Zarxio®
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO ZARZIO®
Composición:
Principio activo: filgrastim (factor estimulante de colonias de granulocitos humanos recombinante);
1 ml de solución contiene 60 millones de UI (600 µg) o 96 millones de UI (960 µg) de filgrastim;
la jeringa precargada (unidad de dosis) contiene 30 millones de UI (300 µg) o 48 millones de UI (480 µg) de filgrastim en 0,5 ml;
Excipientes: ácido glutámico, sorbitol (E 420), polisorbato 80, agua para inyección.
Forma farmacéutica. Solución inyectable o para perfusión.
Principales propiedades físico-químicas: solución transparente incolora o ligeramente amarillenta.
Grupo farmacoterapéutico.
Inmunoestimulantes. Factores estimulantes de colonias. Filgrastim.
Código ATC L03A A02.
Propiedades farmacológicas.
Farmacodinámica.
La sustancia activa del medicamento es filgrastim, un factor estimulante de colonias de granulocitos (FEC-G) humano recombinante. Filgrastim tiene la misma actividad biológica que el FEC-G humano endógeno y solo se diferencia de este último en que es una proteína no glucosilada con un residuo adicional de metionina en el extremo N-terminal. Filgrastim, obtenido mediante tecnología de ADN recombinante, se aísla a partir de células de la bacteria Escherichia coli, cuyo aparato genético ha sido modificado mediante la introducción del gen que codifica la proteína FEC-G.
El factor estimulante de colonias de granulocitos humano – una glicoproteína – regula la formación de granulocitos neutrófilos funcionalmente activos y su liberación a la sangre desde la médula ósea. Filgrastim aumenta significativamente el número de granulocitos neutrófilos en la sangre periférica ya durante las primeras 24 horas tras la administración, y simultáneamente provoca un ligero aumento en el número de monocitos. El incremento en la cantidad de granulocitos neutrófilos con el uso del medicamento dentro del rango de dosis recomendadas depende de la magnitud de la dosis. Sus propiedades funcionales son normales o potenciadas, como lo demuestran los resultados de estudios sobre quimiotaxis y fagocitosis. Tras finalizar el tratamiento con el medicamento, el número de granulocitos neutrófilos en la sangre periférica disminuye en un 50 % en el plazo de 1–2 días y regresa a niveles normales en 1–7 días.
El uso de filgrastim reduce significativamente la frecuencia, gravedad y duración de la neutropenia en pacientes tras quimioterapia con citostáticos o tras terapia mieloablativa seguida de trasplante de médula ósea. La administración de filgrastim, tanto primaria como tras quimioterapia, activa las células progenitoras hematopoyéticas de la sangre periférica (CPHP). Estas CPHP autólogas pueden recolectarse del paciente y administrárselas posteriormente tras tratamiento con citostáticos en dosis altas, ya sea en lugar del trasplante de médula ósea o como complemento al mismo. La administración de CPHP acelera la recuperación de la hematopoyesis, reduce el riesgo de complicaciones hemorrágicas y la necesidad de transfusiones de concentrado de plaquetas. En niños y adultos con neutropenia congénita severa (NCS), filgrastim aumenta de forma estable el número de granulocitos neutrófilos en la sangre periférica y reduce la frecuencia de complicaciones infecciosas.
Farmacocinética.
Tanto tras la administración intravenosa como subcutánea del medicamento, se observa una relación lineal positiva entre la concentración plasmática del fármaco y la dosis administrada. Tras la administración subcutánea de las dosis recomendadas, la concentración en suero sanguíneo supera los 10 ng/ml durante 8–16 horas; el volumen de distribución en sangre es de aproximadamente 150 ml/kg. Tanto tras la administración subcutánea como intravenosa, la eliminación del medicamento del organismo sigue una cinética de primer orden. El valor medio del período de semivida de eliminación de filgrastim en suero es de aproximadamente 3,5 horas, y la velocidad de depuración (clearance) es de aproximadamente 0,6 ml/min por kg de peso. La administración continua mediante infusión durante 28 días en pacientes en recuperación tras un trasplante autólogo de médula ósea no se asoció con signos de acumulación (cumulación) ni con un aumento del período de semivida del medicamento.
Características clínicas.
Indicaciones.
Reducción de la duración y gravedad de la neutropenia en pacientes que reciben quimioterapia mielosupresora intensiva con agentes citotóxicos por neoplasias malignas (excepto leucemia mieloide crónica y síndrome mielodisplásico), y reducción de la duración de la neutropenia en pacientes que reciben quimioterapia de alta dosis con agentes citotóxicos seguida de trasplante autólogo o alogénico de médula ósea.
La seguridad y eficacia del filgrastim son similares en adultos y niños que reciben quimioterapia con agentes citotóxicos.
-
El medicamento está indicado para la movilización de células madre periféricas (CMP).
-
El tratamiento prolongado está indicado en niños y adultos con neutropenia congénita grave, cíclica o idiopática y neutropenia con un recuento absoluto de neutrófilos <0,5×109/l, con el fin de aumentar el número de neutrófilos y reducir la frecuencia de infecciones.
-
Tratamiento de la neutropenia persistente (recuento absoluto de neutrófilos ≤1,0×109/l) en pacientes con infección por VIH en fase avanzada para reducir el riesgo de infecciones bacterianas, cuando otros métodos de tratamiento de la neutropenia no son adecuados.
Contraindicaciones.
- Hipersensibilidad al filgrastim, a los factores estimulantes de colonias, a Escherichia coli o a cualquiera de los excipientes.
- Neutropenia hereditaria grave (síndrome de Kostmann) con alteraciones citogenéticas y neutropenia autoinmune.
- Estadio terminal de insuficiencia renal crónica (IRC).
- Leucemia mieloide crónica y síndrome mielodisplásico.
Interacción con otros medicamentos y otras formas de interacción.
No se ha establecido la seguridad y eficacia de la administración del medicamento Zarzio® el mismo día que los agentes quimioterapéuticos citotóxicos mielosupresores. Debido a la sensibilidad de las células mieloides de división rápida a la quimioterapia citotóxica mielosupresora, no se recomienda administrar el medicamento Zarzio® dentro de un intervalo de 24 horas antes o después de la administración de estos agentes.
Cuando se administra conjuntamente el medicamento Zarzio® y 5-fluorouracilo, la gravedad de la neutropenia puede aumentar. La interacción con otros factores de crecimiento hematopoyéticos y citoquinas es desconocida.
Dado que el litio estimula la liberación de neutrófilos, es posible un efecto potenciado del medicamento Zarzio® cuando se administra en combinación, aunque no se han realizado estudios al respecto.
Debido a la incompatibilidad farmacéutica, no se debe mezclar el medicamento con solución de cloruro de sodio al 0,9 %.
Características de uso.
Hipersensibilidad
Se han observado reacciones de hipersensibilidad, incluyendo reacciones anafilácticas, al inicio o durante el tratamiento, en pacientes que recibieron filgrastim. Debe interrumpirse el uso del medicamento Zarzio® en pacientes con reacciones de hipersensibilidad clínicamente significativas.
No se debe utilizar el medicamento Zarzio® en pacientes con antecedentes de hipersensibilidad al filgrastim o al pegfilgrastim.
Efectos adversos en el pulmón
Existen datos sobre casos raros de efectos adversos en órganos respiratorios, particularmente el desarrollo de neumonía intersticial tras la administración de G-CSF. Los pacientes que recientemente han sufrido enfermedad pulmonar infiltrativa o neumonía pueden tener un riesgo elevado. La aparición de síntomas como tos, fiebre, disnea, combinada con infiltrados pulmonares detectados mediante radiografía y signos de insuficiencia respiratoria progresiva, permite sospechar el síndrome de dificultad respiratoria del adulto (SDRA). En caso de detectarse SDRA, debe interrumpirse el uso de filgrastim y administrarse el tratamiento adecuado.
Durante el período poscomercialización, se han recibido informes sobre casos muy raros de efectos adversos pulmonares (hemoptisis, hemorragias pulmonares, infiltración pulmonar, disnea e insuficiencia de oxígeno), particularmente en donantes sanos. En caso de sospecha o confirmación de un efecto adverso pulmonar, debe interrumpirse el uso de filgrastim y proporcionarse la asistencia médica adecuada.
Glomerulonefritis
La glomerulonefritis se ha registrado en pacientes que reciben filgrastim y pegfilgrastim. Por lo general, la glomerulonefritis remite tras la reducción de la dosis o la suspensión de filgrastim y pegfilgrastim. Se recomienda realizar periódicamente análisis de orina.
Síndrome de pérdida capilar
Se han notificado casos de síndrome de pérdida capilar, potencialmente mortal si no se trata a tiempo, tras la administración de G-CSF, caracterizado por hipotensión arterial, hipoalbuminemia, edema y hemocentração. Los pacientes que presenten signos de este síndrome requieren vigilancia estrecha y tratamiento sintomático, incluyendo medidas de reanimación.
Esplenomegalia y ruptura del bazo
Tras la administración de filgrastim, se han observado casos de esplenomegalia asintomática y ruptura del bazo en donantes sanos y pacientes. El aumento del tamaño del bazo es una consecuencia directa de la administración de filgrastim. En el 31 % de los pacientes participantes en estudios clínicos se observó esplenomegalia detectada mediante palpación. Se han registrado varios casos de ruptura fatal del bazo. Por lo tanto, es necesario realizar un monitoreo cuidadoso del tamaño del bazo (examen clínico y ecográfico). Se debe descartar el diagnóstico de ruptura del bazo en donantes y/o pacientes que presenten dolor en la parte superior izquierda del abdomen o en el hombro izquierdo. La reducción de la dosis puede frenar o detener la progresión del agrandamiento del bazo; en el 3 % de los pacientes fue necesaria una esplenectomía.
Crecimiento de células malignas
Dado que se sabe que el G-CSF favorece el crecimiento de células mieloides in vitro, efectos similares podrían observarse en algunas células no mieloides in vitro.
Síndrome mielodisplásico o leucemia mielógena crónica
No se han establecido la seguridad y eficacia del filgrastim en pacientes con síndrome mielodisplásico o leucemia mielógena crónica; por lo tanto, el filgrastim no está indicado en estas condiciones. Debe tenerse especial cuidado en el diagnóstico diferencial entre leucemia mieloide aguda y transformación blástica de la leucemia mielógena crónica.
Leucemia mieloide aguda (LMA)
Dado que los datos sobre la seguridad y eficacia del filgrastim en pacientes con leucemia mieloide aguda secundaria (LMA) son limitados, el medicamento debe administrarse con precaución.
No se han establecido la seguridad y eficacia del filgrastim en pacientes con LMA de novo menores de 55 años con pronóstico citogenético favorable [t(8;21), t(15;17) e inv(16)].
Trombocitopenia
La trombocitopenia se observó muy frecuentemente en pacientes que recibieron filgrastim. Es necesario realizar un monitoreo cuidadoso del recuento de plaquetas, especialmente durante las primeras semanas de tratamiento con filgrastim. En caso de desarrollarse trombocitopenia en pacientes con neutropenia congénita severa (NCS), es decir, disminución persistente del recuento de plaquetas a niveles < 100 × 10⁹/l, el tratamiento con filgrastim debe suspenderse temporalmente o reducirse la dosis.
Leucocitosis
El recuento de leucocitos en sangre alcanza o supera 100 × 10⁹/l en menos del 5 % de los pacientes que reciben una dosis diaria del medicamento superior a 0,3 millones de UI/kg (3 µg/kg) de peso corporal. No hay datos sobre efectos adversos directamente relacionados con una leucocitosis de esta gravedad. Sin embargo, debido al posible riesgo asociado con una leucocitosis severa, durante el tratamiento con filgrastim debe controlarse regularmente el recuento de leucocitos. Si el recuento de leucocitos supera 50 × 10⁹/l tras alcanzar el nivel esperado, el medicamento debe suspenderse inmediatamente. En caso de uso del medicamento para la movilización de CPCS, debe suspenderse o ajustarse la dosis cuando el recuento de leucocitos alcance > 70 × 10⁹/l.
Inmunogenicidad
Como con todos los medicamentos proteicos, existe la posibilidad de desarrollo de inmunogenicidad. La frecuencia de aparición de anticuerpos contra el filgrastim es generalmente baja. Se han detectado anticuerpos de unión, como ocurre con todos los medicamentos biológicos; sin embargo, hasta ahora no se han asociado con actividad neutralizante.
Advertencias y precauciones especiales relacionadas con enfermedades concomitantes
Precauciones especiales en pacientes con rasgos o enfermedad falciforme
Se han notificado crisis falciformes, en algunos casos fatales, tras la administración de filgrastim en pacientes con rasgos falciformes o enfermedad falciforme. Los médicos deben tener precaución al prescribir filgrastim a pacientes con rasgos o enfermedad falciforme.
En pacientes con anemia falciforme se han observado casos de crisis hemolítica aguda (aumento del número de glóbulos falciformes), a veces con desenlace fatal. A estos pacientes debe administrárseles filgrastim con precaución y debe vigilarse estrechamente los parámetros clínicos y de laboratorio, prestando especial atención al posible aumento del tamaño del bazo y al desarrollo de trombosis vascular.
Osteoporosis
En pacientes con patología ósea concomitante y osteoporosis, se recomienda controlar periódicamente la densidad mineral ósea durante el uso prolongado (más de 6 meses) de filgrastim.
Precauciones especiales en pacientes con enfermedades oncológicas
El filgrastim no debe utilizarse para aumentar la dosis de quimioterapia citotóxica más allá de los regímenes de dosificación establecidos.
Riesgo asociado con el aumento de la dosis de quimioterapia
Debe tenerse especial precaución en el tratamiento de pacientes con neoplasias malignas que reciben altas dosis de agentes citotóxicos, ya que la eficacia del tratamiento no ha sido establecida en estos casos. Se sabe que dosis más altas de agentes quimioterapéuticos pueden provocar una toxicidad más pronunciada, conduciendo a reacciones adversas cardiovasculares, pulmonares, neurológicas y dermatológicas (véanse las instrucciones para uso médico de los agentes citotóxicos concomitantes).
Efecto de la quimioterapia sobre eritrocitos y plaquetas
El tratamiento con filgrastim solo no previene la trombocitopenia ni la anemia inducidas por quimioterapia mielosupresora. Al usar dosis más altas de agentes quimioterapéuticos (por ejemplo, dosis completas según los regímenes indicados), el riesgo de trombocitopenia y anemia severas aumenta.
Se recomienda controlar periódicamente parámetros del hemograma como hematocrito y recuento de plaquetas. Debe tenerse especial precaución al usar agentes quimioterapéuticos simples o combinados que puedan provocar trombocitopenia severa.
Con el uso de filgrastim para movilización de CPCS, se ha observado una reducción en la gravedad y duración de la trombocitopenia inducida por quimioterapia mielosupresora o mieloablativa.
Otras precauciones especiales
No se ha estudiado la eficacia del medicamento en pacientes con recuento muy bajo de células precursoras mieloides. El filgrastim aumenta el número de neutrófilos actuando principalmente sobre las células precursoras de neutrófilos. Por lo tanto, en pacientes con recuento bajo de células precursoras (por ejemplo, tras radioterapia intensiva, quimioterapia o infiltración de la médula ósea por células tumorales), el número de neutrófilos generados puede reducirse.
En ocasiones, en pacientes que recibieron quimioterapia de alta dosis seguida de trasplante de médula ósea autólogo, se han observado trastornos vasculares, como oclusión venosa y alteraciones en el equilibrio hídrico.
Existen datos sobre el desarrollo de reacción de injerto contra huésped y casos fatales en pacientes que reciben G-CSF tras trasplante alogénico de médula ósea.
La estimulación de la hematopoyesis en la médula ósea en respuesta al tratamiento con factores de crecimiento se asocia con la aparición de cambios patológicos transitorios detectables mediante gammagrafía ósea. Esto debe tenerse en cuenta al interpretar imágenes diagnósticas óseas.
Se han notificado casos de aortitis tras la administración de filgrastim, tanto en personas sanas como en pacientes con cáncer. Los síntomas incluyen fiebre, dolor abdominal, malestar general, dolor de espalda y marcadores inflamatorios elevados (por ejemplo, aumento del nivel de proteína C-reactiva y leucocitosis). En la mayoría de los casos, la aortitis se diagnosticó mediante tomografía computarizada y generalmente remitió tras la suspensión de filgrastim.
Precauciones especiales para pacientes que requieren movilización de CPCS
Movilización
No existe comparación aleatorizada entre los dos métodos recomendados de movilización (filgrastim solo o en combinación con quimioterapia mielosupresora) dentro de una misma población de pacientes. La variabilidad entre pacientes individuales y entre laboratorios en el análisis de células CD34+ dificulta la comparación directa entre estudios. Por lo tanto, es difícil recomendar un método óptimo. La elección del método de movilización debe considerarse en relación con los objetivos generales del tratamiento para cada paciente.
Efecto previo de agentes citotóxicos
En pacientes previamente sometidos a terapia mielosupresora intensiva, el uso de filgrastim para movilización de CPCS puede no provocar un aumento del número de CPCS hasta el nivel mínimo recomendado (≥ 2,0 × 10⁶ células CD34+/kg) o una mejora en la velocidad de recuperación de plaquetas.
Algunos agentes citotóxicos son particularmente tóxicos para las células precursoras hematopoyéticas y afectan negativamente su movilización. El uso prolongado de melfalano, carboplatino o carmustina (BCNU) antes de la movilización celular puede empeorar los resultados. Sin embargo, el uso combinado de melfalano, carboplatino o BCNU con filgrastim es eficaz para la movilización de CPCS. Si se planea un trasplante de CPCS, se recomienda realizar la movilización de células madre en una etapa temprana del tratamiento del paciente. Debe prestarse especial atención al número de células precursoras activadas en estos pacientes antes de la administración de quimioterapia de alta dosis. Si los resultados de movilización no son suficientes según los criterios anteriores, debe considerarse el uso de métodos alternativos de tratamiento que no requieran células precursoras.
Evaluación del número de células precursoras
Al evaluar el número de CPCS movilizadas en pacientes tratados con filgrastim, debe prestarse especial atención al método de cuantificación. Los resultados del análisis citométrico de flujo del número de células CD34+ varían considerablemente según la metodología utilizada; por lo tanto, debe tenerse precaución con los resultados obtenidos en otros laboratorios.
Los análisis estadísticos de la relación entre el número de células CD34+ administradas y la velocidad de normalización del recuento de plaquetas tras quimioterapia de alta dosis muestran una dependencia compleja pero constante. Las recomendaciones sobre la necesidad de un contenido mínimo de ≥ 2,0 × 10⁶ células CD34+/kg se basan en datos publicados sobre la recuperación hematológica adecuada. Por encima del nivel mínimo recomendado, la normalización es más rápida; por debajo, es más prolongada.
Precauciones especiales para donantes sanos que se someten a movilización de CPCS
La movilización de CPCS en donantes sanos afecta su estado de salud y se utiliza exclusivamente para obtener células madre alogénicas para trasplante.
Los donantes que se someten a movilización de CPCS para trasplante deben cumplir con los requisitos estándar de indicadores clínicos y de laboratorio para donantes de células madre. Debe prestarse especial atención a los parámetros de análisis de sangre y a la presencia de enfermedades infecciosas. No se han evaluado la seguridad y eficacia del filgrastim en donantes menores de 16 años ni mayores de 60 años.
La trombocitopenia transitoria (recuento de plaquetas < 100 × 10⁹/l) tras la administración de filgrastim y leucoféresis se observó en el 35 % de los pacientes. Entre ellos, se notificaron dos casos con recuento de plaquetas < 50 × 10⁹/l, atribuidos al procedimiento de leucoféresis.
Si es necesario realizar más de una sesión de leucoféresis, debe prestarse especial atención a los donantes cuyo recuento de plaquetas antes del procedimiento sea < 100 × 10⁹/l; generalmente, no se recomienda realizar leucoféresis si el recuento de plaquetas es < 75 × 10⁹/l.
No debe realizarse leucoféresis en donantes que requieran terapia anticoagulante o que presenten trastornos de la hemostasia.
Debe continuarse el monitoreo de donantes que reciben G-CSF para movilización de CPCS hasta la normalización de los parámetros hematológicos.
En donantes sanos, tras la administración de G-CSF, se han observado cambios citogenéticos transitorios. El significado de estos cambios no está determinado.
La evaluación de la seguridad a largo plazo del medicamento en donantes continúa. No se excluye el riesgo de promoción de clones malignos de células mieloides. Se recomienda que los centros de aféresis realicen exámenes sistemáticos de donantes de células madre durante al menos 10 años para monitorear la seguridad a largo plazo.
Precauciones especiales para receptores que se someten a movilización de CPCS con filgrastim
Los datos indican que la interacción inmunológica entre CPCS alogénicas y el receptor conlleva un mayor riesgo de reacción aguda y crónica de injerto contra huésped en comparación con el trasplante de médula ósea.
Precauciones especiales para pacientes con NCS
El filgrastim no debe administrarse a pacientes con neutropenia congénita severa que hayan desarrollado leucemia ni a pacientes con signos de transformación leucémica.
Análisis de composición sanguínea
Pueden ocurrir otros cambios en el perfil sanguíneo, incluyendo anemia y aumento temporal del número de células precursoras mieloides; es necesario un monitoreo cuidadoso del perfil sanguíneo.
Transformación a leucemia o preleucemia
Debe tenerse especial precaución al diagnosticar NCS para diferenciarla de otras enfermedades hematológicas, como anemia aplásica, mielodisplasia y leucemia. Antes de iniciar el tratamiento, debe realizarse un análisis sanguíneo completo con determinación del perfil leucocitario y recuento de plaquetas, así como evaluación morfológica de la médula ósea y cariotipo.
La aparición de síndrome mielodisplásico (SMD) o leucemia en pacientes con neutropenia crónica severa que participaron en estudios clínicos con filgrastim es rara (aproximadamente en el 3 % de los casos). Estos trastornos solo se observaron en pacientes con neutropenia congénita. El SMD y la leucemia son complicaciones frecuentes de la enfermedad; su relación con el tratamiento con filgrastim es incierta. Aproximadamente en el 12 % de los pacientes (sin alteraciones citogenéticas antes del tratamiento) se observaron alteraciones en análisis posteriores, incluyendo monosomía 7. En caso de desarrollarse alteraciones citogenéticas en un paciente con neutropenia crónica severa, debe evaluarse cuidadosamente la relación riesgo-beneficio de continuar con filgrastim. Debe suspenderse el uso de filgrastim en caso de desarrollarse SMD o leucemia. No se sabe si el tratamiento prolongado con filgrastim en pacientes con neutropenia crónica severa aumenta el riesgo de alteraciones citogenéticas, SMD o transformación leucémica. Debe realizarse evaluación morfológica y citogenética de la médula ósea de forma periódica, aproximadamente cada 12 meses.
Otras precauciones especiales
Debe descartarse otras causas de neutropenia, como infecciones virales.
La hematuria se observó frecuentemente, mientras que la proteinuria se observó en un número pequeño de pacientes. Para detectar oportunamente estos eventos, es necesario realizar análisis de orina de forma periódica.
No se han establecido la seguridad y eficacia del medicamento en recién nacidos ni en pacientes con neutropenia autoinmune.
Precauciones especiales para pacientes con infección por VIH
Perfil sanguíneo
Es necesario un monitoreo cuidadoso del recuento absoluto de neutrófilos (RAN), especialmente durante las primeras semanas de tratamiento con filgrastim. En algunos pacientes se observa una respuesta muy rápida con aumento significativo del número de neutrófilos tras la primera dosis de filgrastim. Se recomienda determinar el RAN diariamente durante los primeros 2-3 días de tratamiento con filgrastim. Posteriormente, durante las primeras 2 semanas, se recomienda determinar el RAN al menos 2 veces por semana, y luego 1 vez por semana y cada 2 semanas durante la terapia de mantenimiento. Durante la administración periódica de filgrastim con dosis individualizadas y con dosis de 30 millones de UI/día (300 µg/día), pueden ocurrir mayores fluctuaciones en el valor del RAN. Para determinar los valores mínimos (más bajos del RAN), se recomienda obtener muestras de sangre justo antes de la administración programada de filgrastim.
Riesgo asociado con el uso de medicamentos mielosupresores en dosis elevadas
La administración de filgrastim solo no excluye la posibilidad de trombocitopenia y anemia inducidas por quimioterapia mielosupresora. Debido al uso de agentes quimioterapéuticos en dosis más altas o combinaciones más intensas con filgrastim, el riesgo de trombocitopenia y anemia puede aumentar. Se recomienda controlar periódicamente los parámetros del perfil sanguíneo.
Enfermedades infecciosas y malignas que causan mielosupresión
El desarrollo de neutropenia puede ser consecuencia de infiltración de la médula ósea por patógenos de infecciones oportunistas, como Mycobacterium avium, o por neoplasias malignas, como linfoma. En caso de tener infecciones o neoplasias que afecten la médula ósea, para corregir la neutropenia, además de administrar filgrastim, debe realizarse la terapia adecuada para tratar la enfermedad subyacente. No se ha determinado el efecto del filgrastim sobre la corrección de la neutropenia causada por infecciones o neoplasias que afectan la médula ósea.
Sustancias auxiliares
Zarzio® contiene sorbitol, por lo que no debe administrarse a pacientes con intolerancia hereditaria rara a la fructosa.
La tapa protectora contiene derivados del látex natural. Aunque el medicamento en sí no contiene látex natural. No se ha evaluado la seguridad del producto en pacientes sensibles al látex.
Con el fin de mejorar la trazabilidad de G-CSF, el nombre del medicamento administrado debe registrarse claramente en el historial del paciente.
Este medicamento contiene menos de 1 mmol (23 mg)/dosis de sodio, es decir, prácticamente libre de sodio.
Uso durante el embarazo o la lactancia.
No se ha establecido la seguridad del filgrastim en mujeres embarazadas. Existen datos sobre la penetración de filgrastim a través de la barrera placentaria. No se han obtenido datos sobre teratogenicidad del filgrastim en estudios en animales. En estudios en animales se observó toxicidad reproductiva. En animales que recibieron filgrastim se observó una mayor frecuencia de abortos espontáneos.
No se recomienda el uso del medicamento Zarzio® durante el embarazo.
No se sabe si el filgrastim y sus metabolitos pasan a la leche materna humana; por lo tanto, no puede excluirse el riesgo para el lactante. Por ello, debe tomarse una decisión sobre si suspender la lactancia o el tratamiento con filgrastim, considerando el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
En estudios en animales se ha demostrado que el filgrastim no afecta el sistema reproductivo ni la fertilidad.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
El filgrastim puede tener un efecto leve sobre la capacidad de reacción al conducir vehículos o manejar maquinaria, específicamente, puede causar mareo.
Vía de administración y dosis.
El tratamiento con el medicamento Zarzio® puede realizarse en centros médicos que dispongan del equipamiento diagnóstico necesario. Los médicos deben tener experiencia en el uso de medicamentos que contienen factor estimulante de colonias de granulocitos (G-CSF) y en el tratamiento de pacientes con enfermedades hematológicas.
Los procedimientos de movilización y aféresis deben realizarse en colaboración con médicos que tengan la experiencia adecuada y la posibilidad de realizar el monitoreo necesario de las células precursoras de la hematopoyesis.
Neutropenia en pacientes que reciben quimioterapia citotóxica por enfermedades malignas.
La dosis diaria recomendada del medicamento es de 0,5 millones de UDI/kg (5 µg/kg) de peso corporal, una vez al día. La primera dosis del medicamento debe administrarse no antes de 24 horas después del curso de quimioterapia citotóxica. El medicamento debe administrarse hasta que la cantidad total de neutrófilos en el análisis de sangre clínico supere el nivel esperado y alcance valores normales. Tras la quimioterapia por tumores sólidos, linfomas y linfoleucosis, la duración del tratamiento hasta alcanzar estos valores es de hasta 14 días. Tras la terapia de inducción y consolidación en la leucemia mieloide aguda, la duración del tratamiento puede prolongarse considerablemente (hasta 38 días), dependiendo del tipo, dosis y esquema de la quimioterapia citotóxica empleada.
En los pacientes que reciben quimioterapia citotóxica, suele observarse un aumento transitorio del número de neutrófilos a las 1-2 días después del inicio del tratamiento con Zarzio®. Sin embargo, para lograr un efecto terapéutico estable, es necesario continuar el tratamiento hasta que la cantidad de neutrófilos supere el mínimo esperado y alcance valores normales. No se recomienda suspender prematuramente el tratamiento con el medicamento antes de que la cifra de neutrófilos supere el mínimo esperado.
Vía de administración
El medicamento Zarzio® se administra mediante inyecciones subcutáneas o infusiones intravenosas (diluido en solución de glucosa al 5 %) durante 30 minutos, una vez al día. En la mayoría de los casos, se prefiere la vía subcutánea. Al administrar una dosis única por vía intravenosa, la duración del efecto del medicamento puede reducirse. La relevancia clínica de estos datos respecto al uso de dosis múltiples no ha sido establecida. La elección de la vía de administración depende de las características específicas de cada situación clínica y se determina individualmente para cada paciente.
Pacientes que reciben terapia mieloablativa seguida de trasplante de médula ósea.
La dosis inicial recomendada del medicamento Zarzio® es de 1 millón de UDI/kg (10 µg/kg) de peso corporal por día. La primera dosis debe administrarse no antes de 24 horas después de la quimioterapia citotóxica y no antes de 24 horas después del trasplante de médula ósea.
Después del descenso máximo del número de neutrófilos (nadir), la dosis diaria del medicamento Zarzio® debe ajustarse según los cambios en la cifra de neutrófilos (véase la tabla).
Selección de la dosis de Zarzio® en respuesta al logro del nadir.
| Recuento absoluto de neutrófilos (ANC) |
Ajuste de la dosis del medicamento ZARZIO® |
| ANC > 1 × 109/l durante 3 días consecutivos |
Reducción de la dosis a 0,5 millones de UI/kg (5 µg/kg) de peso corporal al día |
| ANC > 1 × 109/l durante los siguientes 3 días consecutivos |
Supresión del medicamento |
| Si durante el tratamiento el ANC disminuye hasta niveles < 1 × 109/l, la dosis del medicamento ZARZIO® debe aumentarse de acuerdo con el esquema indicado anteriormente. |
|
Vía de administración
El medicamento debe disolverse en 20 ml de solución de glucosa al 5 % y administrarse como una infusión intravenosa breve durante 30 minutos o como una infusión subcutánea o intravenosa prolongada durante 24 horas.
Movilización de células progenitoras hematopoyéticas periféricas (CPHP) en pacientes que reciben terapia mielosupresora o mieloablativa seguida de trasplante autólogo de CPHP
Pacientes que reciben terapia mielosupresora o mieloablativa seguida de trasplante autólogo de CPHP.
Para la movilización de CPHP, cuando se utiliza Zarsio® como monoterapia, la dosis recomendada es de 1 millón de UI/kg (10 µg/kg) de peso corporal al día durante 5-7 días consecutivos mediante infusión subcutánea continua durante 24 horas. Habitualmente son suficientes 1-2 sesiones de leucoaféresis en los días 5 y 6. En algunos casos, puede ser necesario realizar una sesión adicional de leucoaféresis. No se debe modificar la dosis del medicamento antes de la leucoaféresis final.
Para la movilización de CPHP tras quimioterapia mielosupresora, la dosis recomendada de Zarsio® es de 0,5 millones de UI/kg (5 µg/kg) de peso corporal al día, comenzando el primer día tras finalizar el ciclo de quimioterapia, hasta que el recuento de neutrófilos pase el mínimo esperado y alcance valores normales. La leucoaféresis debe realizarse durante el período de aumento del recuento absoluto de neutrófilos (RAN) desde < 0,5 × 10⁹/l hasta > 5 × 10⁹/l. En pacientes que no han recibido quimioterapia intensiva, habitualmente es suficiente una sesión de leucoaféresis. En casos individuales, se recomienda realizar sesiones adicionales de leucoaféresis.
Vía de administración
Filgrastim para la movilización de CPHP, cuando se utiliza solo, puede administrarse como infusión subcutánea continua durante 24 horas o mediante inyección subcutánea. Para las infusiones, filgrastim debe diluirse en 20 ml de solución de glucosa al 5 %.
Filgrastim para la movilización de CPHP tras quimioterapia mielosupresora debe administrarse por vía subcutánea.
Movilización de CPHP en donantes sanos antes del trasplante alogénico de CPHP.
Para la movilización de CPHP antes del trasplante alogénico de CPHP en donantes sanos, la dosis recomendada de Zarsio® es de 1 millón de UI/kg (10 µg/kg) de peso corporal al día durante 4-5 días consecutivos. La leucoaféresis debe comenzar en el día 5 y, si es necesario, continuar hasta el día 6, con el fin de obtener 4 × 10⁶ células CD34+/kg de peso corporal del receptor.
Vía de administración
Filgrastim debe administrarse mediante inyección subcutánea.
En pacientes con neutropenia crónica severa (NCS)
Neutropenia hereditaria
La dosis inicial recomendada es de 1,2 millones de UI/kg (12 µg/kg) de peso corporal al día mediante una inyección subcutánea única o fraccionada.
Neutropenia idiopática y periódica
La dosis inicial recomendada es de 0,5 millones de UI/kg (5 µg/kg) de peso corporal al día, en una sola dosis o fraccionada.
Ajuste de la dosis
Zarsio® debe administrarse diariamente mediante inyección subcutánea hasta alcanzar y mantener estable un recuento de neutrófilos superior a 1,5 × 10⁹/l. Tras alcanzar el efecto terapéutico, debe determinarse la dosis mínima eficaz para mantener este nivel. Para mantener el recuento adecuado de neutrófilos, es necesario el uso diario continuo del medicamento. Después de 1-2 semanas de tratamiento, la dosis inicial puede duplicarse o reducirse a la mitad según la eficacia del tratamiento. Posteriormente, se debe realizar un ajuste individual de la dosis cada 1-2 semanas para estabilizar el recuento medio de neutrófilos en un rango de 1,5 × 10⁹/l a 10 × 10⁹/l. En pacientes con infecciones graves, puede utilizarse un esquema con aumento más rápido de la dosis. No se ha establecido la seguridad del uso prolongado de filgrastim en dosis superiores a 2,4 millones de UI (24 µg/kg) al día.
Vía de administración
Filgrastim debe administrarse mediante inyección subcutánea.
En pacientes con infección por VIH
Recuperación del recuento de neutrófilos
La dosis inicial recomendada del medicamento es de 0,1 millones de UI/kg (1 µg/kg) de peso corporal al día, aumentando hasta 0,4 millones de UI (4 µg/kg) de peso corporal al día mediante una inyección subcutánea única hasta la normalización del recuento de neutrófilos (RAN > 2,0 × 10⁹/l). La normalización del recuento de neutrófilos generalmente se alcanza en 2 días. En un número reducido de casos (< 10 % de los pacientes), para recuperar el recuento de neutrófilos, la dosis del medicamento puede aumentarse hasta 1 millón de UI/kg (10 µg/kg de peso corporal al día).
Mantenimiento del recuento normal de neutrófilos
Cuando se ha corregido la neutropenia, debe establecerse la dosis mínima eficaz para mantener niveles normales de neutrófilos. Tras alcanzar el efecto terapéutico, la dosis de mantenimiento es de 300 µg/día 2-3 veces por semana según un esquema alternativo (cada otro día). Posteriormente, puede ser necesaria una corrección individual de la dosis y el uso prolongado del medicamento para mantener un recuento medio de neutrófilos > 2,0 × 10⁹/l.
Vía de administración
Filgrastim debe administrarse mediante inyección subcutánea.
Categorías especiales de pacientes
No se requiere ajuste de dosis en pacientes con insuficiencia hepática o renal grave, ya que los parámetros farmacocinéticos y farmacodinámicos resultaron similares a los de voluntarios sanos.
No existen recomendaciones especiales para el uso de Zarsio® en pacientes de edad avanzada.
Pacientes pediátricos.
En la práctica pediátrica, el perfil de seguridad de Zarsio® en pacientes con NCS y enfermedades oncológicas no difiere del observado en adultos. No se ha establecido la seguridad ni la eficacia del medicamento en recién nacidos.
Las recomendaciones de dosificación en pacientes pediátricos son las mismas que en adultos que reciben quimioterapia citotóxica mielosupresora.
Recomendaciones antes de la administración
Antes de la administración del medicamento, debe realizarse un control visual del contenido de la jeringa precargada. La solución debe ser transparente y sin partículas. La exposición breve a bajas temperaturas no afecta negativamente la estabilidad del medicamento. El medicamento no contiene conservantes. Para evitar la contaminación microbiana, debe tenerse en cuenta que Zarsio® en jeringa precargada está destinado únicamente para uso único. Durante el almacenamiento y para uso ambulatorio, el medicamento puede sacarse del refrigerador y mantenerse a temperatura ambiente (no superior a 25 °C) una sola vez durante un máximo de 8 días. Tras finalizar este período, no debe volver a refrigerarse y debe eliminarse.
Recomendaciones sobre la dilución del medicamento.
Zarsio® puede administrarse diluido en solución de glucosa al 5 %. No se recomienda diluirlo hasta una concentración inferior a 0,2 millones de UI/ml (2 µg/ml). Cuando se diluya a una concentración < 1,5 millones de UI/ml (15 µg/ml), debe añadirse albúmina humana hasta alcanzar una concentración de 2 mg/ml. Por ejemplo, para obtener un volumen de solución de 20 ml y una dosis total de Zarsio® de 30 millones de UI (300 µg), es necesario añadir 0,2 ml de solución de albúmina (solución al 20 %).
Al diluirse en solución de glucosa, el medicamento puede adsorberse al vidrio y a otros materiales utilizados para la administración por infusión. Está prohibido utilizar solución de cloruro sódico para la dilución del medicamento.
Se ha confirmado la estabilidad química y física de la solución diluida para infusión durante 24 horas tras la apertura del envase, siempre que se almacene a una temperatura de 2 a 8 °C. Desde el punto de vista microbiológico, el medicamento debe usarse inmediatamente. Si no se utiliza inmediatamente, el usuario será responsable del tiempo y condiciones de almacenamiento tras la apertura del envase hasta su administración. La dilución debe realizarse en condiciones asépticas controladas y autorizadas.
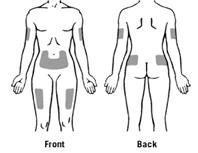
Las zonas preferidas del cuerpo para la administración subcutánea de Zarsio® se muestran en la figura:
Instrucciones para la autoinyección del medicamento por el paciente
Esta sección contiene información sobre la autoinyección de Zarsio® por el paciente. ¡Importante! No intente administrarse la inyección por sí mismo si no ha sido entrenado previamente por un médico o enfermera en la técnica de inyección. Zarsio® se suministra con un dispositivo protector para prevenir lesiones por aguja tras su uso, y su médico o enfermera le mostrarán cómo usar la jeringa. Si tiene dudas sobre su capacidad para realizar la inyección o tiene preguntas, consulte a su médico o enfermera para obtener ayuda.
¡Atención! No utilice la jeringa si ha caído sobre una superficie dura o si ha caído después de retirar la tapa de la aguja.
- Lávese las manos.
- Saque una jeringa del envase y retire el dispositivo protector para prevenir lesiones por aguja de la aguja de inyección. Las jeringas tienen marcas de graduación en relieve para el caso de que sea necesario llenar parcialmente la jeringa. Cada marca graduada equivale a 0,1 ml. Si no se necesita llenar completamente la jeringa, elimine el exceso de solución antes de la inyección.
- Limpie la piel en el lugar de inyección con una toallita con alcohol.
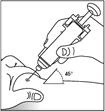
- Forme un pliegue cutáneo pellizcando la piel con el pulgar y el dedo índice.
- Introduzca la aguja en el pliegue cutáneo con un movimiento rápido y seguro. Inyecte la solución de Zarsio® tal como le haya mostrado su médico. Si tiene dudas, debe consultar a su médico o farmacéutico.
|
|
La siguiente información está destinada únicamente a profesionales médicos.
El dispositivo de protección para prevenir lesiones por punción con aguja tras su uso cubre la aguja tras la inyección, con el fin de prevenir lesiones por punción con aguja. Esto no afecta al funcionamiento normal de la jeringa. Presione lentamente y de forma uniforme el émbolo hasta que se haya administrado toda la dosis y el émbolo no pueda avanzar más. Manteniendo la presión sobre el émbolo, retire la aguja de la piel. El dispositivo de protección para prevenir lesiones por punción con aguja tras su uso cubrirá la aguja tras soltar el émbolo.
Eliminación
Los medicamentos no utilizados o los residuos deben eliminarse de acuerdo con los requisitos locales.
Sobredosificación.
Los síntomas de sobredosis con ZARZIO® son desconocidos. De uno a dos días después de la interrupción del tratamiento con el medicamento, la cantidad de granulocitos neutrófilos circulantes generalmente disminuye en un 50 %, y vuelve a la normalidad entre uno y siete días después.
Reacciones adversas.
La frecuencia de aparición de reacciones adversas se clasifica de la siguiente manera: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100, < 1/10); poco frecuentes (≥ 1/1000, < 1/100); raras (≥ 1/10000, < 1/1000); muy raras (< 1/10000).
Las reacciones adversas más graves que pueden ocurrir durante el tratamiento con filgrastim incluyen: reacciones anafilácticas, efectos adversos pulmonares graves (incluyendo neumonitis intersticial y síndrome de dificultad respiratoria aguda), síndrome de fuga capilar, esplenomegalia grave/rotura del bazo, síndrome mielodisplásico o leucemia en pacientes con neutropenia congénita grave, reacción de rechazo del injerto en pacientes que reciben trasplante alogénico de médula ósea o trasplante de células precursoras de sangre periférica y crisis de células falciformes en pacientes con enfermedad de células falciformes.
Los efectos adversos más comunes durante la terapia con filgrastim son fiebre, dolor óseo y muscular (incluyendo dolor óseo, dolor de espalda, artralgia, mialgia, dolor en extremidades, dolor músculo-óseo, dolor músculo-óseo en el tórax, dolor de cuello), anemia, náuseas y vómitos. En pacientes con enfermedades oncológicas, el dolor músculo-óseo de intensidad baja a moderada se observó en el 10 % de los pacientes y de intensidad alta en el 3 %. El dolor óseo y muscular generalmente se alivia con analgésicos estándar.
Durante la movilización de CPCP en donantes sanos, la reacción adversa más frecuente fue el dolor músculo-esquelético.
En pacientes con NC grave, las reacciones adversas más frecuentes durante el tratamiento con filgrastim fueron dolor óseo, dolor músculo-esquelético generalizado, aumento de tamaño y rotura del bazo. El síndrome mielodisplásico (SMD) o leucemia se observaron en pacientes con neutropenia congénita que recibieron filgrastim.
El síndrome de fuga capilar, que puede poner en peligro la vida si no se trata inmediatamente, se ha notificado con frecuencia poco frecuente (≥1/1000 a <1/100) en pacientes con neoplasias malignas sometidos a quimioterapia y en donantes sanos durante el procedimiento de movilización de células madre periféricas tras la administración de factores estimulantes de colonias de granulocitos humanos.
En estudios clínicos realizados en pacientes con infección por VIH, las reacciones adversas consideradas relacionadas con el uso de filgrastim fueron únicamente dolor músculo-esquelético, dolor óseo y mialgia.
La administración de filgrastim no incrementó la frecuencia de efectos adversos provocados por la quimioterapia citotóxica. Entre los efectos adversos observados con igual frecuencia en pacientes que recibieron filgrastim/quimioterapia y en aquellos que recibieron placebo/quimioterapia, se incluyen náuseas y vómitos, alopecia, diarrea, fatiga, anorexia, mucositis, cefalea, tos, erupciones cutáneas, dolor torácico, debilidad general, dolor de garganta, estreñimiento y dolor no especificado.
En pacientes sometidos a quimioterapia con altas dosis seguida de trasplante autólogo de médula ósea, se observaron trastornos del sistema vascular.
A continuación se presenta una lista de reacciones adversas descritas en estudios clínicos y reportadas espontáneamente.
Infecciones e infestaciones
Frecuentes: sepsis, bronquitis, infecciones de las vías respiratorias superiores, infecciones del tracto urinario.
Del sistema sanguíneo y linfático
Muy frecuentes: trombocitopenia, anemia¹.
Frecuentes: esplenomegalia¹, disminución del nivel de hemoglobina⁵.
Poco frecuentes: leucocitosis¹, alteraciones funcionales del bazo.
Rare: rotura del bazo¹, crisis de células falciformes.
Del sistema inmunitario
Frecuentes: reacciones alérgicas, erupciones cutáneas, urticaria, angioedema.
Poco frecuentes: reacciones de hipersensibilidad, reacciones de hipersensibilidad al medicamento¹, reacciones de injerto contra huésped².
Rare: reacciones anafilácticas.
Del metabolismo y nutrición
Frecuentes: aumento del nivel de lactato deshidrogenasa (LDH) en sangre, disminución del apetito⁵.
Poco frecuentes: aumento de ácido úrico en sangre, hiperuricemia.
Rare: pseudogota¹, disminución del nivel de glucosa en sangre, alteraciones en el equilibrio hídrico.
Del sistema psíquico
Frecuentes: insomnio.
Del sistema nervioso
Muy frecuentes: cefalea¹.
Frecuentes: mareo, hipostesia, parestesia.
Del sistema vascular
Frecuentes: hipertensión arterial, hipotensión arterial.
Poco frecuentes: enfermedad veno-oclusiva⁴.
Rare: síndrome de fuga capilar¹, aortitis.
Del sistema respiratorio, órganos torácicos y mediastino
Frecuentes: hemoptisis, disnea, tos¹, dolor orofaríngeo¹,⁵, epistaxis, falta de aire.
Poco frecuentes: síndrome de distrés respiratorio agudo¹, insuficiencia respiratoria¹, edema pulmonar¹, enfermedad intersticial pulmonar¹, formación de infiltrados pulmonares¹, hemorragia pulmonar, hipoxia.
Del tracto gastrointestinal
Muy frecuentes: diarrea¹,⁵, vómitos¹,⁵, náuseas¹.
Frecuentes: dolor en la boca, estreñimiento.
Del sistema hepatobiliar
Frecuentes: hepatomegalia, aumento del nivel de fosfatasa alcalina en sangre.
Poco frecuentes: aumento del nivel de aspartato aminotransferasa (AST), aumento del nivel de gamma-glutamil transferasa (GGT).
De la piel y tejidos subcutáneos
Muy frecuentes: alopecia¹.
Frecuentes: erupciones cutáneas¹, eritema.
Poco frecuentes: erupciones máculo-papulosas, síndrome de Sweet, vasculitis cutánea¹.
Del sistema osteoarticular y tejidos conectivos
Muy frecuentes: dolor muscular y óseo³.
Frecuentes: espasmo muscular.
Poco frecuentes: osteoporosis.
Rare: empeoramiento de la artritis reumatoide y síntomas artríticos, disminución de la densidad ósea.
De los riñones y vías urinarias
Frecuentes: disuria, hematuria.
Poco frecuentes: proteinuria.
Rare: alteraciones patológicas en el análisis de orina, glomerulonefritis.
Trastornos generales y en el sitio de administración
Muy frecuentes: fatiga¹, debilidad, mucositis¹, pirexia.
Frecuentes: dolor torácico¹, dolor¹, astenia¹, malestar⁵, edema periférico⁵, dolor en el sitio de inyección.
Poco frecuentes: reacciones en el sitio de inyección.
Lesiones, intoxicaciones y complicaciones procedimentales
Frecuentes: reacción transfusional⁵.
¹ Véase descripción de reacciones adversas específicas.
² Se han notificado casos de reacciones de injerto contra huésped y muertes en pacientes que recibieron G-CSF tras trasplante alogénico de médula ósea (véase descripción de reacciones adversas específicas).
³ Incluye dolor óseo, dolor de espalda, artralgia, mialgia, dolor en extremidades, dolor músculo-óseo, dolor músculo-óseo torácico, dolor de cuello.
⁴ Se han notificado casos durante el periodo poscomercialización en pacientes que recibieron trasplante alogénico de médula ósea o movilización de CPCP.
⁵ Los eventos adversos se presentaron con mayor frecuencia en pacientes que recibieron filgrastim en comparación con el grupo placebo y están relacionados con las consecuencias de la enfermedad maligna subyacente o de la quimioterapia citotóxica.
Descripción de reacciones adversas específicas
Hipersensibilidad
En estudios clínicos y durante el periodo poscomercialización se han registrado reacciones de hipersensibilidad, incluyendo anafilaxia, erupciones cutáneas, urticaria, angioedema de Quincke, disnea e hipotensión arterial, que pueden ocurrir durante la administración inicial o posterior. En general, se notificaron más frecuentemente tras la administración intravenosa. En algunos casos, los síntomas reaparecieron tras una prueba de provocación, lo que sugiere una relación causal. En caso de reacciones alérgicas graves, no se debe continuar el tratamiento con filgrastim.
Del pulmón
Durante estudios clínicos y en el periodo poscomercialización se han observado efectos adversos pulmonares, incluyendo hemoptisis, hemorragia pulmonar, infiltración pulmonar, disnea e hipoxia, que pueden llevar a insuficiencia respiratoria o al síndrome de distrés respiratorio del adulto (SDRA), a veces con desenlace fatal. Se han notificado casos muy raros en donantes sanos.
Esplenomegalia y rotura del bazo
Tras la administración de filgrastim se han notificado casos de aumento del tamaño del bazo y rotura del bazo. Algunos casos de rotura del bazo fueron fatales.
En todos los casos, el grado de aumento del bazo en pacientes con infección por VIH fue leve a moderado según el examen clínico, y el curso clínico del trastorno fue benigno; no se diagnosticó hiperesplenismo en ningún paciente ni se realizó esplenectomía. Dado que el aumento del bazo es una complicación frecuente en pacientes con infección por VIH y se observa con distintos grados de severidad en la mayoría de los pacientes con SIDA, la relación causal con el uso de filgrastim permanece incierta.
Síndrome de fuga capilar
Se han notificado casos de síndrome de fuga capilar asociados con el uso de factores estimulantes de colonias de granulocitos humanos. Estos casos generalmente ocurren en pacientes con enfermedad avanzada, sepsis, aquellos que reciben quimioterapia combinada o que se someten a aféresis.
Vasculitis cutánea
Se han notificado casos de vasculitis cutánea en pacientes que recibieron filgrastim. El mecanismo de la vasculitis en pacientes que reciben filgrastim es desconocido. Durante el tratamiento prolongado, la vasculitis cutánea se registró en el 2 % de los pacientes con NC grave.
Leucocitosis
La leucocitosis (recuento absoluto de leucocitos > 50 × 10⁹/l) se observó en el 41 % de los donantes. Asimismo, tras la administración de filgrastim y leucaféresis, se observó trombocitopenia transitoria (recuento absoluto de plaquetas < 100 × 10⁹/l) en el 35 % de los donantes.
Síndrome de Sweet
Se han notificado casos de síndrome de Sweet (dermatosis febril aguda atípica) en pacientes con enfermedades oncológicas que reciben filgrastim. Sin embargo, dado que la mayoría de estos pacientes padecen leucemia, una enfermedad que frecuentemente conduce al síndrome de Sweet, no se ha confirmado una relación causal con el uso de filgrastim.
Pseudogota (pirofosfato de calcio dihidratado)
Se han notificado casos de pseudogota en pacientes con enfermedades oncológicas que reciben filgrastim.
Reacciones de injerto contra huésped
Se han notificado casos de reacciones de injerto contra huésped y muertes en pacientes que recibieron G-CSF tras trasplante alogénico de médula ósea.
Inmunogenicidad
Según datos de cuatro estudios clínicos en voluntarios sanos y pacientes con enfermedades oncológicas, no se observó formación de anticuerpos anti-rG-CSF tras la administración de filgrastim en ninguno de los sujetos.
Pediatría
Los datos de estudios clínicos en pacientes pediátricos indican que la seguridad y eficacia del filgrastim son comparables en niños y adultos que reciben quimioterapia citotóxica, lo que sugiere ausencia de diferencias relacionadas con la edad en la farmacocinética del filgrastim. La única reacción adversa reportada consistentemente fue el dolor músculo-esquelético, que no difiere del observado en adultos.
Existen datos insuficientes para evaluar el uso de filgrastim en niños.
Otras categorías especiales de pacientes
Pacientes de edad avanzada
No se han observado diferencias generales en seguridad y eficacia en sujetos mayores de 65 años en comparación con adultos jóvenes (a partir de 18 años) que recibieron quimioterapia citotóxica, y la experiencia clínica no ha revelado diferencias en las respuestas entre pacientes de edad avanzada y adultos jóvenes. No existen datos suficientes para evaluar el uso de filgrastim en pacientes de edad avanzada respecto a otras indicaciones aprobadas.
Niños con NC grave
Se han notificado casos de disminución de la densidad mineral ósea y osteoporosis en niños con neutropenia crónica grave que recibieron tratamiento prolongado con filgrastim. La frecuencia de este evento adverso en estudios clínicos se consideró frecuente.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite continuar con el monitoreo de la relación beneficio-riesgo del medicamento.
Los profesionales sanitarios deben informar de cualquier caso de reacción adversa a través del sistema de farmacovigilancia de Ucrania.
Período de validez. 3 años.
Condiciones de conservación.
Conservar a una temperatura de 2-8 °C en el envase original. No congelar.
Mantener fuera del alcance de los niños.
Después de la dilución, la solución es estable durante 24 horas a una temperatura de 2-8 °C.
Desde el punto de vista microbiológico, la solución debe usarse inmediatamente.
Envase.
0,5 ml de solución en jeringa precargada de vidrio transparente incoloro, equipada con émbolo con tapón de goma gris, aguja inyectable, tapón protector de goma gris, tapón externo de polipropileno y dispositivo protector para prevenir lesiones por la aguja tras su uso, en envase blíster.
1 o 5 envases blíster por caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante.
Novartis Pharmaceutical Manufacturing GmbH
o
Sandoz GmbH – Unidad de producción de medicamentos estériles Schaftlach (Medicamentos estériles SL)
Dirección del fabricante y lugar de actividad.
Biochemiestrasse 10, Unterlangkampfen, Langkampfen, 6336, Austria
o
Biochemiestrasse 10, 6336 Langkampfen, Austria