Rezonativ
Ucraina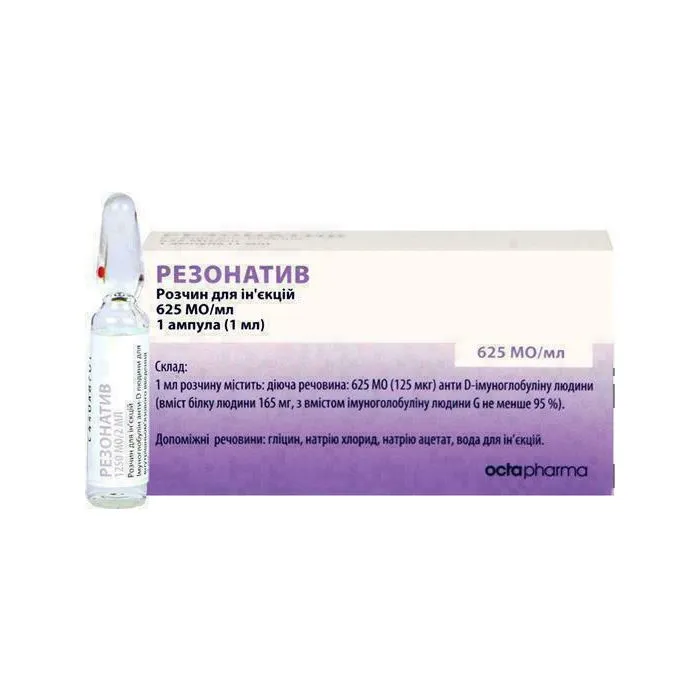
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE REZONATIV
Composizione:
denominazione internazionale non proprietaria: Human anti-D immunoglobulin;
principio attivo: 1 ml di soluzione contiene 625 UI (125 µg) di immunoglobulina anti-D umana. (contenuto proteico umano 165 mg, con contenuto di immunoglobulina umana G non inferiore al 95%);
eccipienti: glicina, cloruro di sodio, acetato di sodio, polisorbato 80, acqua per preparazioni iniettabili.
Il contenuto di immunoglobulina A (IgA) non supera lo 0,05% del contenuto totale di proteine.
Un flaconcino da 1 ml contiene 625 UI (125 µg) di immunoglobulina anti-resso (anti-D) umana.
Un flaconcino da 2 ml contiene 1250 UI (250 µg) di immunoglobulina anti-resso (anti-D) umana.
L'attività è determinata mediante analisi quantitativa secondo la Farmacopea Europea. L'equivalenza in Unità Internazionali del preparato standard internazionale è indicata dall'Organizzazione Mondiale della Sanità.
Distribuzione dei sottoclassi di IgG (valori approssimativi): IgG1 - 70,5%, IgG2 - 26,0%, IgG3 - 2,8%, IgG4 - 0,8%. Contenuto massimo di IgA: 82,5 µg/ml. Prodotto a partire dal plasma sanguigno umano.
Forma farmaceutica. Soluzione iniettabile.
Principali caratteristiche fisico-chimiche: soluzione trasparente o leggermente opalescente, da incolore a giallo pallido o leggermente brunastra. Durante il periodo di conservazione può formarsi una leggera torbidità o una piccola quantità di inclusioni meccaniche.
Gruppo farmacoterapeutico.
Sieri e immunoglobuline immunologici. Immunoglobulina anti-D (Rh). Codice ATC J06BB01.
Proprietà farmacologiche.
Farmacodinamica.
L'immunoglobulina anti-D contiene anticorpi specifici (IgG) contro l'antigene D del fattore Rh (Rh) dei globuli rossi umani.
Durante la gravidanza e in particolare durante il parto, i globuli rossi del feto possono entrare nel circolo sanguigno materno. Se la donna è Rh(D)-negativa e il feto è Rh(D)-positivo, la donna può immunizzarsi contro l'antigene Rh(D) e produrre anticorpi anti-Rh(D), che attraversano la placenta e possono causare la malattia emolitica del neonato. L'immunizzazione passiva con immunoglobulina anti-D previene l'immunizzazione Rh(D) in oltre il 99% dei casi, a condizione che sia stata somministrata una dose sufficiente di immunoglobulina anti-D subito dopo l'esposizione ai globuli rossi Rh(D)-positivi del feto.
Il meccanismo con cui l'immunoglobulina anti-D sopprime l'immunizzazione contro i globuli rossi Rh(D)-positivi non è noto. Tale soppressione potrebbe essere legata alla rimozione dei globuli rossi dalla circolazione prima che raggiungano i siti immunocompetenti oppure, forse, a meccanismi più complessi coinvolti nel riconoscimento dell'antigene estraneo e nella presentazione dell'antigene da parte delle cellule appropriate nei siti pertinenti, in presenza o assenza dell'anticorpo.
Studi sulla profilassi postnatale (studi da 1 a 6) e sulla profilassi antenatale (studio 7) nei pazienti.
Gli studi clinici su Rezonativ sono stati avviati con lo scopo di valutare l'efficacia e la sicurezza del medicinale. Nella tabella sottostante è riportata una panoramica dei dati più importanti relativi all'efficacia:
| Numero dello studio |
Indicazione, numero di persone |
Stato Rh della madre / del bambino |
Presenza di anticorpi anti-D |
Periodo di osservazione |
| 1 |
Rezonativ, n=1937 |
negativo/ positivo |
0,4% |
6 mesi |
| 2 |
Rezonativ, n=2117 Rezonativ, n=723 |
negativo/ positivo successivo bambino Rh positivo |
0,1% 0,7% |
4-6 mesi durante la successiva gravidanza o il parto |
| 3 |
Rezonativ, n=917 |
negativo/ positivo |
0,3% |
6 mesi |
| 4 |
Rezonativ, n=665 |
negativo/ positivo |
0,2% |
6 mesi |
| 5 |
Rezonativ, n=608 IVIG*, n=103 |
negativo/ positivo |
0,3% 0% |
6-8 mesi 8 mesi |
| 6 |
Rezonativ, n=475 |
negativo/ positivo |
0% |
n.d. |
| 7 |
IVIG* & Rezonativ, n=529 |
negativo/ positivo |
0,4% |
8 mesi |
PFP – profilassi post-partum; TUP – profilassi intrauterina; n.r. – non riportato.
* 6-8 settimane prima della data prevista del parto.
Sulla base dei dati di questi studi, si può concludere che il trattamento con Rezonativ assicura una profilassi anti-D affidabile.
Studio sulla trasfusione di componenti ematici Rh-incompatibili.
Nello studio n. 8 è stata valutata l'efficacia di Rezonativ in 21 volontari Rh-negativi ai quali sono stati somministrati globuli rossi Rh-positivi, compatibili per gruppo AB0, in quantità corrispondenti a 10 ml (1 caso), 25 ml (10 casi) e 50 ml (10 casi) di sangue del cordone ombelicale. Dopo 2-3 giorni è stata somministrata per via intramuscolare una dose di 260 mcg di Rezonativ. A 6 mesi (in un caso a 9 mesi) dall'inizio dell'esperimento, non è stata riscontrata alcuna evidenza sierologica di immunizzazione Rh in nessun soggetto. Nel periodo compreso tra 6 mesi e 2,5 anni, 8 soggetti del gruppo 25 ml e tutti i 10 soggetti del gruppo 50 ml hanno ricevuto 5 ml di sangue del cordone ombelicale Rh-positivo e compatibile per gruppo AB0. Dopo 2-3 giorni è stata somministrata una dose rispettivamente di 260 e 333 mcg di Rezonativ. A 6 mesi (in un caso a 8 mesi) non sono state riscontrate anticorpi Rh in nessun soggetto.
I risultati di questo studio dimostrano che la profilassi Rh si ottiene con l'uso di immunoglobulina anti-D alla dose di 10 mcg per 1 ml di sangue fetale. Si è concluso che, poiché l'immunizzazione Rh rappresenta un rischio a causa del sanguinamento feto-placentare alla fine della gravidanza, Rezonativ alla dose di 260 mcg previene l'immunizzazione Rh rilevabile sierologicamente in almeno 998 madri Rh-negative su 1000.
Studio farmacocinetico di Rezonativ.
I principali parametri farmacocinetici e il metabolismo di Rezonativ sono stati studiati in 15 donne Rh-negative in gravidanza, a cui Rezonativ è stato somministrato per via intramuscolare alla 28ª settimana di gestazione. In 8 donne la dose era di 125 mcg, in 7 donne di 250 mcg. Inoltre, 3 donne Rh-negative non in gravidanza hanno ricevuto dosi inferiori.
Il tempo di dimezzamento biologico dell'IgG anti-D dopo iniezione intramuscolare di Rezonativ alla dose di 125 mcg in queste donne è risultato sovrapponibile a quanto descritto in letteratura (vedi sezione «Farmacocinetica»).
Farmacocinetica.
L'immunoglobulina anti-resso (anti-D) umana per somministrazione intramuscolare viene assorbita lentamente nel circolo ematico del ricevente, raggiungendo il picco dopo 2-3 giorni.
Il tempo di dimezzamento nel circolo ematico in soggetti con livelli normali di IgG è di 3-4 settimane. Il tempo di dimezzamento può variare tra pazienti diversi.
Le IgG e i complessi IgG vengono degradati nelle cellule del sistema reticolo-endoteliale.
Caratteristiche cliniche.
Indicazioni.
Prevenzione dell’immunizzazione Rh(D) nelle donne in età fertile Rh(D)-negative
- Profilassi antenatale.
- Profilassi antenatale programmata.
- Profilassi antenatale dopo complicanze della precedente gravidanza, inclusi aborto/spontaneo o minaccia di aborto, gravidanza extrauterina o mola idatiforme, morte fetale intrauterina, emorragia transplacentare dovuta a sanguinamento anteparto, amniocentesi, biopsia dei villi coriali o procedure ostetriche manipolative come versione esterna, interventi invasivi, cordocentesi, trauma addominale contusivo o intervento terapeutico sul feto.
- Profilassi postnatale.
- Nascita di un neonato Rh(D)-positivo (D, D debole, D incompleto).
Trattamento di donne in età fertile Rh(D)-negative dopo trasfusione di sangue Rh-positivo incompatibile o di altri prodotti contenenti eritrociti, ad esempio trombocentratо/trombomassa.
Controindicazioni.
Ipersensibilità ai principi attivi del medicinale o a qualsiasi altro eccipiente.
Ipersensibilità all’immunoglobulina umana, specialmente nei pazienti con anticorpi anti-IgA.
Precauzioni particolari.
Rezonativ non deve essere somministrato per via endovenosa (rischio di shock). Il medicinale deve essere somministrato per via intramuscolare e, prima dell’iniezione, si deve tirare delicatamente lo stantuffo della siringa per assicurarsi che l’ago non sia entrato in un vaso sanguigno.
Nell’uso postnatale, Rezonativ è destinato alla somministrazione alla madre. Non deve essere somministrato al neonato.
Il medicinale non è indicato per persone Rh(D)-positive né per soggetti precedentemente immunizzati contro l’antigene Rh(D).
Dopo la somministrazione del medicinale, il paziente deve essere osservato per almeno 20 minuti e per un’ora in caso di somministrazione accidentale per via endovenosa.
È estremamente raro che l’immunoglobulina anti-D umana possa causare un calo della pressione arteriosa con reazione anafilattica, anche in pazienti precedentemente tolleranti a trattamenti precedenti con immunoglobulina umana.
In caso di comparsa di sintomi di reazione allergica o anafilattica, la somministrazione deve essere immediatamente interrotta.
Ipersensibilità
Le vere reazioni di ipersensibilità sono un evento raro, ma possono verificarsi reazioni allergiche all’immunoglobulina anti-D. I pazienti devono essere informati sui segni precoci di reazioni di ipersensibilità, come orticaria, orticaria generalizzata, senso di costrizione toracica, dispnea, ipotensione e anafilassi. Il trattamento dipende dalla natura e dalla gravità della reazione avversa. In caso di shock, devono essere applicate le moderne procedure mediche per il trattamento dello shock.
Rezonativ contiene una piccola quantità di IgA. Nonostante l’immunoglobulina anti-D sia stata utilizzata con successo nel trattamento di singoli pazienti con deficit di IgA, i pazienti con deficit di IgA possono sviluppare anticorpi anti-IgA e possono manifestare reazioni anafilattiche dopo la somministrazione di medicinali contenenti IgA ottenuti dal plasma umano. Il medico deve valutare attentamente il beneficio rispetto al rischio di possibili reazioni di ipersensibilità.
Raramente, l’immunoglobulina anti-D umana può causare una riduzione della pressione arteriosa con reazione anafilattica, anche in pazienti precedentemente trattati con immunoglobulina umana.
La sospetta comparsa di reazioni allergiche o anafilattoidi richiede l’immediata interruzione dell’iniezione. In caso di shock, deve essere praticato il trattamento medico standard.
Reazioni emolitiche
I pazienti che hanno ricevuto una trasfusione di sangue incompatibile e una dose elevata di immunoglobulina anti-D devono essere sottoposti a un monitoraggio clinico e biologico continuo a causa del rischio di reazioni emolitiche.
Tromboembolia
Complicanze tromboemboliche arteriose e venose, come infarto del miocardio, ictus, trombosi venosa profonda ed embolia polmonare, sono state associate all’uso di immunoglobuline. Nonostante non siano state osservate complicanze tromboemboliche con l’uso di Rezonativ, i pazienti devono essere adeguatamente idratati prima dell’uso di immunoglobuline. È necessaria cautela nel trattamento di pazienti con fattori di rischio preesistenti per eventi trombotici (come ipertensione, diabete mellito, storia di malattie vascolari o complicanze trombotiche, pazienti con trombofilie acquisite o ereditarie, pazienti con periodi prolungati di immobilità, pazienti con grave ipovolemia, pazienti con malattie che aumentano la viscosità del sangue), specialmente quando vengono somministrate dosi più elevate di Rezonativ.
I pazienti devono essere informati sui sintomi precoci di complicanze tromboemboliche, come difficoltà respiratorie (dispnea), dolore e gonfiore agli arti, sintomi neurologici focali e dolore toracico; in caso di comparsa di tali sintomi, devono immediatamente consultare un medico.
Effetto sugli esami sierologici
Dopo la somministrazione di immunoglobulina, l’aumento transitorio di vari anticorpi passivamente trasferiti nel sangue del paziente può portare a risultati falsamente positivi negli esami sierologici.
L’ingresso passivo di anticorpi contro antigeni eritrocitari, ad esempio A, B, D, può influenzare alcuni esami sierologici per gli anticorpi anti-eritrociti, come il test antiglobulinico diretto (DAT, test diretto di Coombs), specialmente nei neonati Rh(D)-positivi le cui madri hanno ricevuto profilassi antenatale.
Pazienti con sovrappeso/obesità
Ai pazienti con sovrappeso/obesità si raccomanda la somministrazione endovenosa del prodotto anti-Rh a causa della possibile inefficacia della somministrazione intramuscolare.
Le misure standard di prevenzione delle infezioni derivanti dall’uso di medicinali ottenuti da sangue o plasma umano comprendono la selezione dei donatori, l’analisi dei singoli campioni di sangue e dei pool di plasma per la presenza di marcatori specifici di infezione e l’implementazione di processi tecnologici efficaci per l’inattivazione o la rimozione dei virus. Tuttavia, nonostante queste misure, non si può escludere completamente la possibilità di trasmissione di agenti infettivi con l’uso di medicinali ottenuti da sangue o plasma umano. Ciò vale anche per virus e altri patogeni sconosciuti o emergenti.
Le misure adottate si considerano efficaci contro virus a capside lipidico come HIV, virus dell’epatite B e C, nonché contro il virus dell’epatite A privo di capside lipidico.
Per alcuni virus privi di capside lipidico, come il parvovirus B19, le misure adottate hanno un’efficacia limitata.
Esiste un’esperienza clinica rassicurante riguardo all’assenza di trasmissione del virus dell’epatite A o del parvovirus B19 con le immunoglobuline, e si ritiene che la presenza di anticorpi sia un fattore importante di protezione antivirale.
Si raccomanda vivamente di registrare ogni volta che Rezonativ viene somministrato, il nome e il numero di lotto del medicinale.
Informazioni importanti su alcuni componenti di Rezonativ
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per 1 ml (625 UI), cioè, in altre parole, è «privo di sodio».
In caso di shock, deve essere praticato il trattamento medico standard.
Interazioni con altri medicinali ed altre forme di interazione.
Vaccini virali vivi attenuati.
La vaccinazione attiva con vaccini virali vivi (ad esempio, morbillo, parotite epidemica o rosolia) deve essere posticipata di 3 mesi dall’ultima somministrazione di immunoglobulina anti-D, poiché l’efficacia del vaccino virale vivo potrebbe essere ridotta.
Se l’immunoglobulina anti-D deve essere somministrata entro 2-4 settimane dalla vaccinazione con vaccini virali vivi, l’efficacia del vaccino virale potrebbe essere ridotta.
Interferenza con gli esami sierologici.
Dopo l’iniezione di immunoglobulina, l’aumento temporaneo dei livelli di vari anticorpi passivamente trasferiti nel sangue del paziente può portare a risultati falsamente positivi nei test sierologici.
Il trasferimento passivo di anticorpi contro antigeni eritrocitari, ad esempio A, B, D, può influire negativamente su alcuni test sierologici per gli anticorpi eritrocitari, come il test antiglobulinico (test di Coombs), specialmente nei neonati Rh(D)-positivi le cui madri hanno ricevuto profilassi antenatale.
Caratteristiche particolari di utilizzo.
Prima dell'uso, il medicinale deve essere portato alla temperatura ambiente o alla temperatura corporea.
La soluzione deve essere limpida o leggermente opalescente, incolore o di colore giallo pallido o marrone chiaro.
Non utilizzare la soluzione se presenta torbidità evidente o un precipitato significativo.
Il medicinale non utilizzato o i rifiuti devono essere smaltiti secondo le norme locali.
Uso durante la gravidanza o l’allattamento.
Rezonativ è indicato per l’uso durante la gravidanza.
Fertilità.
Non sono stati condotti studi sull’effetto di Rezonativ sulla fertilità negli animali. Tuttavia, in base all’esperienza clinica con l’immunoglobulina anti-Rh umana, si ritiene che non influisca sulla fertilità.
Allattamento.
Rezonativ può essere utilizzato durante l’allattamento.
Le immunoglobuline vengono escrete nel latte materno. Non sono stati riportati effetti indesiderati nei neonati nati da oltre 450 donne trattate con dosi standard di Rezonativ nel periodo post-partum.
Capacità riproduttiva.
Non sono stati condotti studi sulla tossicità riproduttiva con Rezonativ negli animali. L’esperienza clinica con l’immunoglobulina anti-Rh indica che l’uso del medicinale non comporta effetti dannosi sulla capacità riproduttiva.
Effetti sulla capacità di guidare veicoli o di usare macchinari.
Il medicinale Rezonativ non influenza la capacità di guidare veicoli o di utilizzare macchinari.
Modalità di somministrazione e dosaggio
Rezonativ deve essere somministrato per via intramuscolare.
Se è necessaria una dose totale elevata (> 5 ml negli adulti), si raccomanda di suddividerla in dosi più piccole e di iniettarla in siti diversi.
Nei casi di disturbi emorragici, quando le iniezioni intramuscolari sono controindicate, Rezonativ può essere somministrato per via sottocutanea, qualora non siano disponibili formulazioni per somministrazione endovenosa. Dopo l’iniezione, si deve applicare delicatamente una fasciatura compressiva morbida sul sito di iniezione.
La dose di immunoglobulina anti-D deve essere determinata in base al livello di esposizione agli eritrociti Rh(D)-positivi, considerando che circa 10 mcg (50 Unità Internazionali, UI) di immunoglobulina anti-D sono necessari per neutralizzare 0,5 ml di massa eritrocitaria Rh(D)-positiva o 1 ml di sangue Rh(D)-positivo.
Sulla base dei risultati degli studi clinici con Rezonativ, si raccomandano le seguenti dosi.
Prevenzione dell’immunizzazione Rh(D) nelle donne Rh(D)-negative
- Profilassi antenatale:
in accordo con le raccomandazioni generali, attualmente vengono prescritte dosi comprese tra 50-330 mcg o 250-1650 UI (oppure 0,4-2,64 ml).
- Profilassi antenatale pianificata:
una dose singola (ad esempio, 250 mcg o 1250 UI (oppure 2 ml)) viene somministrata alla 28a-30a settimana di gravidanza oppure due dosi alla 28a e alla 34a settimana di gravidanza.
- Profilassi antenatale dopo complicanze della gravidanza precedente:
una dose singola (ad esempio, 125 mcg o 625 UI (oppure 1 ml)) prima della 12a settimana di gravidanza e 250 mcg o 1250 UI (oppure 2 ml) dopo la 12a settimana di gravidanza) deve essere somministrata il più presto possibile entro 72 ore e, se necessario, ripetuta a intervalli di 6-12 settimane durante la gravidanza.
Dopo amniocentesi e biopsia del corion, deve essere somministrata una dose singola (ad esempio, 250 mcg o 1250 UI (oppure 2 ml)).
- Profilassi postnatale:
in accordo con le raccomandazioni generali, attualmente vengono prescritte dosi comprese tra 100-300 mcg o 500-1500 UI (oppure 0,8-2,4 ml). Se viene prescritta una dose inferiore (100 mcg o 500 UI), si deve determinare la quantità di emorragia fetomaterno.
Dose standard: 1250 UI (250 mcg o 2 ml).
Nel caso di somministrazione postnatale, il farmaco deve essere somministrato alla madre il più presto possibile entro 72 ore dalla nascita di un neonato Rh-positivo (D, Ddebole, Dparziale). Anche se sono trascorse più di 72 ore, il farmaco deve comunque essere somministrato il prima possibile.
Il farmaco deve essere somministrato postnatalmente anche se è stata effettuata una profilassi antenatale, nonché nei casi in cui nel siero materno siano ancora presenti tracce di attività della profilassi antenatale.
Se si sospetta una grave emorragia feto-placentare (> 4 ml (0,7 % - 0,8 % delle donne), ad esempio in caso di anemia fetale/neonatale o morte intrauterina del feto), la sua entità deve essere determinata con un metodo appropriato, come il test di eluizione acida di Kleihauer-Betke per la rilevazione dell'emoglobina fetale (HbF) o la citometria a flusso per la determinazione specifica delle cellule Rh D-positive. In base a ciò, devono essere somministrate dosi aggiuntive di immunoglobulina anti-D (10 mcg o 50 UI per 0,5 ml di eritrociti fetali).
Trasfusione di eritrociti incompatibili (RBCs)
La dose raccomandata è di 20 mcg (100 UI) di immunoglobulina anti-D per 2 ml di sangue Rh(D)-positivo trasfuso o 1 ml di concentrato eritrocitario.
Si raccomanda la consultazione con uno specialista in ematotrasfusione per valutare l'opportunità di un'eventuale trasfusione scambiata per ridurre il carico di eritrociti Rh-positivi nel circolo e per determinare la dose di immunoglobulina anti-D necessaria per inibire l'immunizzazione. Ogni 48 ore devono essere effettuati ulteriori test per rilevare la presenza di eritrociti Rh D-positivi e deve essere mantenuta la somministrazione di immunoglobulina anti-D fino a quando tutti gli eritrociti Rh D-positivi non saranno stati eliminati dalla circolazione. In ogni caso, a causa del possibile rischio di emolisi, si raccomanda di non superare la dose massima di 3000 mcg (15000 UI). Si raccomanda l'uso di un farmaco alternativo per somministrazione endovenosa, poiché raggiunge immediatamente il livello plasmatico desiderato. In assenza di un farmaco endovenoso, si deve somministrare un volume molto elevato per via intramuscolare nell'arco di diversi giorni.
Bambini
La sicurezza e l'efficacia del farmaco nei bambini non sono state stabilite.
Pazienti con sovrappeso
Nei pazienti con sovrappeso, si deve considerare l'uso di immunoglobulina anti-D per somministrazione endovenosa.
Sovradosaggio
Non sono disponibili dati riguardo al sovradosaggio. I pazienti che hanno ricevuto una trasfusione di sangue incompatibile e una dose elevata di immunoglobulina anti-D devono essere sottoposti a un monitoraggio clinico e biologico costante e continuo a causa del rischio di reazioni emolitiche.
In altri soggetti Rh(D)-negativi, il sovradosaggio non provoca effetti indesiderati più frequenti o gravi rispetto all'uso delle dosi normali.
Effetti indesiderati
Descrizione sintetica del profilo di sicurezza
Possono occasionalmente verificarsi effetti indesiderati come brividi, cefalea, vertigini, febbre, reazioni allergiche, nausea, artralgia (dolore alle articolazioni), ipotensione e dolore lieve nella parte inferiore della schiena.
Raramente le immunoglobuline umane possono causare un'improvvisa diminuzione della pressione arteriosa e, in singoli casi, shock anafilattico, anche quando il paziente non ha mostrato ipersensibilità in seguito a precedenti somministrazioni.
Reazioni locali nei siti di somministrazione del medicinale: edema, sensazione dolorosa, arrossamento, indurimento (ispessimento), sensazione di calore, prurito, ematoma, dolore locale, sensibilità e eruzione cutanea; alcune di queste reazioni possono essere prevenute suddividendo le dosi più elevate in somministrazioni effettuate in più siti.
Per informazioni sulla sicurezza riguardo al rischio di trasmissione di infezioni, vedere il paragrafo "Particolari precauzioni di impiego".
Non sono stati ottenuti dati attendibili sulla frequenza degli effetti indesiderati dagli studi clinici. Sono state segnalate le seguenti reazioni avverse:
La tabella riportata di seguito segue la classificazione per sistemi e organi (SOC) e termini preferenziali del Medical Dictionary for Regulatory Activities (MedDRA).
La frequenza è stata stimata secondo le seguenti convenzioni: molto frequente (≥ 1/10); frequente (≥ 1/100, < 1/10); non frequente (≥ 1/1.000, < 1/100); raro (≥ 1/10.000, < 1/1.000); molto raro (< 1/10.000); non noto (non può essere stimato sulla base dei dati disponibili).
| Classe di sistema organo secondo MedDRA |
Reazione avversa |
Frequenza |
| Patologie del sistema emolinfopoietico |
Reazione emolitica |
non nota |
| Disturbi del sistema immunitario |
Shock anafilattico, reazione anafilattica/ anafilattoide, ipersensibilità |
non nota |
| Disturbi del sistema nervoso |
Cefalea |
non nota |
| Disturbi cardiaci |
Tachicardia |
non nota |
| Disturbi vascolari |
Eventi trombolitici Ipotensione |
non nota |
| Disturbi del sistema respiratorio, toracico e mediastinico |
Respiro sibilante |
non nota |
| Disturbi gastrointestinali |
Vomito, nausea |
non nota |
| Disturbi della cute e del tessuto sottocutaneo |
Dermatite allergica, Reazione cutanea, eritema, prurito cutaneo, prurito, orticaria; |
non nota |
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Artralgia (dolore articolare) |
non nota |
| Patologie sistemiche e condizioni in sede di somministrazione |
Febbre, malessere al torace, malessere (senso di malessere generale) brividi, In sede di somministrazione: edema, dolore, eritema (arrossamento), indurimento (consistenza aumentata), sensazione di calore, prurito cutaneo, eruzione cutanea, prurito |
non nota |
Segnalazione di sospette reazioni avverse
La segnalazione di sospette reazioni avverse dopo la registrazione del medicinale è importante. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Si prega il personale sanitario di segnalare qualsiasi sospetta reazione avversa.
Durata della validità.
2,5 anni.
La fiala aperta deve essere utilizzata immediatamente.
Condizioni di conservazione.
Conservare a una temperatura compresa tra 2 e 8 °C (in frigorifero).
Conservare le fiale nell’imballaggio originale per proteggerle dalla luce. Non congelare.
Conservare in un luogo inaccessibile ai bambini.
Entro la durata di validità, il medicinale può essere conservato a una temperatura fino a 25 °C (senza frigorifero) per un massimo di 1 mese. Se il medicinale non è stato utilizzato entro 1 mese, deve essere smaltito.
Incompatibilità.
Rezonativ non deve essere miscelato con altri preparati.
Confezione.
Soluzione iniettabile da 1 ml o 2 ml in fiala di vetro di tipo I, Farmacopea Europea, con punto di rottura di colore rosso, 1 fiala in una confezione blister di plastica.
1 confezione blister di plastica in una scatola di cartone.
Categoria di rilascio. Sotto prescrizione medica.
Produttore.
Octapharma AB, Svezia/Octapharma AB, Sweden.
Sede del produttore e indirizzo del luogo di attività.
Lars Forssells gata 23, Stoccolma, 11275, Svezia/
Lars Forssells gata 23, Stockholm, 11275, Sweden.