Zarzio
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE ZARZIO®
Composizione:
Principio attivo: filgrastim (fattore stimolante delle colonie di granulociti ricombinante umano);
1 ml di soluzione contiene 60 milioni UI (600 µg) oppure 96 milioni UI (960 µg) di filgrastim;
la siringa preriempita (siringa monodose) contiene 30 milioni UI (300 µg) oppure 48 milioni UI (480 µg) di filgrastim in 0,5 ml;
Eccipienti: acido glutammico, sorbitolo (E 420), polisorbato 80, acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile o per infusione.
Principali caratteristiche fisico-chimiche: soluzione trasparente incolore o leggermente giallastra.
Gruppo farmacoterapeutico.
Immunostimolanti. Fattori stimolanti delle colonie. Filgrastim.
Codice ATC L03A A02.
Proprietà farmacologiche
Farmacodinamica
Il principio attivo del medicinale è il filgrastim, un fattore stimolante le colonie di granulociti umano ricombinante (G-CSF). Il filgrastim ha la stessa attività biologica del G-CSF umano endogeno, differenziandosi da quest'ultimo soltanto per il fatto di essere una proteina non glicosilata con un residuo aggiuntivo di metionina all'estremità N-terminale. Il filgrastim ottenuto mediante tecnologia del DNA ricombinante viene isolato da cellule del batterio Escherichia coli, al cui apparato genetico è stato inserito il gene che codifica per la proteina G-CSF.
Il fattore stimolante le colonie di granulociti umano, un glicoproteina, regola la formazione di granulociti neutrofili funzionalmente attivi e il loro rilascio nel sangue dal midollo osseo. Il filgrastim aumenta in modo significativo il numero di granulociti neutrofili nel sangue periferico già entro le prime 24 ore dall'applicazione, determinando contemporaneamente un lieve aumento del numero di monociti. L'aumento del numero di granulociti neutrofili con l'uso del medicinale, entro il range delle dosi raccomandate, è dose-dipendente. Le loro proprietà funzionali sono normali o potenziate, come dimostrato dai risultati degli studi su chemotassi e fagocitosi. Dopo la sospensione del trattamento con il medicinale, il numero di granulociti neutrofili nel sangue periferico si riduce del 50% nell'arco di 1-2 giorni e ritorna ai livelli normali entro 1-7 giorni.
L'uso del filgrastim riduce in modo significativo la frequenza, la gravità e la durata della neutropenia nei pazienti dopo chemioterapia con citostatici o dopo terapia mieloablativa seguita da trapianto di midollo osseo. L'uso del filgrastim, sia in forma primaria che dopo chemioterapia, attiva le cellule progenitrici ematopoietiche del sangue periferico (CPSP). Queste CPSP autologhe possono essere prelevate dal paziente e reinfuse dopo trattamento con citostatici ad alte dosi, al posto del trapianto di midollo osseo o come integrazione allo stesso. L'infusione di CPSP accelera il recupero ematopoietico, riduce il rischio di complicanze emorragiche e la necessità di trasfusioni di massa piastrinica. In bambini e adulti con neutropenia congenita grave (TCH), il filgrastim aumenta stabilmente il numero di granulociti neutrofili nel sangue periferico e riduce la frequenza delle complicanze infettive.
Farmacocinetica
Sia dopo somministrazione endovenosa che sottocutanea del medicinale, si osserva una relazione lineare positiva tra la concentrazione plasmatica e la dose. Dopo somministrazione sottocutanea delle dosi raccomandate, la concentrazione nel siero supera i 10 ng/ml per un periodo di 8-16 ore; il volume di distribuzione nel sangue è di circa 150 ml/kg. Sia dopo somministrazione sottocutanea che endovenosa, l'eliminazione del medicinale dall'organismo segue una cinetica di primo ordine. Il valore medio del tempo di dimezzamento del filgrastim nel siero è di circa 3,5 ore, mentre la velocità di clearance è di circa 0,6 ml/min per 1 kg di peso corporeo. La somministrazione continua mediante infusione per 28 giorni in pazienti in fase di recupero dopo trapianto autologo di midollo osseo non è stata associata a segni di cumulo o allungamento del tempo di dimezzamento del medicinale.
Caratteristiche cliniche.
Indicazioni.
Riduzione della durata e della gravità della neutropenia in pazienti sottoposti a chemioterapia mielosoppressiva intensiva con agenti citotossici per neoplasie maligne (ad eccezione della leucemia mieloide cronica e del sindrome mielodisplastico), e riduzione della durata della neutropenia in pazienti sottoposti a chemioterapia ad alto dosaggio con agenti citotossici seguita da trapianto autologo o allogenico di midollo osseo.
La sicurezza e l'efficacia dell'uso di filgrastim sono simili negli adulti e nei bambini che ricevono chemioterapia con agenti citotossici.
-
Il medicinale è indicato per la mobilizzazione delle cellule staminali emopoietiche periferiche (CSHP).
-
L'uso prolungato è indicato nei bambini e negli adulti con neutropenia congenita grave, ciclica o idiopatica e neutropenia con conteggio assoluto di neutrofili <0,5 × 109/l, al fine di aumentare il numero di neutrofili e ridurre la frequenza delle infezioni.
-
Trattamento della neutropenia persistente (conteggio assoluto di neutrofili ≤1,0 × 109/l) in pazienti con infezione da HIV in fase avanzata per ridurre il rischio di infezioni batteriche, quando altri trattamenti per la neutropenia non sono appropriati.
Controindicazioni.
- Ipersensibilità al filgrastim, ai fattori stimolanti la colonia, Escherichia coli o a qualsiasi eccipiente.
- Neutropenia ereditaria grave (sindrome di Kostmann) con alterazioni citogenetiche e neutropenia autoimmune.
- Stadio terminale di insufficienza renale cronica (IRC).
- Leucemia mieloide cronica e sindrome mielodisplastico.
Interazioni con altri medicinali e altre forme di interazioni.
La sicurezza e l'efficacia dell'amministrazione del medicinale Zarzio® nello stesso giorno dei farmaci chemioterapici citotossici mielosoppressivi non sono state stabilite. A causa della sensibilità delle cellule mieloidi in rapida divisione alla chemioterapia citotossica mielosoppressiva, non si raccomanda di somministrare il medicinale Zarzio® entro un intervallo di 24 ore prima o dopo l'infusione di tali agenti.
Quando il medicinale Zarzio® viene somministrato contemporaneamente al 5-fluorouracile, la gravità della neutropenia può aumentare. L'interazione con altri fattori di crescita emopoietici e citochine non è nota.
Poiché il litio stimola il rilascio di neutrofili, è possibile un potenziamento dell'effetto del medicinale Zarzio® in caso di somministrazione combinata, anche se studi specifici non sono stati condotti.
A causa dell'incompatibilità farmaceutica, non è consentito mescolare il medicinale con soluzione di sodio cloruro allo 0,9%.
Caratteristiche particolari di utilizzo.
Ipersensibilità
Sono state osservate reazioni di ipersensibilità, comprese reazioni anafilattiche, all'inizio o durante il trattamento, in pazienti che assumevano filgrastim. Il trattamento con Zarzio® deve essere interrotto nei pazienti che sviluppano reazioni di ipersensibilità clinicamente significative.
Zarzio® non deve essere utilizzato nei pazienti con anamnesi di ipersensibilità al filgrastim o al pegfilgrastim.
Effetti avversi sui polmoni
Sono disponibili dati su rari casi di effetti indesiderati sugli organi respiratori, in particolare lo sviluppo di polmonite interstiziale in seguito all'uso di G-CSF. I pazienti che hanno recentemente sofferto di malattia polmonare infiltrativa o di polmonite possono avere un rischio maggiore. L'insorgenza di sintomi come tosse, aumento della temperatura corporea e dispnea, in combinazione con infiltrati polmonari evidenziati alla radiografia e segni di insufficienza respiratoria progressiva, suggerisce la possibile presenza di sindrome da distress respiratorio dell'adulto (ARDS). In caso di diagnosi di ARDS, l'uso di filgrastim deve essere interrotto e deve essere avviato un trattamento appropriato.
Durante il periodo post-marketing sono state segnalate segnalazioni molto rare di effetti avversi sui polmoni (emottisi, emorragie polmonari, infiltrazione polmonare, dispnea e insufficienza di ossigeno), in particolare in donatori sani. In caso di sospetto o conferma di un effetto avverso sui polmoni, l'ulteriore uso di filgrastim deve essere interrotto e deve essere fornita un'adeguata assistenza medica.
Glomerulonefrite
La glomerulonefrite è stata osservata in pazienti in trattamento con filgrastim e pegfilgrastim. Generalmente, la glomerulonefrite regredisce dopo la riduzione della dose o l'interruzione del filgrastim e del pegfilgrastim. Si raccomanda di effettuare periodicamente analisi delle urine.
Sindrome da perdita capillare
Sono stati riportati casi di sindrome da perdita capillare, potenzialmente letale se non trattata tempestivamente, dopo l'uso di G-CSF, caratterizzata da ipotensione arteriosa, ipoalbuminemia, edema e emococoncentrazione. I pazienti che sviluppano segni di questa sindrome richiedono un attento monitoraggio e terapia sintomatica, inclusa la rianimazione.
Splenomegalia e rottura del milza
Dopo l'assunzione di filgrastim, sono stati osservati casi di splenomegalia asintomatica e rottura del milza sia in donatori sani che in pazienti. L'ingrandimento del milza è un effetto diretto dell'amministrazione di filgrastim. Nel 31% dei pazienti coinvolti in uno studio è stata osservata splenomegalia alla palpazione. Sono stati riportati alcuni casi di rottura letale del milza. Pertanto, è necessario monitorare attentamente le dimensioni del milza (esame clinico ed ecografia). È necessario escludere la diagnosi di rottura del milza in donatori e/o pazienti che lamentano dolore nell'ipocondrio sinistro o nella spalla sinistra. La riduzione della dose può rallentare o arrestare la progressione dell'ingrandimento del milza; nel 3% dei pazienti è stata necessaria una splenectomia.
Crescita di cellule maligne
È noto che il G-CSF favorisce la crescita di cellule mieloidi in vitro; effetti simili possono essere osservati in vitro anche per alcune cellule non mieloidi.
Sindrome mielodisplastica o leucemia mieloide cronica
La sicurezza e l'efficacia del filgrastim nei pazienti con sindrome mielodisplastica o leucemia mieloide cronica non sono state stabilite; pertanto, il filgrastim non è indicato in queste condizioni. È necessario prestare particolare attenzione alla diagnosi differenziale tra leucemia mieloide acuta e trasformazione blastica della leucemia mieloide cronica.
Leucemia mieloide acuta (LMA)
Poiché i dati sulla sicurezza e sull'efficacia del filgrastim nei pazienti con leucemia mieloide acuta secondaria (LMA) sono limitati, il farmaco deve essere somministrato con cautela.
La sicurezza e l'efficacia dell'uso di filgrastim de novo nei pazienti con LMA di età < 55 anni con prognosi citogenetica favorevole [t(8;21), t(15;17) e inv(16)] non sono state stabilite.
Trombocitopenia
La trombocitopenia è stata osservata molto frequentemente nei pazienti in trattamento con filgrastim. È necessario monitorare attentamente il numero di piastrine, specialmente durante le prime settimane di terapia con filgrastim. Nei casi di sviluppo di trombocitopenia nei pazienti con neutropenia cronica grave (NCG), ovvero una riduzione persistente del numero di piastrine a livelli < 100 × 109/l, il trattamento con filgrastim deve essere temporaneamente sospeso o la dose ridotta.
Leucocitosi
Il numero di leucociti nel sangue raggiunge o supera 100 × 109/l in meno del 5% dei pazienti che ricevono una dose giornaliera superiore a 0,3 milioni di UI/kg (3 µg/kg) di peso corporeo. Non ci sono dati su effetti avversi direttamente correlati a una leucocitosi di tale gravità. Tuttavia, considerando il possibile rischio associato a una grave leucocitosi, durante il trattamento con filgrastim è necessario monitorare regolarmente il numero di leucociti. Se il numero di leucociti supera 50 × 109/l dopo aver raggiunto il livello atteso, il farmaco deve essere immediatamente sospeso. Nel caso di utilizzo del farmaco per la mobilizzazione delle cellule staminali CD34+, il farmaco deve essere sospeso o la dose aggiustata se il numero di leucociti aumenta a > 70 × 109/l.
Immunogenicità
Come con tutti i farmaci proteici terapeutici, esiste la possibilità di sviluppare immunogenicità. La frequenza di comparsa di anticorpi contro il filgrastim è generalmente bassa. Sono stati rilevati anticorpi leganti, come per tutti i farmaci biologici; tuttavia, attualmente non sono associati a un'azione neutralizzante.
Avvertenze e precauzioni speciali relative a patologie concomitanti
Precauzioni speciali in presenza di caratteristiche o malattia a cellule falciformi
Sono stati riportati episodi di crisi falciformi, in alcuni casi letali, con l'uso di filgrastim in pazienti con caratteristiche a cellule falciformi o malattia falciforme. I medici devono prestare cautela quando prescrivono filgrastim a pazienti con caratteristiche a cellule falciformi o malattia falciforme.
Nei pazienti con anemia falciforme sono stati osservati casi di crisi emolitica acuta (aumento del numero di cellule alterate), talvolta con esito fatale. A tali pazienti il filgrastim deve essere somministrato con cautela e durante il trattamento devono essere attentamente monitorati i parametri clinici e di laboratorio, prestando particolare attenzione al possibile ingrandimento del milza e allo sviluppo di trombosi vascolare.
Osteoporosi
Nei pazienti con patologia ossea concomitante e osteoporosi, durante un uso prolungato (oltre 6 mesi) di filgrastim, si raccomanda di monitorare regolarmente la densità minerale ossea.
Precauzioni speciali nei pazienti con neoplasie
Il filgrastim non deve essere utilizzato per aumentare la dose di chemioterapia citotossica oltre i regimi di dosaggio stabiliti.
Rischio associato all'aumento della dose di chemioterapia
È necessaria particolare cautela nel trattamento di pazienti con neoplasie maligne che ricevono dosi elevate di agenti citostatici, poiché l'efficacia del trattamento in questi casi non è stata stabilita. È noto che dosi elevate di agenti chemioterapici possono causare una tossicità più marcata, portando a reazioni avverse cardiovascolari, polmonari, neurologiche e dermatologiche (vedere il foglio illustrativo degli agenti chemioterapici concomitanti).
Effetto della chemioterapia su globuli rossi e piastrine
La monoterapia con filgrastim non previene la trombocitopenia e l'anemia indotte da chemioterapia mielosoppressiva. Quando vengono utilizzate dosi più elevate di agenti chemioterapici (ad esempio dosi complete secondo gli schemi prescritti), il rischio di trombocitopenia e anemia grave aumenta.
Si raccomanda di monitorare regolarmente parametri ematici come ematocrito e numero di piastrine. È necessaria particolare cautela nell'uso di agenti chemioterapici singoli o combinati che possono causare trombocitopenia grave.
L'uso di filgrastim per la mobilizzazione delle cellule staminali CD34+ ha dimostrato di ridurre gravità e durata della trombocitopenia indotta da chemioterapia mielosoppressiva o mieloablativa.
Altre precauzioni speciali
L'efficacia del farmaco in pazienti con un numero fortemente ridotto di precursori mieloidi non è stata studiata. Il filgrastim aumenta il numero di neutrofili agendo principalmente sui precursori dei neutrofili. Pertanto, in pazienti con un numero ridotto di precursori (ad esempio, a seguito di radioterapia intensiva, chemioterapia o infiltrazione del midollo osseo da cellule tumorali), il numero di neutrofili prodotti può essere ridotto.
Talvolta, in pazienti sottoposti a chemioterapia ad alto dosaggio seguita da trapianto di midollo osseo autologo, sono stati osservati disturbi vascolari, come occlusione venosa e alterazioni dell'equilibrio idrico.
Sono disponibili dati sullo sviluppo della reazione "trapianto contro ospite" e casi fatali in pazienti che ricevono G-CSF dopo trapianto allogenico di midollo osseo.
L'incremento dell'ematopoiesi nel midollo osseo in risposta alla terapia con fattori di crescita è associato all'insorgenza di alterazioni patologiche transitorie rilevabili con scintigrafia ossea. Ciò deve essere considerato nell'interpretazione delle immagini diagnostiche ossee.
Sono stati riportati casi di aortite dopo somministrazione di filgrastim, sia in soggetti sani che in pazienti oncologici. I sintomi osservati comprendevano febbre, dolore addominale, malessere, dolore alla schiena e marcatori infiammatori elevati (ad esempio, aumento della proteina C-reattiva e leucocitosi). Nella maggior parte dei casi, l'aortite è stata diagnosticata mediante TC e di solito regrediva dopo l'interruzione del filgrastim.
Precauzioni speciali per pazienti che richiedono mobilizzazione delle cellule staminali CD34+
Mobilizzazione
Non esiste alcun confronto randomizzato tra i due metodi raccomandati di mobilizzazione (filgrastim da solo o in combinazione con chemioterapia mielosoppressiva) all'interno di una stessa popolazione di pazienti. La variabilità tra pazienti e tra laboratori nell'analisi delle cellule CD34+ rende difficile il confronto diretto tra studi diversi. Pertanto, è difficile raccomandare un metodo ottimale. La scelta del metodo di mobilizzazione deve essere considerata in relazione agli obiettivi terapeutici generali per il singolo paziente.
Effetto pregresso di sostanze citotossiche
In pazienti precedentemente sottoposti a terapia mielosoppressiva intensiva, l'uso di filgrastim per la mobilizzazione delle cellule staminali CD34+ potrebbe non determinare un aumento del numero di cellule staminali CD34+ al livello minimo raccomandato (≥ 2,0 × 106 cellule CD34+/kg) o un miglioramento della velocità di recupero delle piastrine.
Alcuni agenti citotossici mostrano una tossicità particolare nei confronti dei precursori ematopoietici e influiscono negativamente sulla loro mobilizzazione. Un uso prolungato di melphalan, carboplatino o carmustina (BCNU) prima della mobilizzazione delle cellule staminali può compromettere i risultati. Tuttavia, l'uso contemporaneo di melphalan, carboplatino o BCNU con filgrastim è efficace nella mobilizzazione delle cellule staminali CD34+. Se si prevede un trapianto di cellule staminali CD34+, si raccomanda di effettuare la mobilizzazione delle cellule staminali in una fase precoce del trattamento del paziente. Particolare attenzione deve essere prestata al numero di precursori attivati in questi pazienti prima dell'uso di agenti chemioterapici ad alta dose. Se i risultati della mobilizzazione non sono adeguati secondo i criteri sopra indicati, si deve considerare l'uso di metodi alternativi di trattamento che non richiedano l'uso di precursori.
Valutazione del numero di precursori
Nella valutazione del numero di cellule staminali CD34+ mobilizzate in pazienti trattati con filgrastim, particolare attenzione deve essere prestata al metodo di quantificazione. I risultati dell'analisi citometrica delle cellule CD34+ variano notevolmente a seconda della metodologia utilizzata; pertanto, si deve prestare cautela nell'interpretare i risultati ottenuti in altri laboratori.
I risultati dell'analisi statistica della relazione tra il numero di cellule CD34+ infuse e la velocità di normalizzazione del numero di piastrine dopo chemioterapia ad alto dosaggio indicano una dipendenza complessa ma costante. Le raccomandazioni riguardo alla necessità di garantire un contenuto minimo di ≥ 2,0 × 106 cellule CD34+/kg si basano su dati pubblicati sull'esperienza di un adeguato recupero ematologico. A livelli superiori al minimo raccomandato si osserva un recupero più rapido, mentre a livelli inferiori il recupero è più prolungato.
Precauzioni speciali per donatori sani sottoposti a mobilizzazione delle cellule staminali CD34+
La mobilizzazione delle cellule staminali CD34+ in donatori sani influisce sulla loro salute e viene utilizzata esclusivamente per ottenere cellule staminali allogeniche per trapianto.
I donatori sottoposti a mobilizzazione delle cellule staminali CD34+ per trapianto devono soddisfare i requisiti standard per i parametri clinici e di laboratorio richiesti per i donatori di cellule staminali. Particolare attenzione deve essere prestata ai parametri ematici e alla presenza di malattie infettive. La sicurezza e l'efficacia dell'uso di filgrastim in donatori sani di età inferiore ai 16 anni e superiore ai 60 anni non sono state valutate.
Una trombocitopenia transitoria (numero di piastrine < 100 × 109/l) dopo somministrazione di filgrastim e leucocitoaferesi è stata osservata nel 35% dei pazienti. Tra questi, due casi di riduzione del numero di piastrine < 50 × 109/l sono stati riportati e attribuiti alla procedura di leucocitoaferesi.
Nel caso in cui sia necessaria più di una procedura di leucocitoaferesi, particolare attenzione deve essere prestata ai donatori il cui numero di piastrine prima dell'inizio della leucocitoaferesi è < 100 × 109/l; generalmente, la leucocitoaferesi non è raccomandata se il numero di piastrine è < 75 × 109/l.
La leucocitoaferesi non deve essere effettuata in donatori che richiedono terapia anticoagulante o che presentano alterazioni dell'omeostasi.
Il monitoraggio dello stato dei donatori che ricevono G-CSF per la mobilizzazione delle cellule staminali CD34+ deve proseguire fino alla normalizzazione dei parametri ematologici.
In donatori sani, dopo somministrazione di G-CSF, sono state osservate alterazioni citogenetiche transitorie. Il significato di queste alterazioni non è definito.
La valutazione della sicurezza a lungo termine del farmaco nei donatori è in corso. Il rischio di favorire la formazione di cloni maligni di cellule mieloidi non è escluso. Ai centri di aferesi si raccomanda di effettuare controlli sistematici dei donatori di cellule staminali per un periodo di almeno 10 anni per garantire il monitoraggio della sicurezza a lungo termine.
Precauzioni speciali per i riceventi sottoposti a mobilizzazione delle cellule staminali CD34+ con filgrastim
I dati indicano che l'interazione immunologica tra cellule staminali CD34+ allogeniche e ricevente comporta un rischio maggiore di sviluppare reazioni "trapianto contro ospite" acute e croniche rispetto al trapianto di midollo osseo.
Precauzioni speciali per pazienti con NCG
Il filgrastim non deve essere somministrato a pazienti con neutropenia congenita grave che hanno sviluppato leucemia o che presentano segni di trasformazione leucemica.
Analisi del sangue
Possono verificarsi altre modifiche della formula ematica, inclusa anemia e aumento transitorio del numero di precursori mieloidi; è necessario un attento monitoraggio della formula ematica.
Trasformazione in leucemia o pre-leucemia
È necessaria particolare cautela nella diagnosi della NCG per differenziarla da altre malattie ematologiche, come anemia aplastica, mielodisplasia e leucemia mieloide. Prima dell'inizio del trattamento, deve essere effettuato un esame ematico completo con determinazione della formula leucocitaria e del numero di piastrine, nonché l'analisi morfologica del midollo osseo e il cariotipo.
L'insorgenza di sindrome mielodisplastica (MDS) o leucemia in pazienti con neutropenia cronica grave che partecipavano a studi clinici sull'uso di filgrastim è rara (circa nel 3% dei casi). Tali alterazioni sono state osservate solo in pazienti con neutropenia congenita. La MDS e la leucemia sono complicanze comuni della malattia; il loro legame con la terapia con filgrastim è incerto. Circa nel 12% dei pazienti (senza alterazioni citogenetiche prima dell'inizio della terapia) sono state osservate alterazioni nei successivi esami, inclusa monosomia 7. In caso di alterazioni citogenetiche in un paziente con neutropenia cronica grave, si deve attentamente valutare il rapporto rischio/beneficio dell'ulteriore uso di filgrastim. L'ulteriore somministrazione di filgrastim in caso di sviluppo di MDS o leucemia deve essere interrotta. Non è noto se un trattamento prolungato con filgrastim in pazienti con neutropenia cronica grave aumenti il rischio di alterazioni citogenetiche, MDS o trasformazione della malattia in leucemia. L'esame morfologico e citogenetico del midollo osseo dei pazienti deve essere effettuato regolarmente, con intervalli di circa 12 mesi.
Altre precauzioni speciali
È necessario escludere altre cause di neutropenia, come infezioni virali.
L'ematuria è stata osservata frequentemente, mentre la proteinuria è stata osservata in un numero ridotto di pazienti. Per una diagnosi precoce di questi fenomeni, è necessario effettuare regolarmente analisi delle urine.
La sicurezza e l'efficacia nell'uso in neonati e pazienti con neutropenia autoimmune non sono state stabilite.
Precauzioni speciali per pazienti con infezione da HIV
Formula ematica
È necessario un attento monitoraggio del valore assoluto dei neutrofili (ANC), specialmente durante le prime settimane di terapia con filgrastim. In alcuni pazienti si osserva una risposta molto rapida con un marcato aumento del numero di neutrofili in risposta alla prima dose di filgrastim. Si raccomanda la determinazione giornaliera dell'ANC durante i primi 2-3 giorni di somministrazione di filgrastim. Successivamente, durante le prime 2 settimane, si raccomanda di determinare l'ANC almeno 2 volte alla settimana, poi 1 volta alla settimana e ogni 2 settimane durante la terapia di mantenimento. Durante la somministrazione periodica di filgrastim con dose individuale e dose di 30 milioni UI/giorno (300 µg/giorno), sono possibili maggiori fluttuazioni del valore di ANC. Per determinare i valori minimi (valori più bassi di ANC), si raccomanda di prelevare campioni di sangue per l'analisi dell'ANC immediatamente prima della somministrazione programmata di filgrastim.
Rischio associato all'uso di farmaci mielosoppressivi a dosi elevate
La somministrazione di filgrastim da solo non esclude la possibilità di sviluppare trombocitopenia e anemia indotte da chemioterapia mielosoppressiva. A causa dell'uso di agenti chemioterapici a dosi più elevate o di un numero maggiore di tali agenti in combinazione con filgrastim, il rischio di sviluppare trombocitopenia e anemia nel paziente può aumentare. Si raccomanda di monitorare regolarmente i parametri ematici.
Malattie infettive e maligne che causano mielosoppressione
Lo sviluppo di neutropenia può essere conseguenza dell'infiltrazione del midollo osseo da parte di agenti infettivi opportunistici, come Mycobacterium avium, o da lesioni maligne, ad esempio linfoma. In caso di infezioni o neoplasie maligne che infiltrano il midollo osseo, per correggere la neutropenia, oltre alla somministrazione di filgrastim, è necessario effettuare un trattamento specifico per la malattia. L'effetto del filgrastim sulla correzione della neutropenia causata da infezioni o neoplasie maligne che infiltrano il midollo osseo non è stato determinato.
Sostanze ausiliarie
Zarzio® contiene sorbitolo; pertanto, non deve essere utilizzato in pazienti con rara intolleranza ereditaria al fruttosio.
Il tappo protettivo contiene derivati del lattice naturale del caucciù. Sebbene il medicinale stesso non contenga lattice naturale del caucciù, la sicurezza del prodotto in pazienti sensibili al lattice non è stata valutata.
Al fine di migliorare la tracciabilità del G-CSF, il nome del medicinale somministrato deve essere chiaramente indicato nel fascicolo del paziente.
Questo medicinale contiene meno di 1 mmol (23 mg)/dose di sodio, cioè è praticamente privo di sodio.
Uso durante la gravidanza o l'allattamento.
La sicurezza del filgrastim durante la gravidanza non è stata stabilita. Esistono dati sulla capacità del filgrastim di attraversare la barriera placentare. Non sono stati ottenuti dati sulla teratogenicità del filgrastim negli studi sugli animali. Negli studi sugli animali è stata osservata tossicità riproduttiva. Negli animali trattati con filgrastim è stata osservata una maggiore frequenza di aborti.
Zarzio® non è raccomandato durante la gravidanza.
Non è noto se il filgrastim e i suoi metaboliti passino nel latte materno umano; pertanto, il rischio per il neonato non può essere escluso. È necessario prendere una decisione riguardo all'interruzione dell'allattamento o del trattamento con filgrastim, considerando i benefici dell'allattamento per il bambino e i benefici della terapia per la madre.
Negli studi sugli animali è stato dimostrato che il filgrastim non influenza il sistema riproduttivo o la fertilità.
Capacità di influire sulla velocità di reazione nella guida di automezzi o nell'uso di macchinari.
Il filgrastim può avere un lieve effetto sulla capacità di guidare automezzi o di usare macchinari, in particolare vertigini.
Modalità e dosi di somministrazione.
La terapia con Zarzio® può essere effettuata presso strutture sanitarie dotate delle necessarie attrezzature diagnostiche. I medici devono avere esperienza nell'uso di medicinali contenenti fattore stimolante le colonie di granulociti (G-CSF) e nel trattamento di pazienti con malattie ematologiche.
Le procedure di mobilizzazione e di aferesi devono essere effettuate in collaborazione con medici esperti e in grado di effettuare il necessario monitoraggio delle cellule progenitrici ematopoietiche.
Neutropenia in pazienti sottoposti a chemioterapia citotossica per neoplasie maligne.
La dose giornaliera raccomandata è di 0,5 milioni di UI/kg (5 µg/kg) di peso corporeo, una volta al giorno. La prima dose di Zarzio® deve essere somministrata non prima di 24 ore dopo il completamento del ciclo di chemioterapia citotossica. Il trattamento con il medicinale deve proseguire finché il numero totale di neutrofili nell’emocromo non superi il livello previsto e non raggiunga valori normali. Dopo chemioterapia per tumori solidi, linfomi e linfoleucosi, la durata del trattamento per raggiungere tali valori è generalmente fino a 14 giorni. Dopo terapia induttiva e di consolidamento per leucemia mieloide acuta, la durata del trattamento può essere significativamente prolungata (fino a 38 giorni), in base al tipo, alla dose e allo schema di chemioterapia citotossica impiegata.
Nei pazienti sottoposti a chemioterapia citotossica, un aumento transitorio del numero di neutrofili si osserva generalmente dopo 1-2 giorni dall’inizio del trattamento con Zarzio®. Tuttavia, per ottenere un effetto terapeutico stabile, è necessario continuare il trattamento finché il numero di neutrofili non superi il minimo previsto e non raggiunga la normalità. Non è raccomandato interrompere prematuramente il trattamento con Zarzio® prima che il numero di neutrofili superi tale minimo.
Modalità di somministrazione
Zarzio® deve essere somministrato per via sottocutanea o per infusione endovenosa (diluito in soluzione glucosata al 5%) per 30 minuti, una volta al giorno. Nella maggior parte dei casi, la via sottocutanea è preferibile. Quando la dose singola viene somministrata per via endovenosa, la durata dell’effetto del medicinale può essere ridotta. L’importanza clinica di questi dati relativamente all’uso di dosi multiple non è stata stabilita. La scelta della via di somministrazione dipende dalle caratteristiche specifiche della situazione clinica e deve essere determinata caso per caso.
Pazienti sottoposti a terapia mieloablativa seguita da trapianto di midollo osseo.
La dose iniziale raccomandata di Zarzio® è di 1 milione di UI/kg (10 µg/kg) di peso corporeo al giorno. La prima dose deve essere somministrata non prima di 24 ore dopo il completamento della chemioterapia citotossica e non prima di 24 ore dopo il trapianto di midollo osseo.
Dopo il calo massimo del numero di neutrofili (nadir), la dose giornaliera di Zarzio® deve essere aggiustata in base alle variazioni del numero di neutrofili (vedere tabella).
Adattamento della dose di Zarzio® in risposta al raggiungimento del nadir.
| Numero assoluto di neutrofili (ANC) |
Aggiustamento della dose di Zarzio® |
| ANC > 1 × 109/l per 3 giorni consecutivi |
Ridurre la dose a 0,5 milioni di UI/kg |
| ANC > 1 × 109/l per i successivi 3 giorni consecutivi |
Sospendere il trattamento |
| Se durante il trattamento l'ANC diminuisce fino a livelli < 1 × 109/l, aumentare la dose di Zarzio® secondo lo schema sopra indicato. |
|
Modalità di somministrazione
Il medicinale va disciolto in 20 ml di soluzione glucosata al 5% ed utilizzato come infusione endovenosa breve della durata di 30 minuti oppure come infusione sottocutanea o endovenosa prolungata della durata di 24 ore.
Mobilizzazione delle cellule staminali emopoietiche periferiche (CSPE) in pazienti sottoposti a terapia mielosoppressiva o mieloablative seguita da trapianto autologo di CSPE
Pazienti sottoposti a terapia mielosoppressiva o mieloablative seguita da trapianto autologo di CSPE.
Per la mobilizzazione delle CSPE, quando il medicinale Zarzio® viene utilizzato come monoterapia, la dose raccomandata è di 1 milione UI/kg (10 mcg/kg) di peso corporeo al giorno per 5-7 giorni consecutivi, sotto forma di infusione sottocutanea prolungata della durata di 24 ore. Generalmente sono sufficienti 1-2 sessioni di leucocitaferesi al quinto e al sesto giorno. In alcuni casi può essere necessaria un’ulteriore sessione di leucocitaferesi. La dose del medicinale non deve essere modificata prima dell’ultima sessione di leucocitaferesi.
Per la mobilizzazione delle CSPE dopo chemioterapia mielosoppressiva, la dose raccomandata di Zarzio® è di 0,5 milioni UI/kg (5 mcg/kg) di peso corporeo al giorno, da iniziare dal primo giorno successivo al termine del ciclo chemioterapico, fino a quando il numero dei neutrofili non supera il minimo atteso e raggiunge valori normali. La leucocitaferesi deve essere effettuata durante il periodo di incremento dell’ANC da < 0,5 × 109/l a > 5 × 109/l. Nei pazienti che non hanno ricevuto chemioterapia intensiva, generalmente è sufficiente una singola sessione di leucocitaferesi. In singoli casi possono essere raccomandate sessioni aggiuntive di leucocitaferesi.
Modalità di somministrazione
Il filgrastim per la mobilizzazione delle CSPE, quando utilizzato da solo, può essere somministrato come infusione sottocutanea continua per 24 ore oppure mediante iniezione sottocutanea. Per le infusioni, il filgrastim deve essere diluito in 20 ml di soluzione glucosata al 5%.
Il filgrastim per la mobilizzazione delle CSPE dopo chemioterapia mielosoppressiva deve essere somministrato per via sottocutanea.
Mobilizzazione delle CSPE in donatori sani prima di un trapianto allogenico di CSPE
Per la mobilizzazione delle CSPE prima di un trapianto allogenico di CSPE in donatori sani, la dose raccomandata di Zarzio® è di 1 milione UI/kg (10 mcg/kg) di peso corporeo al giorno per 4-5 giorni consecutivi. La leucocitaferesi deve essere effettuata a partire dal quinto giorno e, se necessario, protratta fino al sesto giorno, al fine di ottenere 4 × 106 cellule CD34+/kg di peso corporeo del ricevente.
Modalità di somministrazione
Il filgrastim deve essere somministrato come iniezione sottocutanea.
In pazienti con neutropenia cronica grave (NCG)
Neutropenia ereditaria
Dose iniziale raccomandata: 1,2 milioni UI/kg (12 mcg/kg) di peso corporeo al giorno, mediante iniezione sottocutanea singola o in dosi frazionate.
Neutropenia idiopatica e periodica
Dose iniziale raccomandata: 0,5 milioni UI/kg (5 mcg/kg) di peso corporeo al giorno, singola o in dosi frazionate.
Adattamento della dose
Zarzio® deve essere somministrato giornalmente per via sottocutanea fino al raggiungimento e al mantenimento stabile di un conteggio dei neutrofili superiore a 1,5 × 109/l. Dopo aver raggiunto l’effetto terapeutico, va determinata la dose efficace minima necessaria per mantenere tale livello. Per mantenere il numero adeguato di neutrofili è richiesta una somministrazione giornaliera prolungata del medicinale. Dopo 1-2 settimane di trattamento, la dose iniziale può essere raddoppiata o dimezzata in base all’efficacia della terapia. Successivamente, ogni 1-2 settimane va effettuata una regolazione individuale della dose al fine di stabilizzare il conteggio medio dei neutrofili in un intervallo compreso tra 1,5 × 109/l e 10 × 109/l. Nei pazienti con infezioni gravi può essere adottato un regime con aumento più rapido della dose. La sicurezza dell’uso di filgrastim in trattamenti prolungati con dosi di Zarzio® superiori a 2,4 milioni UI (24 mcg/kg) al giorno non è stata stabilita.
Modalità di somministrazione
Il filgrastim deve essere somministrato come iniezione sottocutanea.
In pazienti con infezione da HIV
Ripristino del numero di neutrofili
Dose iniziale raccomandata del medicinale: 0,1 milioni UI/kg (1 mcg/kg) di peso corporeo al giorno, aumentabile fino a 0,4 milioni UI/kg (4 mcg/kg) di peso corporeo al giorno, mediante singola iniezione sottocutanea, fino alla normalizzazione del numero di neutrofili (ANC > 2,0 × 109/l). La normalizzazione del numero di neutrofili si verifica generalmente entro 2 giorni. In un numero limitato di casi (< 10% dei pazienti), per ripristinare il numero di neutrofili la dose del medicinale può essere aumentata fino a 1 milione UI/kg (10 mcg/kg di peso corporeo al giorno).
Mantenimento del numero normale di neutrofili
Una volta superata la neutropenia, va stabilita la dose efficace minima necessaria per mantenere livelli normali di neutrofili. Dopo il raggiungimento dell’effetto terapeutico, la dose di mantenimento è di 300 mcg/giorno, 2-3 volte alla settimana secondo uno schema alternato (ogni altro giorno). Successivamente può essere necessaria una regolazione individuale della dose e un uso prolungato del medicinale per mantenere un conteggio medio di neutrofili > 2,0 × 109/l.
Modalità di somministrazione
Il filgrastim deve essere somministrato come iniezione sottocutanea.
Categorie speciali di pazienti
Non è necessario alcun aggiustamento della dose nei pazienti con grave insufficienza epatica o renale, poiché i parametri farmacocinetici e farmacodinamici si sono dimostrati simili a quelli osservati nei volontari sani.
Non vi sono raccomandazioni specifiche per l’uso di Zarzio® nei pazienti di età avanzata.
Pediatria
Nella pratica pediatrica, nei pazienti con NCG e malattie oncologiche, il profilo di sicurezza di Zarzio® non si è differenziato da quello osservato negli adulti. La sicurezza ed efficacia del medicinale nei neonati non sono state stabilite.
Le raccomandazioni per il dosaggio nei pazienti pediatrici sono le stesse previste per gli adulti sottoposti a chemioterapia citotossica mielosoppressiva.
Raccomandazioni prima della somministrazione
Prima dell’uso, ispezionare visivamente il contenuto della siringa preriempita. La soluzione deve essere limpida e priva di particelle. Un breve esposizione a basse temperature non compromette la stabilità del medicinale. Il medicinale non contiene conservanti. Per evitare contaminazione microbica, si deve considerare che Zarzio® nella siringa preriempita è destinato solo per uso monouso. Per conservazione e uso ambulatoriale, il medicinale può essere prelevato dal frigorifero e conservato a temperatura ambiente (non superiore a 25 °C) per un solo periodo di 8 giorni. Trascorso tale periodo, non deve essere riposto nuovamente in frigorifero, ma deve essere eliminato.
Raccomandazioni per la diluizione del medicinale
Zarzio® può essere somministrato in forma diluita in soluzione glucosata al 5%. La diluizione a una concentrazione inferiore a 0,2 milioni UI/ml (2 mcg/ml) non è raccomandata. Quando si diluisce a una concentrazione < 1,5 milioni UI/ml (15 mcg/ml), è necessario aggiungere albumina umana fino al raggiungimento di una concentrazione di 2 mg/ml. Ad esempio, per ottenere un volume di soluzione di 20 ml e una dose totale di Zarzio® di 30 milioni UI (300 mcg), è necessario aggiungere 0,2 ml di soluzione di albumina (soluzione al 20%).
Quando diluito in soluzione glucosata, il medicinale viene adsorbito dal vetro e da altri materiali utilizzati per la somministrazione per infusione. È vietato utilizzare soluzione fisiologica per la diluizione del medicinale.
La stabilità chimica e fisica della soluzione diluita per infusione dopo l’apertura della confezione è stata confermata per 24 ore, se conservata a una temperatura compresa tra 2 e 8 °C. Dal punto di vista microbiologico, il medicinale deve essere utilizzato immediatamente. Se non utilizzato immediatamente, il responsabile dell’uso è tenuto a rispettare tempi e condizioni di conservazione dall’apertura della confezione fino alla somministrazione. La diluizione deve avvenire in condizioni asettiche controllate e approvate.
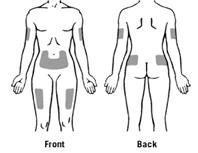
Le aree preferite del corpo per la somministrazione sottocutanea di Zarzio® sono illustrate nella figura:
Istruzioni per l’autoiniezione del medicinale da parte del paziente
Questa sezione contiene informazioni relative all’autoiniezione del medicinale Zarzio® da parte del paziente. Importante! Non tentare di eseguire l’iniezione autonomamente se non si è stati istruiti dal medico o dall’infermiere sulla tecnica di somministrazione. Zarzio® è fornito con un dispositivo di protezione per prevenire lesioni da ago dopo l’uso, e il medico o l’infermiere vi mostreranno come utilizzare la siringa. Se non siete sicuri di poter effettuare l’iniezione o avete domande, rivolgetevi al medico o all’infermiere per assistenza.
Attenzione! Non utilizzare la siringa se è caduta su una superficie dura o se è caduta dopo aver rimosso il tappo dell’ago.
- Lavarsi le mani.
- Estrarre una siringa dalla confezione e rimuovere il dispositivo di protezione per prevenire lesioni da ago dall’ago di iniezione. Le siringhe sono dotate di divisioni graduate in rilievo, nel caso in cui sia necessario riempire parzialmente la siringa con il medicinale. Ogni divisione graduata corrisponde a un volume di 0,1 ml. Se la siringa deve essere riempita solo parzialmente, eliminare l’eccesso di soluzione prima della somministrazione.
- Pulire la cute nel sito di iniezione con una salvietta alcolica.
- Formare una piega cutanea pizzicando la pelle con il pollice e l’indice.
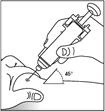
- Inserire l’ago nella piega cutanea con un movimento rapido e deciso. Iniettare la soluzione di Zarzio® come dimostrato dal medico. Se non siete sicuri, rivolgetevi al medico o al farmacista per assistenza.
|
|
Le informazioni riportate di seguito sono destinate esclusivamente al personale medico.
Il dispositivo di protezione per la prevenzione delle lesioni da aghi dopo l'uso copre l'ago dopo l'iniezione per prevenire lesioni da punta d'ago. Questo non influenza il normale funzionamento della siringa. Premere lentamente e uniformemente lo stantuffo fino a quando tutta la dose è stata iniettata e lo stantuffo non può essere spinto oltre. Continuando a premere sullo stantuffo, rimuovere l'ago dalla pelle. Il dispositivo di protezione per la prevenzione delle lesioni da aghi dopo l'uso coprirà l'ago una volta rilasciato lo stantuffo.
Smaltimento
Eventuali farmaci non utilizzati o rifiuti derivati dal loro utilizzo devono essere smaltiti in conformità con i requisiti locali.
Sovradosaggio.
I sintomi da sovradosaggio con Zarzio® non sono noti. Entro 1-2 giorni dopo l'interruzione del trattamento con il medicinale, il numero di granulociti neutrofili circolanti di solito si riduce del 50% e torna alla normalità entro 1-7 giorni.
Effetti indesiderati.
La frequenza degli effetti indesiderati è classificata come segue: molto comune (≥ 1/10); comune (≥ 1/100, < 1/10); non comune (≥ 1/1000, < 1/100); raro (≥ 1/10000, < 1/1000); molto raro (< 1/10000).
Le reazioni avverse più gravi che possono verificarsi durante il trattamento con filgrastim includono: reazioni anafilattiche, gravi effetti indesiderati polmonari (compresa pneumonite interstiziale e sindrome da distress respiratorio acuto), sindrome da perdita capillare, splenomegalia grave/rottura del milza, sindrome mielodisplastica o leucemia in pazienti con neutropenia congenita grave, reazione di rigetto del trapianto in pazienti sottoposti a trapianto allogenico di midollo osseo o trapianto di cellule progenitrici del sangue periferico e crisi falcemica in pazienti con anemia falciforme.
Gli effetti collaterali più comuni associati al trattamento con filgrastim sono febbre, dolore osseo e muscolare (compreso dolore osseo, dolore alla schiena, artralgia, mialgia, dolore agli arti, dolore muscolo-scheletrico, dolore muscolo-scheletrico al torace, dolore al collo), anemia, nausea e vomito. Nei pazienti oncologici, il dolore muscolo-scheletrico di grado da lieve a moderato è stato osservato nel 10% dei pazienti, di grado elevato nel 3%. Il dolore osseo e muscolare generalmente si risolve con l’assunzione di analgesici standard.
Nella mobilizzazione delle CPCP in donatori sani, la reazione avversa più comune è stata il dolore muscolo-scheletrico.
Nei pazienti con NC grave, le reazioni avverse più comuni durante il trattamento con filgrastim sono state dolore osseo, dolore muscolo-scheletrico generalizzato, aumento di volume e rottura della milza. Sindrome mielodisplastica (SMD) o leucemia sono state osservate in pazienti con neutropenia congenita in trattamento con filgrastim.
La sindrome da perdita capillare, potenzialmente letale in assenza di trattamento immediato, è stata riportata con frequenza non comune (≥1/1000 a <1/100) in pazienti con neoplasie maligne sottoposti a chemioterapia e in donatori sani durante la procedura di mobilizzazione delle cellule staminali periferiche dopo somministrazione di fattori stimolanti le colonie di granulociti umani.
Negli studi clinici condotti in pazienti con infezione da HIV, gli effetti indesiderati ritenuti correlati all’uso di filgrastim sono stati esclusivamente dolore muscolo-scheletrico, dolore osseo e mialgia.
L’uso di filgrastim non ha aumentato la frequenza degli effetti indesiderati indotti dalla chemioterapia citotossica. Gli effetti collaterali osservati con uguale frequenza nei pazienti trattati con filgrastim/chemioterapia e nei pazienti trattati con placebo/chemioterapia includono nausea e vomito, alopecia, diarrea, affaticamento, anoressia, mucosite, cefalea, tosse, eruzioni cutanee, dolore toracico, debolezza generale, dolore alla gola, stitichezza e dolore non specificato.
Nei pazienti sottoposti a chemioterapia ad alte dosi seguita da trapianto autologo di midollo osseo, sono stati osservati disturbi del sistema vascolare.
Di seguito è riportato un elenco di reazioni avverse descritte negli studi clinici e segnalate spontaneamente.
Infezioni e infestazioni
Comune: sepsi, bronchite, infezioni delle vie respiratorie superiori, infezioni delle vie urinarie.
Dal sistema emolinfopoietico
Molto comune: trombocitopenia, anemia1.
Comune: splenomegalia1, riduzione dei livelli di emoglobina5.
Non comune: leucocitosi1, alterazioni della funzione della milza.
Raro: rottura della milza1, crisi delle cellule falciformi.
Dal sistema immunitario
Comune: reazioni allergiche, eruzioni cutanee, orticaria, edema angioneurotico.
Non comune: reazioni di ipersensibilità, reazioni di ipersensibilità al farmaco1, reazioni di "trapianto contro l'ospite"2.
Raro: reazioni anafilattiche.
Dal sistema metabolico e nutrizionale
Comune: aumento dei livelli ematici di lattato deidrogenasi (LDH), riduzione dell'appetito5.
Non comune: aumento dell'acido urico ematico, iperuricemia.
Raro: pseudogotta1, riduzione dei livelli ematici di glucosio, alterazioni dell'equilibrio idrico.
Dal sistema psichico
Comune: insonnia.
Dal sistema nervoso
Molto comune: cefalea1.
Comune: capogiri, ipoestesia, parestesia.
Dal sistema vascolare
Comune: ipertensione arteriosa, ipotensione arteriosa.
Non comune: malattia da occlusione venosa4.
Raro: sindrome da perdita capillare1, aortite.
Dal sistema respiratorio, toracico e mediastinico
Comune: emottisi, dispnea, tosse1, dolore alla faringe1,5, epistassi, affanno.
Non comune: sindrome da distress respiratorio acuto1, insufficienza respiratoria1, edema polmonare1, malattia polmonare interstiziale1, formazione di infiltrati polmonari1, emorragia polmonare, insufficienza di ossigeno.
Dal sistema gastrointestinale
Molto comune: diarrea1,5, vomito1,5, nausea1.
Comune: dolore orale, stitichezza.
Dal sistema epatobiliare
Comune: epatomegalia, aumento dei livelli ematici di fosfatasi alcalina.
Non comune: aumento dei livelli ematici di aspartato aminotransferasi (AST), aumento dei livelli ematici di gamma-glutammiltransferasi (GGT).
Da cute e tessuti sottocutanei
Molto comune: alopecia1.
Comune: eruzione cutanea1, eritema.
Non comune: eruzioni maculopapulari, sindrome di Sweet, vasculite cutanea1.
Dal sistema muscoloscheletrico e connettivo
Molto comune: dolore muscolare e osseo3.
Comune: spasmo muscolare.
Non comune: osteoporosi.
Raro: peggioramento dell'artrite reumatoide e dei sintomi articolari, riduzione della densità ossea.
Da reni e vie urinarie
Comune: disuria, ematuria.
Non comune: proteinuria.
Raro: alterazioni patologiche nell'analisi delle urine, glomerulonefrite.
Disturbi generali e reazioni nel sito di somministrazione
Molto comune: affaticamento1, debolezza, mucosite1, piressia.
Comune: dolore toracico1, dolore1, astenia1, malessere5, edemi periferici5, dolore nel sito di iniezione.
Non comune: reazioni nel sito di iniezione.
Traumi, avvelenamenti e complicanze da procedure
Comune: reazione trasfusionale5.
1 Vedi descrizione delle singole reazioni avverse.
2 Sono stati segnalati casi di reazione di "trapianto contro l'ospite" e decessi tra i pazienti trattati con G-CSF dopo trapianto allogenico di midollo osseo (vedi descrizione delle singole reazioni avverse).
3 Include dolore osseo, dolore alla schiena, artralgia, mialgia, dolore agli arti, dolore muscolo-scheletrico, dolore muscolo-scheletrico al torace, dolore al collo.
4 I casi sono stati riportati nel periodo post-marketing in pazienti sottoposti a trapianto allogenico di midollo osseo o mobilizzazione delle CPCP.
5 Gli eventi avversi si sono verificati con maggiore frequenza nei pazienti trattati con filgrastim rispetto al gruppo placebo e sono correlati alle conseguenze della malattia maligna di base o della chemioterapia citotossica.
Descrizione delle singole reazioni avverse
Ipersensibilità
Negli studi clinici e nel periodo post-marketing sono state registrate reazioni di ipersensibilità, compresa anafilassi, eruzioni cutanee, orticaria, edema di Quincke, dispnea e ipotensione arteriosa, che si verificano all’inizio o in fasi successive del trattamento. In generale, le segnalazioni sono state più frequenti dopo somministrazione endovenosa. In alcuni casi, i sintomi sono ricomparsi durante test provocativi, indicando un rapporto di causa-effetto. Nei casi di gravi reazioni allergiche, il filgrastim non deve essere ulteriormente somministrato al paziente.
Effetti polmonari
Negli studi clinici e nel periodo post-marketing sono stati osservati effetti indesiderati polmonari, inclusi emottisi, emorragie polmonari, infiltrati polmonari, dispnea e insufficienza di ossigeno, che hanno portato a insufficienza respiratoria o sindrome da distress respiratorio dell’adulto (ARDS), talvolta con esito fatale. Sono state segnalate rare reazioni polmonari in donatori sani.
Splenomegalia e rottura della milza
Dopo l’assunzione di filgrastim sono stati riportati aumento della milza e rottura della milza. Alcuni casi di rottura della milza sono stati fatali.
In tutti i casi, nei pazienti con infezione da HIV, l’ingrandimento della milza era da lieve a moderato all’esame clinico e l’evoluzione clinica del disturbo era benigna; in nessun paziente è stata diagnosticata ipersplenismo e nessun paziente è stato sottoposto a splenectomia. Poiché l’ingrandimento della milza è una complicanza comune nei pazienti con infezione da HIV e si verifica con diversi gradi di gravità nella maggior parte dei pazienti con AIDS, il rapporto causale con l’uso di filgrastim rimane incerto.
Sindrome da perdita capillare
Sono stati riportati casi di sindrome da perdita capillare in pazienti in trattamento con fattori stimolanti le colonie di granulociti umani. Tali casi si sono verificati generalmente in pazienti con stadi avanzati di malattia, sepsi, in trattamento con chemioterapia multi-agente o sottoposti ad aferesi.
Vasculite cutanea
È stata riportata vasculite cutanea in pazienti in trattamento con filgrastim. Il meccanismo della vasculite nei pazienti in trattamento con filgrastim non è noto. Durante un trattamento prolungato, la vasculite cutanea è stata osservata nel 2% dei pazienti con NC.
Leucocitosi
Leucocitosi (conteggio assoluto di leucociti > 50 × 109/l) è stata osservata nel 41% dei donatori. Inoltre, dopo somministrazione di filgrastim e leucaferesi, nel 35% dei donatori si è osservata una trombocitopenia transitoria (conteggio assoluto di piastrine < 100 × 109/l).
Sindrome di Sweet
Sono stati riportati casi di sindrome di Sweet (dermatosi neutrofila acuta febbrile) in pazienti oncologici in trattamento con filgrastim. Tuttavia, poiché la maggior parte di questi pazienti aveva leucemia, una malattia spesso associata alla sindrome di Sweet, un rapporto causale con l’uso di filgrastim non è stato confermato.
Pseudogotta (pirofosfato di condrocalcinosi)
Sono stati riportati casi di pseudogotta in pazienti oncologici in trattamento con filgrastim.
Reazioni di "trapianto contro l'ospite"
Sono stati riportati casi di reazione di "trapianto contro l'ospite" e decessi tra i pazienti trattati con G-CSF dopo trapianto allogenico di midollo osseo.
Immunogenicità.
Dati di quattro studi clinici condotti su volontari sani e pazienti oncologici indicano che nessun soggetto ha sviluppato anticorpi anti-rG-CSF in seguito alla somministrazione di filgrastim.
Pediatria.
I dati degli studi clinici nei pazienti pediatrici indicano che la sicurezza e l’efficacia dell’uso di filgrastim sono simili in adulti e bambini sottoposti a chemioterapia citotossica, suggerendo l’assenza di differenze legate all’età nella farmacocinetica di filgrastim. L’unico effetto indesiderato riportato costantemente è stato il dolore muscolo-scheletrico, che non differisce da quello osservato negli adulti.
I dati disponibili sono insufficienti per una valutazione ulteriore dell’uso di filgrastim nei bambini.
Altre categorie di pazienti particolari.
Pazienti anziani
Non sono state osservate differenze generali di sicurezza ed efficacia nei soggetti di età superiore a 65 anni rispetto ai giovani adulti (dai 18 anni in su) sottoposti a chemioterapia citotossica, e l’esperienza clinica non ha evidenziato differenze nelle risposte tra pazienti anziani e giovani adulti. I dati sono insufficienti per valutare l’uso di filgrastim nei pazienti anziani per altri indicazioni approvate.
Bambini con NC
Sono stati riportati casi di riduzione della densità minerale ossea e osteoporosi in bambini con neutropenia cronica grave in trattamento prolungato con filgrastim. La frequenza di questo effetto indesiderato negli studi clinici è stata considerata comune.
Segnalazione delle reazioni avverse sospette.
La segnalazione delle reazioni avverse sospette dopo l’autorizzazione del medicinale è importante. Permette un monitoraggio continuo del rapporto beneficio/rischio del farmaco.
I professionisti sanitari devono segnalare qualsiasi reazione avversa attraverso il sistema di farmacovigilanza dell’Ucraina.
Durata della validità. 3 anni.
Condizioni di conservazione.
Conservare a 2-8 °C nella confezione originale. Non congelare.
Conservare fuori dalla portata dei bambini.
Dopo la ricostituzione, la soluzione è stabile per 24 ore a 2-8 °C.
Dal punto di vista microbiologico, la soluzione deve essere utilizzata immediatamente.
Confezionamento.
0,5 ml di soluzione in siringa preriempita in vetro incolore trasparente, dotata di stantuffo con guarnizione in gomma grigia, ago per iniezione, tappo protettivo in gomma grigia, tappo esterno in polipropilene e dispositivo di protezione per prevenire lesioni da ago dopo l’uso, in confezione blister.
1 o 5 confezioni blister in scatola di cartone.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore.
Novartis Pharmaceuticals Manufacturing GmbH
oppure
Sandoz GmbH – Unità Produttiva Farmaci Aseptici Schaffhausen (FAS)
Indirizzo del produttore e sede operativa.
Biochemiestrasse 10, Unterlängkampfen, Längkampfen, 6336, Austria
oppure
Biochemiestrasse 10, 6336 Längkampfen, Austria