BRIUMVI
Włochy
Spis treści
- Ulotka: informacje dla pacjenta
- Briumvi 150 mg stężenie do sporządzenia roztworu do infuzji
- 1. Co to jest Briumvi i do czego służy
- 2. Co należy wiedzieć przed podaniem Briumvi
- 3. Jak Briumvi jest podawane
- 4. Możliwe działania niepożądane
- 5. Jak przechowywać Briumvi
- 6. Zawartość opakowania i inne informacje
- Poniżej zamieszczone informacje przeznaczone są wyłącznie dla personelu medycznego:
Ulotka: informacje dla pacjenta
Briumvi 150 mg stężenie do sporządzenia roztworu do infuzji
ublituksimab

Lek objęty dodatkowym nadzorem. Umożliwia to szybkie wykrywanie nowych informacji dotyczących bezpieczeństwa. Możesz pomóc, zgłaszając wszelkie zaobserwowane działania niepożądane podczas przyjmowania tego leku. Zobacz koniec punktu 4, aby uzyskać informacje na temat sposobu zgłaszania działań niepożądanych.
Przed podaniem tego leku należy dokładnie przeczytać ulotkę,
ponieważ zawiera ona ważne informacje dla Ciebie.
- Zachowaj tę ulotkę. Może się okazać konieczne ponowne zapoznanie się z jej treścią.
- W przypadku jakichkolwiek wątpliwości skontaktuj się z lekarzem.
- Jeśli wystąpi jakiekolwiek działanie niepożądane, w tym te niewymienione w tej ulotce, skontaktuj się z lekarzem lub pielęgniarką. Zobacz punkt 4.
Zawartość ulotki
- Co to jest Briumvi i do czego służy
- Co należy wiedzieć przed podaniem Briumvi
- Jak stosuje się Briumvi
- Możliwe działania niepożądane
- Jak przechowywać Briumvi
- Zawartość opakowania i inne informacje
1. Co to jest Briumvi i do czego służy
Co to jest Briumvi
Briumvi zawiera substancję czynną ublituksimab, rodzaj białka zwanego przeciwciałem monoklonalnym.
Przeciwciała działają wiążąc się do określonych celów w organizmie.
Do czego służy Briumvi
Briumvi stosuje się w leczeniu dorosłych pacjentów z odnowionymi postaciami stwardnienia rozsianego (RMS), w których występują nawroty (przypady nasilenia objawów) naprzemiennie z okresami mniejszych objawów lub ich brakiem.
Co to jest stwardnienie rozsiane
Stwardnienie rozsiane (SM) dotyczy ośrodkowego układu nerwowego, szczególnie nerwów w mózgu i rdzeniu kręgowym. W SM niektóre białe krwinki zwane komórkami B, które są częścią układu odpornościowego (mechanizmu obronnego organizmu), działają nieprawidłowo i atakują ochronną warstwę (tzw. osłonkę mielinową) otaczającą komórki nerwowe, powodując ich uszkodzenie i stan zapalny. Zniszczenie osłonki mielinowej uniemożliwia prawidłowe funkcjonowanie nerwów i powoduje objawy SM. Objawy SM zależą od obszaru ośrodkowego układu nerwowego, który jest zaangażowany, i mogą obejmować problemy z równowagą i chodzeniem, osłabienie mięśni, mrowienie, podwójne widzenie, zamazanie widzenia, słabszą koordynację oraz problemy z pęcherzem moczowym.
W odnowionych postaciach SM pacjent doświadcza powtarzających się ataków objawów (nawrotów), które mogą pojawić się nagle, w ciągu kilku godzin, lub powoli, w ciągu kilku dni. Między nawrotami objawy ustępują lub poprawiają się, ale uszkodzenia mogą się kumulować i prowadzić do trwałej niepełnosprawności.
Jak działa Briumvi?
Briumvi działa wiążąc się do celu znajdującego się na powierzchni komórek B, zwanego CD20. Komórki B to rodzaj białych krwinek, które są częścią układu odpornościowego. W stwardnieniu rozsianym układ odpornościowy atakuje ochronną warstwę otaczającą komórki nerwowe. Komórki B są zaangażowane w ten proces. Briumvi atakuje i eliminuje komórki B, co zmniejsza ryzyko nawrotu, łagodzi objawy i spowalnia postęp choroby.
2. Co należy wiedzieć przed podaniem Briumvi
Nie należy podawać Briumvi:
- jeśli jest uczulony na ublituksimab lub którykolwiek z innych składników tego leku (wymienionych w punkcie 6),
- jeśli ma ciężkie zakażenie,
- jeśli lekarz poinformował o poważnych problemach z układem odpornościowym,
- lub jeśli ma nowotwór.
Jeśli nie jest pewien, skonsultuj się z lekarzem przed podaniem Briumvi.
Ostrzeżenia i środki ostrożności
Skonsultuj się z lekarzem przed podaniem Briumvi, jeśli dotyczy go którykolwiek z poniższych stanów. Lekarz może postanowić odłożyć leczenie Briumvi lub stwierdzić, że nie może otrzymać Briumvi, jeśli:
- ma zakażenie. Lekarz będzie czekać, aż zakażenie ustąpi, zanim poda Briumvi;
- miał wcześniej zapalenie wątroby typu B lub jest nosicielem wirusa zapalenia wątroby typu B. Leki takie jak Briumvi mogą bowiem spowodować reaktywację wirusa zapalenia wątroby typu B. Przed leczeniem Briumvi lekarz sprawdzi, czy nie istnieje ryzyko rozwoju zakażenia wirusem zapalenia wątroby typu B. Pacjenci, którzy mieli zapalenie wątroby typu B lub są nosicielami wirusa zapalenia wątroby typu B, będą poddawani badaniom krwi i będą monitorowani przez lekarza w celu wykrycia ewentualnych oznak zakażenia wirusem zapalenia wątroby typu B;
- jeśli w ostatnim czasie otrzymał jakąkolwiek szczepionkę lub może otrzymać szczepionkę w najbliższym czasie;
- ma nowotwór lub miał go wcześniej. Lekarz może zdecydować o odłożeniu leczenia.
Reakcje związane z wlewaniem
- Reakcje związane z wlewaniem są najczęstszym niepożądaniem leczenia Briumvi. Są to reakcje alergiczne, które pojawiają się podczas podawania leku lub krótko po jego podaniu. Mogą one być poważne.
- Objawy reakcji związanej z wlewaniem mogą obejmować:
- swędzenie skóry
- pokrzywkę
- zaczerwienienie twarzy lub skóry
- podrażnienie gardła
- trudności w oddychaniu
- obrzęk języka lub gardła
- świsty oddechowe
- dreszcze
- gorączkę
- ból głowy
- zawroty głowy
- uczucie omdlenia
- nudności
- ból brzucha (ból żołądka)
- przyspieszone bicie serca.
- Natychmiast powiadomić lekarza lub pielęgniarkę, jeśli wystąpi lub podejrzewa się reakcję związaną z wlewaniem. Reakcje związane z wlewaniem mogą wystąpić podczas wlewania lub do 24 godzin po wlewaniu.
- Aby zmniejszyć ryzyko reakcji związanych z wlewaniem, lekarz poda inne leki przed każdym wlewaniem Briumvi (patrz punkt 3) i będzie dokładnie obserwował podczas wlewania.
- W przypadku reakcji związanej z wlewaniem może być konieczne przerwanie wlewania lub spowolnienie tempa wlewania.
Zakażenia
- Skonsultuj się z lekarzem przed podaniem Briumvi, jeśli ma lub podejrzewa zakażenie. Lekarz będzie czekał, aż zakażenie ustąpi, zanim poda Briumvi.
- Po podaniu Briumvi może łatwiej dojść do zakażeń, ponieważ komórki odpornościowe, na które działa Briumvi, pomagają również w walce z zakażeniami.
- Natychmiast powiadomić lekarza lub pielęgniarkę, jeśli wystąpi zakażenie lub zauważa się którykolwiek z poniższych objawów zakażenia podczas lub po leczeniu Briumvi:
- gorączka lub dreszcze
- kaszel, który nie ustępuje
- opryszcz (np. pęcherzyk na wardze, św. Antoniego lub opryszcz genitalny)
- Natychmiast powiadomić lekarza lub pielęgniarkę, jeśli uważa się, że SM się nasila lub pojawiają się nowe objawy, ponieważ istnieje bardzo rzadkie i potencjalnie śmiertelne zakażenie mózgu, zwane „postępującą multifokalną leukoenkefalopatią” (PML), które może powodować objawy podobne do objawów SM. PML może wystąpić u pacjentów przyjmujących leki takie jak Briumvi i inne leki stosowane w leczeniu SM.
- Poinformować partnera lub osobę opiekującą się pacjentem o leczeniu Briumvi, ponieważ może on zauważyć objawy PML, których pacjent nie dostrzega, takie jak: lapsusy pamięci, trudności w myśleniu, trudności w chodzeniu, utrata wzroku lub zmiany w mowie, które lekarz może chcieć zbadać.
Szczepienia
- Poinformować lekarza, jeśli w ostatnim czasie otrzymano jakąkolwiek szczepionkę lub może się otrzymać szczepionkę w najbliższym czasie.
- Lekarz sprawdzi, czy potrzebuje szczepień przed rozpoczęciem leczenia Briumvi. Wszelkie szczepienia żywymi lub osłabionymi szczepionkami żywymi należy podać co najmniej 4 tygodnie przed rozpoczęciem leczenia Briumvi. Podczas leczenia Briumvi nie należy podawać szczepionek żywych ani osłabionych szczepionek żywych, dopóki lekarz nie stwierdzi, że układ odpornościowy nie jest już osłabiony.
- Wszelkie szczepienia szczepionkami inaktywowanymi należy podać co najmniej 2 tygodnie przed rozpoczęciem leczenia Briumvi, o ile to możliwe. Jeśli chce się zaszczepić szczepionkami inaktywowanymi podczas leczenia Briumvi, należy skonsultować się z lekarzem.
Dzieci i młodzież
Briumvi nie jest przeznaczone do stosowania u dzieci i młodzieży poniżej 18. roku życia, ponieważ nie zostało jeszcze zbadane w tej grupie wiekowej.
Inne leki i Briumvi
Poinformuj lekarza lub farmaceutę, jeśli przyjmuje, niedawno przyjmował lub może przyjmować inne leki. W szczególności poinformuj lekarza:
- jeśli przyjmuje, niedawno przyjmował lub może przyjmować leki wpływające na układ odpornościowy, takie jak chemioterapeutyki, leki immunosupresyjne (z wyjątkiem glikokortykosteroidów) lub inne leki stosowane w leczeniu SM, ponieważ mogą one mieć addytywny wpływ na układ odpornościowy;
- jeśli planuje szczepienia (patrz powyżej punkt „Ostrzeżenia i środki ostrożności”).
Jeśli którykolwiek z tych stanów dotyczy (lub jeśli nie jest pewien), skonsultuj się z lekarzem przed podaniem Briumvi.
Ciąża i karmienie piersią
- Jeśli jest w ciąży, podejrzewa ciążę lub planuje ciążę, poinformuj lekarza przed podaniem Briumvi. Briumvi może bowiem przenikać przez łożysko i wpływać na dziecko.
- Jeśli jest w ciąży, nie należy stosować Briumvi, chyba że porozmawiano z lekarzem. Lekarz oceni stosunek korzyści z leczenia Briumvi dla pacjentki do ryzyka dla dziecka.
- Jeśli po otrzymaniu Briumvi w czasie ciąży urodziło się dziecko, ważne jest, aby poinformować pediatrę, aby mógł doradzić, kiedy dziecko będzie mogło zostać zaszczepione.
- Nie wiadomo, czy Briumvi przechodzi do mleka matki. Skonsultuj się z lekarzem, aby poznać najlepszy sposób karmienia dziecka podczas stosowania Briumvi.
Antykoncepcja u kobiet
Jeśli może mieć dzieci (czyli jest w stanie zajść w ciążę), należy stosować środki antykoncepcyjne:
- podczas leczenia Briumvi oraz
- przez co najmniej 4 miesiące po ostatnim wlewaniu Briumvi.
Kierowanie pojazdami i obsługa maszyn
Najprawdopodobniej Briumvi nie wpływa na zdolność kierowania pojazdami i obsługi maszyn.
Briumvi zawiera sód
Ten lek zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, co oznacza, że jest zasadniczo „bezsodowy”.
3. Jak Briumvi jest podawane
Briumvi będzie podawane przez lekarza lub doświadczoną pielęgniarkę, którzy będą dokładnie obserwować stan pacjenta podczas podawania leku, na wypadek wystąpienia działań niepożądanych. Lek Briumvi jest zawsze podawany za pomocą wlewu (infuzji dożyłowej).
Leki podawane przed leczeniem Briumvi
Przed podaniem Briumvi będą podawane inne leki w celu zapobiegania lub ograniczania możliwych działań niepożądanych, takich jak reakcje związane z infuzją (informacje dotyczące reakcji związanych z infuzją znajdują się w punktach 2 i 4).
Przed każdą infuzją otrzyma się kortykosteroid oraz lek przeciwhistaminowy, a także mogą zostać podane leki obniżające gorączkę.
W jakich dawkach i jak często będzie podawane Briumvi
- Pierwsza dawka Briumvi wyniesie 150 mg. Infuzja ta potrwa 4 godziny.
- Druga dawka Briumvi wyniesie 450 mg i zostanie podana 2 tygodnie po pierwszej dawce. Infuzja ta potrwa 1 godzinę.
- Kolejne dawki Briumvi będą wynosić 450 mg i będą podawane 24 tygodnie po pierwszej dawce, a następnie co 24 tygodnie. Infuzje te będą trwały po 1 godzinie.
Jak jest podawane Briumvi
- Briumvi będzie podawane przez lekarza lub pielęgniarkę. Przed podaniem lek Briumvi musi zostać rozcieńczony. Rozcieńczenie wykona personel medyczny. Lek będzie podawany za pomocą infuzji do żyły (infuzji dożyłowej).
- Pacjent będzie dokładnie obserwowany podczas infuzji Briumvi oraz przez co najmniej 1 godzinę po podaniu dwóch pierwszych infuzji, na wypadek wystąpienia działań niepożądanych, takich jak reakcje związane z infuzją. W przypadku wystąpienia reakcji związanej z infuzją, tempo podawania może zostać spowolnione, a infuzja może zostać tymczasowo wstrzymana lub całkowicie przerwana, w zależności od jej nasilenia (informacje dotyczące reakcji związanych z infuzją znajdują się w punktach 2 i 4).
Jeśli pacjent opuści infuzję Briumvi
- Jeśli pacjent opuści infuzję Briumvi, powinien jak najszybciej umówić się z lekarzem na jej podanie. Nie należy czekać na następną zaplanowaną infuzję.
- Aby osiągnąć pełny efekt terapeutyczny leku Briumvi, ważne jest, aby każda infuzja była podawana zgodnie z ustalonym harmonogramem.
Jeśli pacjent przerwie leczenie Briumvi
- Ważne jest, aby kontynuować leczenie do momentu, w którym pacjent i lekarz uznają, że nadal przynosi ono korzyści.
- Niektóre działania niepożądane mogą być związane z niskim poziomem komórek B. Mogą one nadal występować nawet po zakończeniu leczenia Briumvi, aż do czasu, gdy poziom komórek B wróci do normy.
- Przed rozpoczęciem przyjmowania jakichkolwiek innych leków należy poinformować lekarza o dacie ostatniej infuzji Briumvi.
Jeśli pacjent ma jakiekolwiek wątpliwości dotyczące stosowania tego leku, powinien skonsultować się z lekarzem.
4. Możliwe działania niepożądane
Tak jak wszystkie leki, ten lek może powodować działania niepożądane, choć nie u wszystkich osób one występują.
Poniżej wymieniono działania niepożądane zgłaszane dla Briumvi:
Działania niepożądane poważne
Reakcje związane z wlewem
- Reakcje związane z wlewem są najczęstszym działaniem niepożądanym leczenia Briumvi (bardzo często: może dotyczyć więcej niż 1 osoby na 10). W większości przypadków są to reakcje łagodne, ale mogą również wystąpić reakcje poważne.
- Natychmiast powiadom lekarza lub pielęgniarkę, jeśli pojawią się u Ciebie jakiekolwiek objawy reakcji związanej z wlewem, podczas wlewu lub w ciągu 24 godzin po jego zakończeniu. Objawy mogą obejmować, ale nie są do tego ograniczone:
- świąd skóry
- pokrzywkę
- zaczerwienienie twarzy lub skóry
- podrażnienie gardła
- trudności w oddychaniu
- obrzęk języka lub gardła
- świsty podczas oddychania
- dreszcze
- gorączkę
- ból głowy
- zawroty głowy
- uczucie omdlenia
- nudności
- ból brzucha (ból żołądka)
- przyśpieszone bicie serca.
- Jeśli wystąpi reakcja związana z wlewem, podane zostaną Ci leki do jej leczenia i może być konieczne spowolnienie lub przerwanie wlewu. Po ustąpieniu reakcji wlew może być wznowiony. Jeśli reakcja związana z wlewem będzie stanowiła zagrożenie dla życia, lekarz całkowicie odstawi leczenie Briumvi.
Infekcje
- Po podaniu Briumvi możesz łatwiej nabawić się infekcji, niektóre z nich mogą być poważne. U pacjentów leczonych Briumvi z powodu SM zaobserwowano następujące infekcje:
- Bardzo często (może dotyczyć więcej niż 1 osoby na 10)
- infekcje dróg oddechowych górnych (infekcje nosa i gardła)
- infekcje dróg oddechowych
- Często (może dotyczyć do 1 osoby na 10)
- infekcje dróg oddechowych dolnych (infekcje płuc, takie jak zapalenie oskrzeli lub zapalenie płuc)
- infekcje spowodowane wirusem Herpes (np. opryszczka wargowa lub półpaśca)
- Nieczoście (może dotyczyć do 1 osoby na 100)
- infekcja opon mózgowo-rdzeniowych (zapalenie opon mózgowych), infekcja mózgu (zapalenie mózgu) lub obu naraz (zapalenie opon mózgowo-rdzeniowych i mózgu)
- Natychmiast powiadom lekarza lub pielęgniarkę, jeśli zauważysz u siebie jakiekolwiek z poniższych objawów infekcji:
- gorączkę lub dreszcze
- kaszel, który nie ustępuje
- objawy opryszczki (np. opryszczka wargowa, półpaśce lub opryszczka narządów płciowych)
- ból głowy towarzyszący gorączce, sztywności szyi, nadwrażliwości na światło, nudnościom, dezorientacji, napadom padaczkowym, zaburzeniom osobowości, niestabilności (ataksji), zaburzeniom świadomości i/lub śpiączce. Mogą to być objawy infekcji opon mózgowo-rdzeniowych (zapalenie opon mózgowych), infekcji mózgu (zapalenie mózgu) lub obu naraz (zapalenie opon mózgowo-rdzeniowych i mózgu), które mogą prowadzić do śmierci.
Lekarz poczeka, aż infekcja ustąpi, zanim poda Ci Briumvi.
Inne działania niepożądane
Często (może dotyczyć do 1 osoby na 10)
- neutropenia (obniżone stężenie neutrofili, rodzaj białych krwinek)
- ból kończyn (ramion lub nóg)
Zgłaszanie działań niepożądanych
Jeśli wystąpi u Ciebie jakiekolwiek działanie niepożądane, w tym te niewymienione w niniejszym ulotce, skontaktuj się z lekarzem lub pielęgniarką. Możesz również zgłaszać działania niepożądane bezpośrednio za pośrednictwem krajowego systemu zgłaszania wymienionego w załączniku V. Zgłaszanie działań niepożądanych może przyczynić się do uzyskania większej liczby informacji na temat bezpieczeństwa tego leku.
5. Jak przechowywać Briumvi
Przechowywać w lodówce (2 °C – 8 °C).
Briumvi będzie przechowywany przez personel medyczny w szpitalu lub klinice w następujących warunkach:
- Nie należy stosować tego leku po dacie wygaśnięcia wskazanej na zewnętrznej opakowaniu i etykiecie fiolki po napisie „Scad”. Data wygaśnięcia odnosi się do ostatniego dnia danego miesiąca.
- Lek należy przechowywać w lodówce (2 °C – 8 °C). Nie wolno go zamrażać. Fiolkę należy trzymać w opakowaniu zewnętrznym, aby chronić lek przed światłem.
Zaleca się stosowanie leku natychmiast po rozcieńczeniu. Jeżeli nie zostanie użyty natychmiast, czasy i warunki przechowywania przed użyciem są odpowiedzialnością personelu medycznego i zazwyczaj nie powinny przekraczać 24 godzin w temperaturze od 2 °C do 8 °C oraz kolejnych 8 godzin w temperaturze pokojowej.
Nie wyrzucać żadnych leków do kanalizacji. To pomoże chronić środowisko.
6. Zawartość opakowania i inne informacje
Co zawiera Briumvi
- Substancją czynną jest ublituximab. Każda fiolka zawiera 150 mg ublituximabu w 6 mL, w stężeniu 25 mg/mL.
- Pozostałe składniki to: chlorek sodu, cytrynian sodu, polisorbat 80, kwas chlorowodorowy i woda do wstrzykiwań.
Opis wyglądu Briumvi i zawartości opakowania
- Briumvi to roztwór od klarownego do mlecznego, od bezbarwnego do lekko żółtego.
- Dostarczany jest jako stężony roztwór do infuzji.
- Lek ten jest dostępny w opakowaniach zawierających 1 lub 3 fiolki (fiolki szklane zawierające 6 mL stężonego roztworu). Nie wszystkie opakowania mogą być wprowadzone do obrotu.
Właściciel pozwolenia na dopuszczenie do obrotu
Neuraxpharm Pharmaceuticals, S.L.
Avda. Barcelona 69
08970 Sant Joan Despí - Barcelona
Hiszpania
Producent
Neuraxpharm Pharmaceuticals, S.L.
Avda. Barcelona 69
08970 Sant Joan Despí
Barcelona - Hiszpania
W celu uzyskania dodatkowych informacji na temat tego leku należy skontaktować się z lokalnym przedstawicielem właściciela pozwolenia na dopuszczenie do obrotu:
België/Belgique/Belgien Lietuva
Neuraxpharm Belgium Neuraxpharm Pharmaceuticals, S.L.
Tél/Tel: +32 (0)2 732 56 95 Tel:+34 93 475 96 00
България Luxembourg/Luxemburg
Neuraxpharm Pharmaceuticals, S.L. Neuraxpharm France
Teл.: +34 93 475 96 00 Tél/Tel: +32 474 62 24 24
Česká republika Magyarország
Neuraxpharm Bohemia s.r.o. Neuraxpharm Hungary Kft.
Tel: +420 739 232 258 Tel.: +3630 464 6834
Danmark Malta
Neuraxpharm Sweden AB Neuraxpharm Pharmaceuticals, S.L.
Tlf.: +46 (0)8 30 91 41 Tel.:+34 93 475 96 00
(Sverige)
Deutschland Nederland
neuraxpharm Arzneimittel GmbH Neuraxpharm Netherlands B.V.
Tel: +49 2173 1060 0 Tel.: +31 70 208 5211
Eesti Norge
Neuraxpharm Pharmaceuticals, S.L. Neuraxpharm Sweden AB
Tel: +34 93 475 96 00 Tlf:+46 (0)8 30 91 41
(Sverige)
Ελλάδα Österreich
Brain Therapeutics IKE Neuraxpharm Austria GmbH
Τηλ: +302109931458 Tel.:+ 43 (0) 1 208 07 40
España Polska
Neuraxpharm Spain, S.L.U. Neuraxpharm Polska Sp. z.o.o.
Tel: +34 93 475 96 00 Tel.: +48 783 423 453
France Portugal
Neuraxpharm France Neuraxpharm Portugal, Unipessoal Lda
Tél: +33 1.53.62.42.90 Tel: +351 910 259 536
Hrvatska România
Neuraxpharm Pharmaceuticals, S.L. Neuraxpharm Pharmaceuticals, S.L.
Tel: +34 93 475 96 00 Tel: +34 93 475 96 00
Ireland Slovenija
Neuraxpharm Ireland Ltd Neuraxpharm Pharmaceuticals, S.L.
Tel: +353 (0)1 428 7777 Tel: +34 93 475 96 00
Ísland Slovenská republika
Neuraxpharm Sweden AB Neuraxpharm Slovakia a.s.
Sími: +46 (0)8 30 91 41 Tel: +421 255 425 562
(Svíþjóð)
Italia Suomi/Finland
Neuraxpharm Italy S.p.A. Neuraxpharm Sweden AB
Tel: +39 0736 980619 Puh/Tel: +46 (0)8 30 91 41
(Ruotsi/Sverige)
Κύπρος Sverige
Brain Therapeutics IKE Neuraxpharm Sweden AB
Τηλ: +302109931458 Tel: +46 (0)8 30 91 41
Latvija
Neuraxpharm Pharmaceuticals, S.L.
Tel: +34 93 475 96 00
Inne źródła informacji
Szczegółowe informacje na temat tego leku dostępne są na stronie internetowej Europejskiej Agencji Leków: https://www.ema.europa.eu .
Poniżej zamieszczone informacje przeznaczone są wyłącznie dla personelu medycznego:
Zapoznać się z charakterystyką produktu leczniczego w celu uzyskania dodatkowych informacji.
Dawkowanie
- Pierwsza i druga dawka
Pierwszą dawkę podaje się w postaci wlewu dożylnego 150 mg (pierwszy wlew), następnie
wlew dożylny 450 mg podaje się 2 tygodnie później (drugi wlew).
- Kolejne dawki
Kolejne dawki Briumvi podaje się w postaci pojedynczego wlewu dożylnego 450 mg
co 24 tygodnie (Tabela 1). Pierwszą kolejną dawkę 450 mg należy podać
24 tygodnie po pierwszym wlewie. Między dawkami Briumvi należy zachować minimalny odstęp 5 miesięcy.
Ryc. 1: Dawkowanie i schemat dawkowania Briumvi
| Pierwsza infuzja | Druga infuzja | Kolejne infuzje |
Dzień 1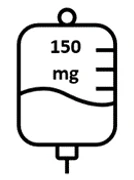 | Dzień 15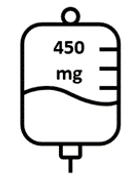 | Co 6 miesięcy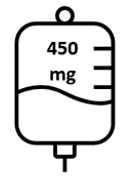 |
Zarządzanie reakcjami związanymi z infuzją (IRR) przed podaniem
- Leczenie Briumvi należy rozpoczynać i nadzorować pod kierunkiem doświadczonego personelu medycznego, posiadającego dostęp do odpowiedniej pomocy medycznej w przypadku wystąpienia ciężkich reakcji, takich jak ciężkie reakcje związane z infuzją (infusion-related reactions, IRR).
- Leczenie profilaktyczne w celu zapobiegania IRR
Tuż przed każdą infuzją Briumvi należy podać następujące dwa leki profilaktyczne w celu zmniejszenia częstości i nasilenia reakcji IRR:
- 100 mg metylprednizolonu lub 10–20 mg dexametazonu (lub odpowiednik) około 30–60 minut przed każdą infuzją Briumvi;
- difenhydrodaminę około 30–60 minut przed każdą infuzją Briumvi. Można również rozważyć podanie leku przeciwpiretycznego (np. paracetamol).
Instrukcje dotyczące rozcieńczania
-
Przygotowanie Briumvi należy przeprowadzić przez personel medyczny, stosując technikę bezpyłową. Nie wstrząsać fiolką.
-
Produkt przeznaczony jest tylko do jednorazowego użytku.
-
Nie należy stosować roztworu, jeśli jego kolor jest zmieniony lub jeśli widoczne są obce cząstki.
-
Lek Briumvi należy rozcieńczyć przed podaniem. Roztwory Briumvi do dożylnej infuzji przygotowuje się poprzez rozcieńczenie leku w worku do infuzji zawierającym izotoniczny roztwór chlorku sodu 0,9%. W przypadku pierwszej infuzji rozcieńczyć jedną fiolkę leku w worku do infuzji (150 mg/250 mL), aby uzyskać końcowe stężenie około 0,6 mg/mL. W przypadku kolejnych infuzji rozcieńczyć trzy fiolki leku w worku do infuzji (450 mg/250 mL), aby uzyskać końcowe stężenie około 1,8 mg/mL.
-
Przed rozpoczęciem infuzji dożylnej zawartość worka do infuzji powinna mieć temperaturę pokojową.
Sposób podania
- Po rozcieńczeniu Briumvi podaje się w formie dożylnej infuzji za pomocą oddzielnego przewodu.
- Infuzji Briumvi nie należy podawać w formie szybkiej infuzji dożylnej ani bolusowo.
Tabela 1: Dawkowanie i schemat dawkowania Briumvi
| Ilość i objętość | Prędkość wlewu | Czas trwania1 | |
| Pierwszy wlew | 150 mg w 250 mL |
| 4 godziny |
| Drugi wlew (2 tygodnie później) | 450 mg w 250 mL |
| 1 godzina |
| Kolejne wlewy (co 24 tygodnie)2 | 450 mg w 250 mL |
| 1 godzina |
Czas trwania wlewu może być dłuższy, jeśli wlew zostanie przerwany lub spowolniony.
Następne wlewy należy podawać 24 tygodnie po pierwszym wlewie.
Zarządzanie reakcjami podczas i po wlewie (IRR)
Pacjentów należy obserwować podczas wlewu oraz przez co najmniej jedną godzinę po zakończeniu dwóch pierwszych wlewów.
Podczas wlewu
- Modyfikacje wlewu w przypadku IRR
W przypadku wystąpienia IRR podczas wlewu należy zastosować następujące modyfikacje.
Potencjalnie śmiertelne IRR
W przypadku wystąpienia objawów potencjalnie śmiertelnej lub powodującej trwałe uszkodzenia IRR podczas wlewu, wlew należy natychmiast przerwać, a pacjentowi należy podać odpowiednie leczenie. U pacjentów tych leczenie Briumvi należy trwale przerwać (patrz punkt 4.3).
Ciężkie IRR
W przypadku wystąpienia ciężkiej IRR wlew należy natychmiast przerwać, a pacjentowi należy podać leczenie objawowe. Wlew może być wznowiony dopiero po ustąpięciu wszystkich objawów. Po wznowieniu wlewu należy rozpocząć podawanie z prędkością równą połowie prędkości, z jaką wlew był prowadzony w momencie wystąpienia IRR. Jeśli IRR nie powtarza się, prędkość wlewu może być następnie zwiększana zgodnie z Tabelą 1.
Lekkie do umiarkowanych IRR
W przypadku wystąpienia lekkiej do umiarkowanej IRR należy zmniejszyć prędkość wlewu o połowę w porównaniu z prędkością, z jaką był prowadzony w momencie wystąpienia zdarzenia. Zmniejszoną prędkość należy utrzymywać przez co najmniej 30 minut. Jeśli IRR nie powtarza się, prędkość wlewu może być następnie zwiększana zgodnie z Tabelą 1.
Po wlewie
- Pacjentów leczonych lekiem Briumvi należy obserwować przez co najmniej jedną godzinę po zakończeniu dwóch pierwszych wlewów w celu wykrycia ewentualnych objawów IRR.
- Lekarze powinni poinstruować pacjentów, że reakcje IRR mogą wystąpić w ciągu 24 godzin po wlewie.
Okres ważności
Fiolka nieotwarta
3 lata
Roztwór do wlewu dożylnego po rozcieńczeniu
- Stabilność chemiczna i fizyczna w warunkach użytkowania została potwierdzona przez 24 godziny w temperaturze od 2 do 8 °C oraz następnie przez 8 godzin w temperaturze pokojowej.
- Z mikrobiologicznego punktu widzenia przygotowany roztwór do wlewu należy stosować natychmiast. Jeśli nie jest stosowany natychmiast, czas i warunki przechowywania przed użyciem są odpowiedzialnością użytkownika i zazwyczaj nie powinny przekraczać 24 godzin w temperaturze od 2 do 8 °C oraz następnie 8 godzin w temperaturze pokojowej, chyba że rozcieńczenie przeprowadzono w warunkach kontrolowanej sterylności i zweryfikowanych.
- W przypadku, gdy nie można ukończyć wlewu dożylnego w tym samym dniu, pozostały roztwór należy usunąć.
Załącznik IV
Wnioski naukowe i podstawy zmiany warunków pozwolenia(-zeń) na dopuszczenie do obrotu
Wnioski naukowe
Na podstawie oceny Komitetu ds. Oceny Ryzyka w Farmakowigilancji (Pharmacovigilance and Risk Assessment Committee, PRAC) okresowego raportu aktualizacyjnego dotyczącego bezpieczeństwa (Periodic Safety Update Report, PSUR) dla ublituksymabu, wnioski naukowe PRAC są następujące:
Na podstawie dostępnych danych dotyczących encefalitu/meningitu/meningoencefalitu z badań klinicznych i zgłoszeń spontanicznych, w tym w siedmiu przypadkach ścisłego związku czasowego, oraz na podstawie możliwego mechanizmu działania, PRAC uznał, że związek przyczynowy między ublituksymabem a encefalitą/meningitą/meningoencefalitą jest co najmniej rozsądnym prawdopodobieństwem i stwierdził zatem, że informacje produktowe leków zawierających ublituksymab należy zaktualizować zgodnie z przedstawionymi zaleceniami.
Po przeanalizowaniu rekomendacji PRAC, Komitet ds. Leków dla Ludzi (Committee for Human Medicinal Products, CHMP) zgadza się z ogólnymi wnioskami i podstawami rekomendacji.
Podstawy zmiany warunków pozwolenia(-eń) na dopuszczenie do obrotu
Na podstawie naukowych wniosków dotyczących ublituksymabu, CHMP uznał, że stosunek korzyści do ryzyka leku(-ków) zawierającego(-cych) ublituksymab pozostaje niezmieniony, przy zastrzeżeniu proponowanych zmian informacji produktowych.
CHMP zaleca zmianę warunków pozwolenia(-eń) na dopuszczenie do obrotu.