraz częstotliwość podawania powinny być zawsze dostosowane do skuteczności klinicznej w każdym konkretnym przypadku.
W poniższych przypadkach krwawień aktywność czynnika VIII nie powinna być niższa niż wskazany poziom aktywności w osoczu krwi (w % normy) w odpowiednim okresie. Tabelę 2 można stosować jako wytyczne dotyczące dawkowania w przypadku krwawień i zabiegów chirurgicznych.
Tabela 2
| Stopień krwawienia / rodzaj zabiegu chirurgicznego |
Wymagany poziom czynnika VIII (%) (j.m./dL) |
Częstotliwość podawania (godziny) / czas trwania leczenia (dni) |
| Krwawienie |
||
| Wczesny krwotok do stawu, krwotok do mięśni lub krwawienie z jamy ustnej |
20–40 |
Podawać co 12–24 godziny przez co najmniej 1 dzień do ustania krwawienia, o czym świadczy ustąpienie bólu lub gojenie. |
| Bardziej nasilony krwotok do stawu, krwotok do mięśni lub siniaka |
30–60 |
Podawać co 12–24 godziny przez 3–4 dni lub dłużej, aż ustąpi ból i ostre niepełnosprawność. |
| Krwawienie zagrażające życiu |
60–100 |
Podawać co 8–24 godziny do czasu wyeliminowania zagrożenia życia. |
| Zabiegi operacyjne |
||
| Małe zabiegi operacyjne, w tym ekstrakcja zęba |
30–60 |
Co 24 godziny przez co najmniej 1 dzień do czasu gojenia. |
| Duże zabiegi operacyjne |
80–100 (przed i po zabiegu operacyjnym) |
Podawać co 8–24 godziny do właściwego gojenia rany; następnie kontynuować leczenie przez co najmniej 7 dni, utrzymując aktywność czynnika VIII na poziomie 30–60% (30–60 j.m./dL). |
Profilaktyka
Długotrwała profilaktyka krwawień u pacjentów z ciężką formą hemofilii A polega na podawaniu dawek od 20 do 40 MI czynnika VIII na 1 kg masy ciała co 2–3 doby. W niektórych przypadkach, szczególnie u młodszych pacjentów, mogą być wymagane krótsze odstępy między dawkami lub wyższe dawki.
Sposób stosowania
Stosować dożylnie w formie wstrzyknięcia lub powolnej infuzji.
W przypadku wstrzyknięcia dożylnego zaleca się podawanie leku w ciągu 3–5 minut, kontrolując jednocześnie częstość pulsu u pacjenta. W przypadku zwiększenia częstości pulsu należy przerwać podawanie lub zmniejszyć jego szybkość.
Szybkość infuzji należy dobierać indywidualnie dla każdego pacjenta.
Odtworzenie proszku za pomocą rozpuszczalnika
- Doprowadzić buteleczkę z proszkiem i buteleczkę z rozpuszczalnikiem do temperatury pokojowej.
- Utrzymywać temperaturę pokojową przez cały czas procesu odtwarzania (rozpuszczania) (maksymalnie 10 minut).
- Usunąć ochronne nakrywki z buteleczki z proszkiem i buteleczki z rozpuszczalnikiem.
- Przetrzeć powierzchnię zatyczek obu buteleczek alkoholem etylowym.
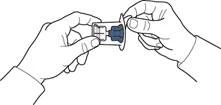
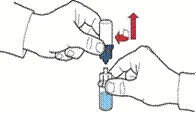
- Otworzyć opakowanie urządzenia zgodnie z rysunkiem A, nie dotykając wewnętrznej części opakowania (rys. A).
- Nie wyjmować urządzenia z opakowania.
- Odwrócić opakowanie z urządzeniem i nakłuć plastikowym kolcem urządzenia zatyczkę buteleczki z rozpuszczalnikiem, tak aby niebieska część urządzenia połączyła się z buteleczką z rozpuszczalnikiem (rys. B).
- Usunąć opakowanie urządzenia, trzymając je za krawędź i nie dotykając samego urządzenia (rys. C).
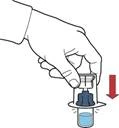
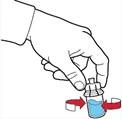
- Upewnić się, że buteleczka z proszkiem znajduje się na stabilnej powierzchni; odwrócić buteleczkę z rozpuszczalnikiem z podłączonym urządzeniem, tak aby buteleczka z rozpuszczalnikiem znalazła się nad urządzeniem; nacisnąć przezroczysty adapter na zatyczce buteleczki z proszkiem, aby plastikowy kolec nakłuł zatyczkę buteleczki z proszkiem; rozpuszczalnik zacznie automatycznie przepływać do buteleczki z proszkiem (rys. D).
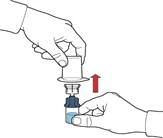
- Po przetoczeniu rozpuszczalnika odkręcić niebieską część systemu, do której podłączona jest buteleczka z rozpuszczalnikiem, i usunąć ją (rys. E).
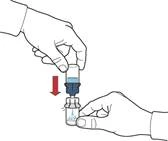
- Delikatnie wstrząsać buteleczką aż do całkowitego rozpuszczenia proszku (rys. F).
- Nie wstrząsać buteleczki zbyt intensywnie, aby uniknąć powstawania piany.
| Rys. A |
Rys. B |
|
|
|
| Rys. B |
Rys. C |
|
|
|
| Rys. D |
Rys. E |
|
|
|
Wprowadzenie roztworu
Po odtworzeniu roztwór może zawierać niewielką ilość drobnych nitek lub cząstek.
Przed podaniem roztwór należy wizualnie sprawdzić pod kątem obecności cząstek lub zmiany barwy. Roztwór powinien być przejrzysty lub lekko opalescencyjny. Nie należy stosować roztworu, jeśli jest on zamglony lub zawiera osad.
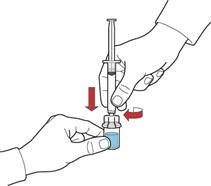
- Napełnij strzykawkę powietrzem, wysuwając tłok; podłącz strzykawkę do urządzenia i wprowadź powietrze ze strzykawki do fiolki z odtworzonym roztworem (rys. E).
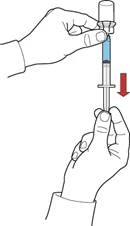
- Trzymając tłok, odwróć układ, tak aby fiolka z odtworzonym roztworem znalazła się nad urządzeniem. Powoli wysuwając tłok, napełnij strzykawkę koncentratem (rys. Ż).
- Odłącz strzykawkę, obracając ją przeciwnie do ruchu wskazówek zegara.
- Sprawdź wizualnie roztwór w strzykawce, który powinien być przejrzysty lub lekko opalescencyjny i nie powinien zawierać cząstek.
- Podłącz do strzykawki igłę motylkową i podaj lek dożylnie za pomocą infuzji lub powolnego wstrzyknięcia.
| Rys. E |
Rys. Ż |
|
|
|
Po otwarciu zawartość fiolki należy natychmiast wykorzystać.
Roztwór po rekonstytucji, napełniony do strzykawki, należy natychmiast wykorzystać.
Zawartość fiolki należy wykorzystać wyłącznie do jednego wstrzyknięcia.
Zabrania się stosowania leku po upływie okresu ważności wskazanego na opakowaniu.
Nie wykorzystany lek lub odpady powstałe po jego użyciu należy zniszczyć zgodnie z lokalnymi wymaganiami.
Dzieci.
Nie ustalono bezpieczeństwa i skuteczności stosowania leku Emoklot u dzieci poniżej 12. roku życia. Dostępne dane opisano w sekcji „Farmakodynamika”, jednak nie można podać zaleceń dotyczących dawkowania.
Dawkowanie dla młodzieży (12–18 lat) dla każdego wskazania oblicza się według masy ciała.
Przedawkowanie.
Nie odnotowano żadnych objawów przedawkowania czynnikiem krzepnięcia krwi człowieka VIII.
Działania niepożądane.
Krótki opis profilu bezpieczeństwa
Zwiększona wrażliwość lub reakcje alergiczne (w tym obrzęk naczynioruchowy, pieczenie i ostry ból w miejscu wlewu, dreszcze, flush, pokrzywka uogólniona, ból głowy, wysypka, hipotensja, letargia, nudności, niepokój, tachykardia, uczucie ściskania w klatce piersiowej, mrowienie, wymioty i świsty w czasie oddychania) występują rzadko i w niektórych przypadkach mogą postępować w kierunku ciężkiej anafilaksji (w tym wstrząsu).
Obserwowano również podwyższenie temperatury ciała.
U pacjentów z hemofilią A leczonych lekami zawierającymi czynnik VIII, w tym Emoklot, mogą rozwijać się przeciwciała neutralizujące (inhibitory). Pojawienie się inhibitorów u pacjentów z hemofilią A objawia się niewystarczającą odpowiedzią kliniczną. W takich przypadkach zaleca się skontaktowanie się ze specjalistycznym ośrodkiem hemofilii.
Informacje dotyczące bezpieczeństwa związanych z czynnikami przenoszonymi drogą zakaźną znajdują się w sekcji „Szczególne ostrzeżenia i środki ostrożności stosowania”.
Lista działań niepożądanych w postaci tabeli
Działania niepożądane, które mogą wystąpić podczas stosowania ludzkiego czynnika krzepnięcia krwi VIII, przedstawiono w tabeli 3 zgodnie z klasyfikacją układów narządów MedDRA [Medyczny Słownik do Celów Regulacyjnych] oraz terminów preferowanych.
Częstość występowania szacowana jest według następujących kategorii umownych: bardzo często (≥ 1/10); często (≥ 1/100 do < 1/10); nieczęsto (≥ 1/1 000 do < 1/100); rzadko (≥ 1/10 000 do < 1/1 000); bardzo rzadko (< 1/10 000); częstość nieznana (niemożliwe do oszacowania na podstawie dostępnych danych).
Potwierdzone dane dotyczące częstości działań niepożądanych z badań klinicznych są nieobecne.
Poniższe dane oparte są na profilu bezpieczeństwa ludzkiego czynnika krzepnięcia krwi VIII i częściowo obserwowano je w okresie pogwarancyjnym (doświadczenie pogwarancyjne z lekiem); ponieważ doniesienia o działaniach niepożądanych w okresie pogwarancyjnym są dobrowolne i dotyczą populacji nieznanej liczby, niemożliwe jest oszacowanie częstości tych reakcji.
Tabela 3
| Klasa układu narządów według MedDRA |
Reakcja niepożądana |
Częstotliwość |
| Zaburzenia układu krwiotwórczego i limfatycznego |
Pojawienie się inhibitorów czynnika VIII |
Nieczęsto (PRL) ** Bardzo często (PRN) ** |
| Zaburzenia układu odpornościowego |
Podwyższona wrażliwość |
Nieznana |
| Reakcje alergiczne (nadwrażliwość)* |
Nieznana |
|
| Reakcja anafilaktyczna |
Nieznana |
|
| Szok anafilaktyczny |
Nieznana |
|
| Zaburzenia psychiczne |
Niepokój |
Nieznana |
| Zaburzenia układu nerwowego |
Ból głowy |
Nieznana |
| Lethargia |
Nieznana |
|
| Paraesthesia |
Nieznana |
|
| Zaburzenia serca |
Tachykardia |
Nieznana |
| Zaburzenia naczyniowe |
Zapłony |
Nieznana |
| Hipotonia |
Nieznana |
|
| Zaburzenia oddechowe, piersiowe i śródpiersia |
Świszczący oddech* |
Nieznana |
| Zaburzenia przewodu pokarmowego |
Nudności |
Nieznana |
| Wymioty |
Nieznana |
|
| Zaburzenia skóry i tkanki podskórnej |
Angioobrzęk |
Nieznana |
| Uogólnione pokrzywki (pokrzywka)* |
Nieznana |
|
| Wysypka (pokrzywka)* |
Nieznana |
|
| Zaburzenia ogólne i reakcje w miejscu podania |
Ból pieczenia w miejscu infuzji (ból w miejscu infuzji)* |
Nieznana |
| Ostry ból w miejscu infuzji (ból w miejscu infuzji)* |
Nieznana |
|
| Drżenie |
Nieznana |
|
| Ściszenie w klatce piersiowej (niedogodność w klatce piersiowej)* |
Nieznana |
|
| Piroksja |
Nieznana |
* Terminy niższego rzędu MedDRA, które dokładniej opisują te działania niepożądane; w nawiasach podano termin preferowanego użycia MedDRA.
** Dane dotyczące częstości oparte są na wynikach badań wszystkich leków zawierających czynnik VIII przeprowadzonych u pacjentów z ciężką hemofilią A. PRL – pacjenci wcześniej leczeni, PRN – pacjenci wcześniej nieleczony.
Dzieci
Brak danych specyficznych dotyczących dzieci.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawowe przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku za pośrednictwem Zautomatyzowanego Systemu Informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
3 lata.
Roztwórnik (woda do wstrzykiwań) – 5 lat.
Preparat należy użyć natychmiast po rozpuszczeniu.
Warunki przechowywania.
Przechowywać w lodówce (w temperaturze od 2 do 8 ℃). Nie zamrażać. Przechowywać we wstępnej opakowaniu w celu ochrony przed światłem, w miejscu niedostępnym dla dzieci.
Przed zastosowaniem leku i w trakcie jego okresu ważności fiolkę z proszkiem można przechowywać w temperaturze pokojowej nie przekraczającej 25 ℃ przez okres do 6 miesięcy z rzędu. Po upływie tego okresu fiolkę z proszkiem należy zniszczyć.
Po przechowywaniu fiolki z proszkiem w temperaturze pokojowej zabrania się dalszego przechowywania jej w lodówce.
Dane dotyczące daty rozpoczęcia przechowywania leku w temperaturze pokojowej należy wpisać na oryginalnym opakowaniu leku w specjalnie do tego przeznaczonym miejscu.
Niezgodność.
Z powodu braku badań dotyczących zgodności ten lek nie może być mieszany z innymi lekami.
Należy używać wyłącznie zestawu do wstrzykiwań/infuzji dołączanego do opakowania, ponieważ leczenie może być nieskuteczne z powodu adsorpcji ludzkiego czynnika VIII krzepnięcia krwi na wewnętrznych powierzchniach innego urządzenia do podania.
Opakowanie.
500 JE lub 1000 JE w fiolce nr 1 w zestawie z rozcieńczalnikiem (woda do wstrzykiwań) 10 ml w fiolce nr 1 oraz zestawem do rozpuszczenia i podania w tekturowym pudełku.
Kategoria wydania. Na receptę.
Producent.
KEDRION S.P.A.
Miejsce produkcji i adres siedziby producenta.
VIA PROVINCIALE (lok. BOLOGNANA) - 55027 GALLICANO (LU), Włochy.