Polayvi®
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE Polayvi® (Polivy®)
Composizione:
Principio attivo: polatuzumab vedotin;
1 flaconcino contiene 140 mg di polatuzumab vedotin;
1 flaconcino contiene 30 mg di polatuzumab vedotin;
1 ml di soluzione ricostituita contiene 20 mg/ml di polatuzumab vedotin;
Eccipienti: acido succinico, idrossido di sodio, saccarosio, polisorbato 20.
Forma farmaceutica. Polvere per concentrato per soluzione per infusione.
Principali caratteristiche fisico-chimiche: massa liofilizzata di colore bianco fino a bianco-grigiastro.
Gruppo farmacoterapeutico. Agenti antineoplastici e immunomodulatori. Agenti antineoplastici. Anticorpi monoclonali e coniugati anticorpo-farmaco. Altri anticorpi monoclonali e coniugati anticorpo-farmaco.
Codice ATC L01F X14.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Polatuzumab vedotin è un coniugato anticorpo-farmaco contenente l'agente antimiotico monometil auristatina E (MMAE), legato covalentemente a un anticorpo monoclonale (immunoglobulina G1 [IgG1] ricombinante umanizzata) diretto contro CD79b. Gli anticorpi monoclonali sono prodotti mediante tecnologia del DNA ricombinante in cellule ovariche del criceto cinese. L'anticorpo monoclonale si lega con elevata affinità e selettività a CD79b, un componente del recettore delle cellule B presente sulla superficie cellulare. L'espressione di CD79b è limitata alle cellule normali della linea di differenziazione delle cellule B (escluso le cellule plasmatiche) e alle cellule B maligne. CD79b è espresso nel > 95% dei linfomi B a grandi cellule diffusi. Dopo il legame con CD79b, il polatuzumab vedotin viene rapidamente internalizzato e il linker viene scisso dalle proteasi lisosomiali, consentendo il rilascio intracellulare di MMAE. MMAE si lega ai microtubuli e uccide le cellule in divisione inibendo la divisione cellulare e inducendo l'apoptosi.
Effetti farmacodinamici
Elettrofisiologia cardiaca
In base ai dati di elettrocardiogramma (ECG) ottenuti in due studi aperti su pazienti con neoplasie maligne delle cellule B precedentemente trattate, il polatuzumab vedotin alla dose raccomandata non ha prolungato clinicamente in modo significativo l'intervallo medio QTc.
Efficacia clinica e sicurezza
DLBCL precedentemente non trattata
L'efficacia di Polayvi® è stata valutata in uno studio internazionale, multicentrico, randomizzato, in doppio cieco, controllato con placebo (POLARIX, GO39942), che ha coinvolto 879 pazienti con DLBCL precedentemente non trattata.
I criteri di inclusione comprendevano pazienti di età compresa tra 18 e 80 anni, con un valore dell'Indice Prognostico Internazionale (IPI) pari a 2–5 e uno stato generale compreso tra 0 e 2 secondo la scala dell'Eastern Cooperative Oncology Group (ECOG). I tipi istologici di tumore includevano DLBCL (non altrimenti specificata [NAS], a cellule B attivate [ABC], a cellule B del centro germinale [GCB]), linfoma B a grande cellula ad alto grado (NAS, double-hit, triple-hit) e altri sottotipi di linfomi B a grandi cellule (positivi per il virus di Epstein-Barr, arricchiti in cellule T/istiociti). I pazienti non dovevano presentare note linfomi del SNC né neuropatia periferica di grado > 1.
I pazienti sono stati randomizzati in rapporto 1:1 per ricevere Polayvi® più R-CHP o R-CHOP per sei cicli di 21 giorni, seguiti da due cicli aggiuntivi di monoterapia con rituximab in entrambi i gruppi. La stratificazione dei pazienti è stata effettuata in base all'indice IPI (2 vs 3–5), presenza o assenza di malattia voluminosa (interessamento ≥ 7,5 cm) e regione geografica.
Polayvi® è stato somministrato per via endovenosa alla dose di 1,8 mg/kg nel giorno 1 dei cicli 1–6. R-CHP o R-CHOP sono stati somministrati dal giorno 1 dei cicli 1–6, seguiti da monoterapia con rituximab nel giorno 1 dei cicli 7–8. La dose in ciascun gruppo di trattamento è stata somministrata secondo le seguenti schemi:
- Gruppo Polayvi® + R-CHP: Polayvi® 1,8 mg/kg, rituximab 375 mg/m², ciclofosfamide 750 mg/m², doxorubicina 50 mg/m² e prednisone 100 mg/giorno nei giorni 1–5 di ogni ciclo per via orale.
- Gruppo R-CHOP: rituximab 375 mg/m², ciclofosfamide 750 mg/m², doxorubicina 50 mg/m², vincristina 1,4 mg/m² e prednisone 100 mg/giorno nei giorni 1–5 di ogni ciclo per via orale.
I due gruppi di trattamento erano complessivamente bilanciati per caratteristiche demografiche e cliniche basali. L'età media dei pazienti era di 65 anni (intervallo da 19 a 80 anni), il 53,6% dei pazienti era di razza caucasica, il 53,8% erano uomini, il 43,8% presentava malattia voluminosa, il 38% aveva un indice IPI pari a 2, il 62% un indice IPI da 3 a 5 e l'88,7% aveva uno stadio di malattia pari a 3 o 4. La maggior parte dei pazienti (84,2%) aveva DLBCL (inclusa NAS, ABC e GCB). Per 211 pazienti non sono stati riportati i risultati dell'analisi del tipo cellulare di origine (COO). Nella popolazione analizzata per COO (n = 668), il 33,1% dei pazienti aveva DLBCL di tipo ABC e il 52,7% DLBCL di tipo GCB.
Il punto finale primario dello studio era la sopravvivenza libera da progressione valutata dall'investigatore. La mediana della durata del periodo di follow-up è stata di 28,2 mesi.
Tabella 1
Riassunto dell'efficacia nei pazienti con DLBCL precedentemente non trattata nello studio GO39942 (POLARIX)
| Polayvi® + R-CHP N = 440 |
R-CHOP N = 439 |
|
| Punto finale primario |
||
| Sopravvivenza libera da progressione1,* |
||
| Numero (%) di pazienti con eventi |
107 (24,3 %) |
134 (30,5 %) |
| RR (IC 95 %) |
0,73 [0,57; 0,95] |
|
| valore p3,** |
0,0177 |
|
| Tasso di SLFP a 2 anni (%) |
76,7 |
70,2 |
| [IC 95 %] |
[72,65; 80,76] |
[65,80; 74,61] |
| Punti finali secondari chiave |
||
| Sopravvivenza libera da eventi (EFSeff)1 |
||
| Numero (%) di pazienti con evento |
112 (25,5 %) |
138 (31,4 %) |
| RR [IC 95%] |
0,75 [0,58; 0,96] |
|
| valore p3,** |
0,0244 |
|
| Frequenza di risposta oggettiva (ORR) al termine del trattamento2 |
||
| Pazienti che hanno risposto al trattamento (%) (risposta completa, risposta parziale) |
376 (85,5%) |
368 (83,8%) |
| Differenza nella frequenza di risposta (%) [IC 95 %] |
1,63 [-3,32; 6,57] |
|
| Frequenza di risposta completa (%) (CR)2,* |
||
| Pazienti che hanno risposto al trattamento (%) |
343 (78%) |
325 (74%) |
| Differenza nella frequenza di risposta (%) [IC 95%] |
3,92 [-1,89; 9,70] |
|
| Risposta parziale (%) (PR) |
33 (7,5%) |
43 (9,8%) |
| IC 95 % con metodo di Clopper-Pearson |
[5,22; 10,37] |
[7,18; 12,97] |
DI – intervallo di confidenza; VR – rapporto dei rischi; VBP – sopravvivenza libera da progressione; EFSeff – indice di sopravvivenza libera da eventi in seguito all’efficacia: utilizzato per rappresentare la sopravvivenza libera da eventi legati all’efficacia, definita come il tempo trascorso dalla data della randomizzazione fino al verificarsi più precoce di uno degli eventi elencati: progressione/ricaduta della malattia, morte per qualsiasi causa, causa primaria di efficacia stabilita dal ricerciatore, diversa dalla progressione/ricaduta della malattia, che abbia portato all’inizio di un qualsiasi trattamento non previsto dal protocollo per il linfoma (NALT), se era stata eseguita una biopsia dopo il completamento del trattamento ed era risultata positiva per malattia residua, indipendentemente dal fatto che fosse stato avviato o meno un trattamento NALT; CMH: Cochran – Mantel – Haenszel.
1 Valutazione del ricerciatore.
2 Valutazione BICR (analisi centralizzata cieca indipendente).
3 Test log-rank stratificato.
* Secondo i criteri di valutazione della risposta Lugano del 2014.
** Stratificato per IPI (2 contro 3–5), presenza o assenza di malattia voluminosa, regione geografica.
Durante l’analisi intermedia, il parametro secondario principale, la sopravvivenza globale, era immaturo e non mostrava differenze statisticamente significative [rapporto dei rischi stratificato 0,94 (IC 95%, 0,65; 1,37); p = 0,7524].
| Probabilità di sopravvivenza libera da progressione (%) |
|
|||||||
| Tempo, mesi |
||||||||
| Pazienti a rischio |
||||||||
| R-CHOP |
439 |
389 |
330 |
296 |
220 |
78 |
3 |
NE |
| Polayvi® + R-CHP |
440 |
404 |
353 |
327 |
246 |
78 |
NE |
NE |
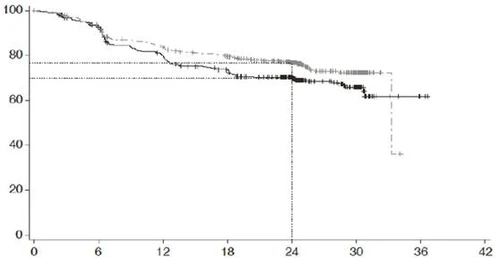
NE – non determinabile
Gruppi di trattamento: R-CHOP (n = 439); Polayvi® + R-CHP; censurato
Valore p (test log-rank) = 0,0177; rapporto di rischio (IC 95%) = 0,73 (0,57 – 0,95)
Fig. 1. Curva di sopravvivenza libera da progressione (PFS) di Kaplan-Meier secondo la valutazione dello sperimentatore nello studio GO39942 (POLARIX).
DLBCL recidivante o refrattaria
L'efficacia di Polayvi® è stata valutata in uno studio internazionale, multicentrico, aperto (GO29365), che includeva una coorte randomizzata di 80 pazienti con linfoma B a grandi cellule diffuso (DLBCL) precedentemente trattato. I pazienti sono stati randomizzati in rapporto 1:1 a ricevere Polayvi® + bendamustina e rituximab (BR) oppure solo BR per sei cicli di 21 giorni ciascuno. I pazienti sono stati stratificati in base alla durata della risposta all'ultimo trattamento precedente, ≤ 12 mesi o > 12 mesi.
I pazienti non erano candidati al trapianto autologo di cellule staminali ematopoietiche (HSCT) e presentavano malattia recidivante o refrattaria dopo almeno un precedente regime di chemioterapia sistemica. Non sono stati inclusi nello studio pazienti con pregresso trapianto allogenico di HSCT, linfoma del sistema nervoso centrale, linfoma indolente trasformato, linfoma follicolare stadio 3b, malattie cardiovascolari o polmonari significative, infezioni attive, livelli di aspartato aminotransferasi (AST) o alanina aminotransferasi (ALT) > 2,5 × limite superiore della norma (LSN) o livello di bilirubina totale ≥ 1,5 × LSN, livello di creatinina > 1,5 × LSN (o clearance della creatinina < 40 ml/min), a meno che l'elevazione non fosse dovuta al linfoma in studio.
Polayvi® è stato somministrato per via endovenosa alla dose di 1,8 mg/kg nel giorno 2 del ciclo 1 e nel giorno 1 dei cicli 2–6. La bendamustina è stata somministrata alla dose di 90 mg/m²/giorno per via endovenosa nei giorni 2 e 3 del ciclo 1 e nei giorni 1 e 2 dei cicli 2–6. Il rituximab è stato somministrato alla dose di 375 mg/m² nel giorno 1 dei cicli 1–6.
Dei 80 pazienti randomizzati a ricevere Polayvi® + BR (n = 40) o solo BR (n = 40), la maggior parte era di razza caucasica (71%) e di sesso maschile (66%). L'età media era di 69 anni (range: 30–86 anni). 64 dei 80 pazienti (80%) avevano un indice di stato generale compreso tra 0 e 1 secondo la scala ECOG (Eastern Cooperative Oncology Group), e 14 dei 80 pazienti (18%) avevano un indice di stato generale pari a 2 secondo la scala ECOG. La maggior parte dei pazienti (98%) aveva DLBCL non altrimenti specificato. Complessivamente, il 48% dei pazienti aveva DLBCL con cellule B attivate e il 40% aveva DLBCL con cellule B del centro germinale. Le principali ragioni per cui i pazienti non erano candidati al trapianto HSCT erano l'età (40%), la mancata risposta alla terapia di salvataggio (26%) e l'insuccesso del precedente trapianto (20%). La mediana del numero di terapie precedenti era di 2 (range: 1–7), con il 29% (n = 23) che aveva ricevuto una terapia precedente, il 25% (n = 20) due terapie precedenti e il 46% (n = 37) tre o più terapie precedenti. Tutti i pazienti, ad eccezione di uno nel braccio Polayvi® + BR dello studio di Fase II randomizzato, non avevano precedentemente ricevuto trattamento con bendamustina. L'80% dei pazienti aveva malattia refrattaria. Nei pazienti trattati con polatuzumab vedotin + BR e nei quali è stato determinato il numero di linfociti CD3+, il numero assoluto di linfociti CD3+ era > 200 cellule/µl rispettivamente nel 95%, 79% e 83% dei soggetti nei quali la determinazione è stata effettuata prima dell'inizio del trattamento (n = 134), alla fine del trattamento (n = 72) e a 6 mesi dalla fine del trattamento (n = 18).
Il criterio primario di efficacia di questo studio era la percentuale di risposte complete (CR) al termine del trattamento (6–8 settimane dopo il giorno 1 del ciclo 6 o dopo l'ultima dose del trattamento sperimentale), valutata mediante PET-TAC da un comitato di revisione indipendente.
Tabella 2
Riassunto dell'efficacia nei pazienti con DLBCL precedentemente trattato (studio GO29365)
| Indice |
Polayvi® + bendamustina + rituximab N = 40 |
Bendamustina + rituximab N = 40 |
| Mediana durata del follow-up 22 mesi |
||
| Endpoint primario |
||
| Frequenza di risposta completa* (valutata da un comitato di revisione indipendente) al termine del trattamento** |
||
| Pazienti che hanno risposto al trattamento (%) |
16 (40,0) |
7 (17,5) |
| Differenza nella frequenza di risposta (%) [IC 95%] |
22,5 [2,6; 40,2] |
|
| Valore p (test del chi-quadro, test di Cochran-Mantel-Haenszel***) |
0,0261 |
|
| Endpoint secondari e endpoint esplorativi |
||
| Durata della risposta (valutata dall'investigatore) |
||
| Numero di pazienti incluso nell'analisi Numero (%) di pazienti con evento |
28 17 (60,7) |
13 11 (84,6) |
| Mediana durata della risposta (IC 95%), mesi HR [IC 95%] |
10,3 (5,6; NA) |
4,1 (2,6; 12,7) |
| 0,44 [0,20; 0,95] |
||
| Valore p (test log-rank, stratificato***) |
0,0321 |
|
| Frequenza di risposta globale* (valutata dall'investigatore) al termine del trattamento** |
||
| Pazienti che hanno risposto al trattamento (%) (RG, RC) |
19 (47,5) |
7 (17,5) |
| Differenza nella frequenza di risposta (%) [IC 95%] |
30,0 [9,5; 47,4] |
|
| Valore p (test del chi-quadro, test di Cochran-Mantel-Haenszel***) |
0,0036 |
|
| Risposta completa (%) (RC) |
17 (42,5) |
6 (15,0) |
| Differenza nella frequenza di risposta (%) [IC 95%] |
27,5 [7,7; 44,7] |
|
| Valore p (test del chi-quadro, test di Cochran-Mantel-Haenszel***) |
0,0061 |
|
| Risposta parziale (%) (RP) IC 95% con metodo di Clopper-Pearson |
2 (5,0) [0,6; 16,9] |
1 (2,5) [0,06; 13,2] |
| Frequenza della migliore risposta globale* (valutata dall'investigatore) |
||
| Pazienti che hanno risposto al trattamento (%) (RC, RP) |
28 (70,0) |
13 (32,5) |
| Differenza nella frequenza di risposta (%) [IC 95%] |
37,5 [15,6; 54,7] |
|
| Risposta completa (%) (RC) |
23 (57,5) |
8 (20,0) |
| IC 95% con metodo di Clopper-Pearson |
[40,9; 73,0] |
[9,1; 35,7] |
| Risposta parziale (%) (RP) IC 95% con metodo di Clopper-Pearson |
5 (12,5) [4,2; 26,8] |
5 (12,5) [4,2; 26,8] |
IC – intervallo di confidenza; RR – rapporto di rischio; NE – non stimabile; CR – risposta completa; PR – risposta parziale.
* Secondo i criteri modificati di Lugano 2014: è richiesta la conferma della risposta completa a livello del midollo osseo mediante tomografia computerizzata a emissione di positroni (PET-TC). Una PR confermata mediante PET-TC deve soddisfare i criteri PET-TC e TC.
** 6-8 settimane dopo il giorno 1 del ciclo 6 o dopo l’ultima dose del trattamento sperimentale.
*** Stratificazione in base alla durata della risposta al trattamento precedente (≤ 12 mesi vs > 12 mesi).
La sopravvivenza globale (SG) era un endpoint secondario non corretto per l’errore di tipo I. La mediana della SG nel gruppo Polayvi® + BR era di 12,4 mesi (IC 95 %: 9,0, NE) rispetto a 4,7 mesi (IC 95 %: 3,7, 8,3) nel gruppo di controllo. La stima non aggiustata del RR per la SG era 0,42. Dopo aggiustamento per l’effetto delle covariate di base, il RR per la SG è stato aggiustato a 0,59. Le covariate includevano lo status di refrattarietà primaria, il numero di precedenti linee di terapia, l’indice prognostico internazionale e la precedente trapianto di cellule staminali.
La sopravvivenza libera da progressione (SLPD) valutata dall’investigatore era un endpoint secondario non corretto per l’errore di tipo I. La mediana della SLPD nel gruppo Polayvi® + BR era di 7,6 mesi (IC 95 %: 6,0, 17,0) rispetto a 2,0 mesi (IC 95 %: 1,5, 3,7) nel gruppo di controllo. La stima non aggiustata del RR per la SLPD era 0,34.
Immunogenicità
Come tutte le proteine con effetto terapeutico, polatuzumab vedotin ha il potenziale di indurre una risposta immunitaria. Negli studi GO39442 (POLARIX) e GO29365, gli anticorpi diretti contro polatuzumab vedotin sono stati rilevati rispettivamente nel 1,4 % (6/427) e nel 5,2 % (12/233) dei pazienti, nessuno dei quali ha mostrato anticorpi neutralizzanti. A causa del numero limitato di pazienti con anticorpi anti-polatuzumab vedotin, non è possibile trarre conclusioni sull’eventuale impatto dell’immunogenicità sull’efficacia o sulla sicurezza.
I risultati della valutazione dell’immunogenicità dipendono fortemente da diversi fattori, tra cui la sensibilità e specificità del metodo analitico, la metodologia, la gestione dei campioni, il momento del prelievo, il trattamento concomitante e la malattia di base. Per questi motivi, il confronto tra la frequenza di rilevamento degli anticorpi anti-polatuzumab vedotin e quella di altri medicinali potrebbe portare a un’interpretazione errata dei dati.
Farmacocinetica.
L’esposizione di MMAE coniugato all’anticorpo (acMMAE) nel plasma aumentava in modo proporzionale alla dose nell’intervallo di dosi di polatuzumab vedotin da 0,1 a 2,4 mg/kg. Dopo la somministrazione della prima dose di polatuzumab vedotin a 1,8 mg/kg, la concentrazione massima media (Cmax) di acMMAE era di 803 (± 233) ng/ml e l’area sotto la curva concentrazione-tempo da 0 all’infinito (AUCinf) era di 1860 (± 966) giorno•ng/ml. Sulla base di un’analisi farmacocinetica di popolazione, l’AUC di acMMAE nel ciclo 3 aumentava di circa il 30 % rispetto all’AUC nel ciclo 1 e raggiungeva oltre il 90 % dell’AUC nel ciclo 6. L’emivita terminale di acMMAE nel ciclo 6 era di circa 12 giorni (IC 95 %: 8,1–19,5 giorni). Sulla base dell’analisi farmacocinetica di popolazione, la concentrazione prevista di acMMAE alla fine del ciclo 6 è di circa l’80 % del valore teorico a stato stazionario. L’esposizione di MMAE non coniugato, il componente citotossico di polatuzumab vedotin, aumentava in modo proporzionale alla dose nell’intervallo di dosi di polatuzumab vedotin da 0,1 a 2,4 mg/kg. Per le concentrazioni di MMAE nel plasma era caratteristica una cinetica limitata dalla velocità di formazione di MMAE.
Dopo la somministrazione della prima dose di polatuzumab vedotin a 1,8 mg/kg, la Cmax era di 6,82 (± 4,73) ng/ml, il tempo per raggiungere la concentrazione massima nel plasma era di circa 2,5 giorni e l’emivita terminale era di circa 4 giorni. L’esposizione di MMAE non coniugato nel plasma rappresenta < 3 % dell’esposizione di acMMAE. Secondo l’analisi farmacocinetica di popolazione, si osserva una riduzione dell’esposizione (AUC) di MMAE non coniugato nel plasma dopo somministrazioni ripetute ogni tre settimane.
Sulla base della modellizzazione della farmacocinetica di popolazione e dei risultati di un’analisi post-hoc, si prevede che l’esposizione di MMAE non coniugato nei pazienti con peso corporeo superiore a 100 kg aumenti di non oltre il 55 %.
Assorbimento
Polayvi® viene somministrato per infusione endovenosa. Non sono stati condotti studi con altri percorsi di somministrazione.
Distribuzione
Il volume centrale di distribuzione stimato per acMMAE nella popolazione era di 3,15 l, valore approssimativamente pari al volume del plasma. In vitro, MMAE si lega in misura moderata (71–77 %) alle proteine plasmatiche umane. In vitro, MMAE non penetra significativamente negli eritrociti umani; il rapporto tra concentrazione nel sangue intero e nel plasma è compreso tra 0,79 e 0,98.
Dati in vitro indicano che MMAE è un substrato di P-gp, ma non inibisce P-gp a concentrazioni clinicamente rilevanti.
Biotrasformazione
Si prevede che nei pazienti polatuzumab vedotin subisca catabolismo, con formazione di piccoli peptidi, amminoacidi, MMAE non coniugato e metaboliti derivati da MMAE non coniugato. I livelli dei metaboliti di MMAE non sono stati determinati nel plasma umano.
Studi in vitro indicano che MMAE è un substrato di CYP3A4/5, ma non stimola gli enzimi CYP principali. MMAE è un inibitore debole e dipendente dal tempo di CYP3A4/5, ma non inibisce in modo competitivo CYP3A4/5 a concentrazioni clinicamente rilevanti.
MMAE non inibisce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 e CYP2D6.
Eliminazione
Sulla base dei dati dell’analisi farmacocinetica di popolazione, il coniugato (acMMAE) viene eliminato principalmente attraverso un chiarimento lineare non specifico pari a 0,9 l/giorno. Studi in vivo su ratti ai quali era stato somministrato polatuzumab vedotin (marcato radioattivamente sull’MMAE) hanno dimostrato che la maggior parte della radioattività viene escreta nelle feci e una parte minore nell’urina.
Gruppi particolari di pazienti
Pazienti pediatrici
Studi di farmacocinetica di polatuzumab vedotin nei bambini (età < 18 anni) non sono stati condotti.
Pazienti anziani
Sulla base dell’analisi farmacocinetica di popolazione in pazienti di età compresa tra 19 e 89 anni, l’età non influenza la farmacocinetica di acMMAE e di MMAE non coniugato. Secondo l’analisi farmacocinetica di popolazione, non sono state osservate differenze clinicamente rilevanti nella farmacocinetica di acMMAE e di MMAE non coniugato tra pazienti di età < 65 anni (n = 394) e pazienti di età ≥ 65 anni (n = 495).
Alterazioni della funzionalità renale
In pazienti con compromissione renale lieve (clearance della creatinina 60–89 ml/min, n = 361) o moderata (clearance della creatinina 30–59 ml/min, n = 163), l’esposizione di acMMAE e di MMAE non coniugato era simile a quella di pazienti con funzionalità renale normale (clearance della creatinina ≥ 90 ml/min, n = 356), sulla base dei risultati dell’analisi farmacocinetica di popolazione. I dati sono insufficienti per valutare l’impatto di una compromissione renale grave (clearance della creatinina 15–29 ml/min, n = 4) sulla farmacocinetica. Non sono disponibili dati per pazienti con insufficienza renale allo stadio terminale e/o pazienti in dialisi.
Alterazioni della funzionalità epatica
Sulla base dell’analisi farmacocinetica di popolazione, in pazienti con compromissione epatica lieve (AST o ALT > 1,0–2,5 × LSN o bilirubina totale > 1,0–1,5 × LSN, n = 133), l’esposizione di acMMAE è simile a quella di pazienti con funzionalità epatica normale (n = 737), mentre l’AUC di MMAE non coniugato è aumentata di non oltre il 40 %.
I dati sono insufficienti per valutare l’impatto di una compromissione epatica moderata (bilirubina totale > 1,5–3 × LSN, n = 11) sulla farmacocinetica. Sono disponibili dati limitati per pazienti con compromissione epatica grave o dopo trapianto epatico.
Caratteristiche cliniche.
Indicazioni.
Il medicinale Polayvi® in combinazione con rituximab, ciclofosfamide, doxorubicina e prednisone (R-CHP) è indicato per il trattamento di adulti con linfoma B a grandi cellule diffuso (DLBCL) precedentemente non trattato.
Il medicinale Polayvi® in combinazione con bendamustina e rituximab è indicato per il trattamento di adulti con linfoma B a grandi cellule diffuso recidivante/refrattario che non sono candidati al trapianto di cellule staminali ematopoietiche.
Controindicazioni.
Ipersensibilità al polatuzumab vedotin o a qualsiasi eccipiente del medicinale.
Infezioni gravi in atto (vedere il paragrafo «Speciali avvertenze e precauzioni di impiego»).
Interazioni con altri medicinali e altre forme di interazione.
Non sono stati condotti studi clinici specifici sull’interazione del polatuzumab vedotin con altri medicinali nell’uomo.
Interazione con l’uso concomitante di medicinali che sono inibitori, substrati o induttori del CYP3A4, nonché con medicinali che sono inibitori della glicoproteina P (P-gp)
Sulla base dei risultati di un modello di simulazione della farmacocinetica del rilascio di MMAE dal polatuzumab vedotin basato su parametri fisiologici, potenti inibitori del CYP3A4 e della P-gp (ad esempio chetoconazolo) possono aumentare l’area sotto la curva concentrazione-tempo (AUC) del MMAE non coniugato del 48%. Si raccomanda cautela nell’uso concomitante di inibitori del CYP3A4. I pazienti che ricevono contemporaneamente potenti inibitori del CYP3A4 (ad esempio boceprevir, claritromicina, cobicistat, indinavir, itraconazolo, nefazodone, nelfinavir, posaconazolo, ritonavir, saquinavir, telaprevir, telitromicina, voriconazolo) devono essere attentamente monitorati per la comparsa di segni di tossicità.
Non si prevede che il MMAE non coniugato alteri l’AUC di altri medicinali somministrati contemporaneamente che sono substrati del CYP3A4 (come il midazolam).
Potenti induttori del CYP3A4 (ad esempio rifampicina, carbamazepina, fenobarbital, fenitoina, erba di San Giovanni [Hypericum perforatum]) possono ridurre l’esposizione al MMAE non coniugato.
Interazione con l’uso di rituximab, bendamustina, ciclofosfamide e doxorubicina in combinazione con polatuzumab vedotin
L’uso concomitante di polatuzumab vedotin non influenza la farmacocinetica di rituximab, bendamustina, ciclofosfamide e doxorubicina. L’uso concomitante di rituximab è associato a un aumento del 24% dell’AUC dell’anticorpo coniugato con MMAE (acMMAE) e a una riduzione del 37% dell’AUC del MMAE non coniugato nel plasma, secondo l’analisi farmacocinetica di popolazione. I valori di AUC di acMMAE e di MMAE non coniugato nel plasma per Polayvi® in combinazione con R-CHP sono in linea con i dati ottenuti in altri studi con Polayvi®. Non è necessaria alcuna modifica della dose.
La bendamustina non influenza l’AUC di acMMAE e del MMAE non coniugato nel plasma.
Caratteristiche di impiego.
Tracciabilità
Al fine di migliorare la tracciabilità dei medicinali biologici, nella documentazione medica del paziente deve essere chiaramente indicato il nome commerciale e il numero di lotto del medicinale somministrato.
Mielosoppressione
Sono stati riportati casi di neutropenia grave e febbrile in pazienti trattati con Polayvi®, già durante il primo ciclo di trattamento. Si deve prendere in considerazione l'uso di fattore stimolante le colonie di granulociti (G-CSF), il cui impiego profilattico era richiesto nel programma di sviluppo clinico. Inoltre, con l'uso di Polayvi® possono verificarsi trombocitopenia o anemia di grado 3 o 4. L'emocromo completo deve essere monitorato prima di ogni somministrazione di Polayvi®. Nei pazienti con neutropenia e/o trombocitopenia di grado 3 o 4 si deve considerare un monitoraggio ematologico più frequente e/o il ritardo o l'interruzione del trattamento con Polayvi® (vedere il paragrafo «Modalità di somministrazione e posologia»).
Neuropatia periferica (NP)
Sono stati riportati casi di NP già durante il primo ciclo di trattamento con Polayvi®; il rischio aumenta con le somministrazioni successive. Nei pazienti con NP preesistente, tale condizione può peggiorare. La NP riportata durante il trattamento con Polayvi® è prevalentemente di tipo sensoriale. Tuttavia, sono stati riportati anche casi di NP motoria e sensorio-motoria. I pazienti devono essere monitorati per la comparsa di sintomi di NP, come ipoestesia, iperestesia, parestesia, disestesia, dolore neuropatico, sensazione di bruciore, debolezza muscolare o alterazioni della deambulazione. Nei pazienti in cui insorge per la prima volta o peggiora la NP, potrebbe essere necessario ritardare la somministrazione, ridurre la dose o interrompere il trattamento con Polayvi® (vedere il paragrafo «Modalità di somministrazione e posologia»).
Infezioni
Nei pazienti trattati con Polayvi® sono state riportate infezioni gravi, potenzialmente letali o letali, comprese infezioni opportunistiche come polmonite (inclusa quella causata da Pneumocystis jirovecii e altre polmoniti fungine), batteriemia, sepsi, infezione erpetica e infezione da citomegalovirus (vedere il paragrafo «Effetti indesiderati»). Sono stati riportati casi di riattivazione di infezioni latenti. I pazienti devono essere attentamente monitorati durante il trattamento per la comparsa di segni di infezioni batteriche, fungine o virali e, in caso di comparsa di tali segni, si deve consultare il medico. Durante il trattamento con Polayvi® si deve considerare la profilassi delle infezioni. Polayvi® non deve essere somministrato in caso di infezione grave attiva. Nei pazienti in cui si sviluppa un'infezione grave, il trattamento con Polayvi® e qualsiasi chemioterapia concomitante devono essere interrotti.
Virus dell'immunodeficienza umana (HIV)
Polayvi® non è stato studiato in pazienti con HIV. Per informazioni sull'uso concomitante con inibitori del CYP3A, vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione».
Immunizzazione
Non devono essere somministrate vaccinazioni vive o vive attenuate contemporaneamente al trattamento. I pazienti che hanno recentemente ricevuto vaccini vivi non sono stati inclusi negli studi clinici.
Leucoencefalopatia multifocale progressiva (LMP)
Durante il trattamento con Polayvi® sono stati riportati casi di LMP (vedere il paragrafo «Effetti indesiderati»). I pazienti devono essere attentamente monitorati per la comparsa di nuovi sintomi o il peggioramento di sintomi neurologici, cognitivi o comportamentali che potrebbero indicare LMP. Il trattamento con Polayvi® e qualsiasi chemioterapia concomitante devono essere interrotti in caso di sospetto di LMP e definitivamente sospesi in caso di conferma della diagnosi.
Sindrome da lisi tumorale (SLT)
Nei pazienti con elevato carico tumorale e tumori con elevata proliferazione, il rischio di SLT può aumentare. Prima dell'inizio del trattamento con Polayvi® devono essere adottate adeguate misure preventive in conformità con le raccomandazioni locali. Durante il trattamento con Polayvi® i pazienti devono essere attentamente monitorati per la comparsa di SLT.
Reazioni da infusione
Polayvi® può causare reazioni da infusione, comprese quelle gravi. Sono state osservate reazioni da infusione ritardate fino a 24 ore dopo la somministrazione di Polayvi®. Prima della somministrazione di Polayvi® devono essere somministrati un antistaminico e un antipiretico e il paziente deve essere attentamente monitorato per tutta la durata dell'infusione. In caso di reazioni da infusione, l'infusione deve essere interrotta e deve essere avviato un appropriato trattamento farmacologico (vedere il paragrafo «Modalità di somministrazione e posologia»).
Tossicità embrio-fetale
Data il meccanismo d'azione e i risultati degli studi preclinici, Polayvi® può arrecare danno al feto se somministrato a una donna in gravidanza. Le donne in gravidanza devono essere informate del rischio per il feto.
Alle donne in età fertile deve essere raccomandato di usare metodi contraccettivi efficaci durante il trattamento con Polayvi® e per almeno 9 mesi dopo l'ultima dose (vedere il paragrafo «Uso in gravidanza o allattamento»). Ai pazienti di sesso maschile con partner di sesso femminile in età fertile deve essere raccomandato di usare metodi contraccettivi efficaci durante il trattamento con Polayvi® e per almeno 6 mesi dopo l'ultima dose (vedere il paragrafo «Uso in gravidanza o allattamento»).
Fertilità
Negli studi preclinici, l'uso di polatuzumab vedotin è stato associato a tossicità testicolare, che potrebbe causare alterazioni della funzione riproduttiva e della fertilità negli uomini. Pertanto, agli uomini in trattamento con Polayvi® si raccomanda di conservare e congelare campioni di sperma prima dell'inizio del trattamento (vedere il paragrafo «Uso in gravidanza o allattamento»).
Pazienti anziani
Nello studio GO39942, su 435 pazienti precedentemente non trattati con LBDCG che hanno ricevuto Polayvi® in combinazione con R-CHP, 227 (52,2%) avevano un'età ≥ 65 anni. Nei pazienti di età ≥ 65 anni, la frequenza di effetti indesiderati gravi era del 39,2%, mentre nei pazienti di età < 65 anni era del 28,4%. Una frequenza simile di effetti indesiderati gravi è stata osservata nei pazienti anziani nel gruppo trattato con R-CHOP.
Su 151 pazienti precedentemente trattati con LBDCG che hanno ricevuto Polayvi® in combinazione con bendamustina e rituximab nello studio GO29365, 103 (68%) avevano un'età ≥ 65 anni. Nei pazienti di età ≥ 65 anni, la frequenza di effetti indesiderati gravi (55%) era simile a quella dei pazienti di età < 65 anni (56%). Gli studi clinici su Polayvi® non hanno incluso un numero sufficiente di pazienti di età ≥ 65 anni per stabilire differenze di risposta rispetto ai pazienti più giovani.
Epatotossicità
Nei pazienti trattati con Polayvi® si sono verificati casi gravi di epatotossicità, indicativi di danno epatocellulare, inclusi aumenti dei livelli di transaminasi e/o bilirubina (vedere il paragrafo «Effetti indesiderati»). Condizioni epatiche preesistenti, livelli iniziali elevati di enzimi epatici e l'uso concomitante di altri farmaci possono aumentare il rischio di epatotossicità. I livelli di enzimi epatici e bilirubina devono essere monitorati (vedere il paragrafo «Modalità di somministrazione e posologia»).
Eccipienti
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose e pertanto è praticamente privo di sodio.
Uso in gravidanza o allattamento.
Donne in età fertile/contraccezione in uomini e donne
Donne
Alle donne in età fertile deve essere raccomandato di usare metodi contraccettivi efficaci durante il trattamento con polatuzumab vedotin e per almeno 9 mesi dopo l'ultima dose.
Uomini
Ai pazienti di sesso maschile con partner di sesso femminile in età fertile deve essere raccomandato di usare metodi contraccettivi efficaci durante il trattamento con polatuzumab vedotin e per almeno 6 mesi dopo l'ultima dose.
Gravidanza
Non sono disponibili dati sull'uso di Polayvi® in donne in gravidanza. Studi sugli animali hanno mostrato tossicità riproduttiva. Data il meccanismo d'azione e i risultati degli studi preclinici, il polatuzumab vedotin può arrecare danno al feto se somministrato a una donna in gravidanza. Le donne in età fertile devono essere sottoposte a test di gravidanza prima del trattamento. Polayvi® non è raccomandato durante la gravidanza e nelle donne in età fertile che non usano contraccettivi, a meno che il beneficio potenziale per la donna non superi il rischio potenziale per il feto.
Allattamento
Non è noto se il polatuzumab vedotin o i suoi metaboliti siano escreti nel latte materno umano. Non può essere escluso un rischio per i neonati allattati al seno. Durante il trattamento con Polayvi® e per almeno 3 mesi dopo l'ultima dose, le donne devono interrompere l'allattamento.
Fertilità
Negli studi preclinici, l'uso di polatuzumab vedotin è stato associato a tossicità testicolare e potrebbe causare alterazioni della funzione riproduttiva e della fertilità nei maschi.
Pertanto, agli uomini in trattamento con questo medicinale si raccomanda di conservare e congelare campioni di sperma prima del trattamento. Agli uomini in trattamento con Polayvi® non è raccomandato procreare durante il trattamento e per 6 mesi dopo l'ultima dose.
Effetti sulla capacità di guidare veicoli o sull'uso di macchinari.
Polayvi® ha un'influenza trascurabile sulla capacità di guidare veicoli o di usare macchinari. Durante il trattamento con Polayvi® possono verificarsi reazioni da infusione, neuropatia periferica, affaticamento e capogiri (vedere i paragrafi «Caratteristiche di impiego» e «Effetti indesiderati»).
Modalità e posologia di somministrazione
Polayvi® deve essere somministrato sotto stretta supervisione di un medico esperto nella diagnosi e nel trattamento di pazienti con malattie oncologiche.
Dosaggio
Blastoma linfocitico B diffuso
Pazienti precedentemente non trattati
La dose raccomandata di Polayvi® è di 1,8 mg/kg sotto forma di infusione endovenosa ogni 21 giorni, in combinazione con rituximab, ciclofosfamide, doxorubicina e prednisone (R-CHP), per un totale di 6 cicli. Polayvi®, rituximab, ciclofosfamide e doxorubicina possono essere somministrati in qualsiasi ordine nel giorno 1, dopo la somministrazione del prednisone. Il prednisone viene somministrato nei giorni 1–5 di ogni ciclo. I cicli 7 e 8 prevedono la somministrazione di rituximab come monoterapia.
Consultare il foglio illustrativo dei farmaci chemioterapici utilizzati in combinazione con Polayvi® nei pazienti con blastoma linfocitico B diffuso precedentemente non trattato.
Pazienti con malattia recidivante o refrattaria
La dose raccomandata di Polayvi® è di 1,8 mg/kg sotto forma di infusione endovenosa ogni 21 giorni, in combinazione con bendamustina e rituximab, per un totale di 6 cicli. Polayvi®, bendamustina e rituximab possono essere somministrati in qualsiasi ordine nel giorno 1 di ogni ciclo. Quando utilizzato in combinazione con Polayvi®, la dose raccomandata di bendamustina è di 90 mg/m²/giorno nel giorno 1 e giorno 2 di ogni ciclo, mentre la dose raccomandata di rituximab è di 375 mg/m² nel giorno 1 di ogni ciclo. A causa dell’esperienza clinica limitata nei pazienti trattati con Polayvi® alla dose di 1,8 mg/kg con una dose cumulativa > 240 mg, non si raccomanda di superare la dose di 240 mg per ciclo.
Pazienti precedentemente non trattati e pazienti con malattia recidivante o refrattaria
Se non è stata effettuata una premedicazione precedente, prima della somministrazione di Polayvi® deve essere effettuata una premedicazione con antistaminici e farmaci antipiretici.
Ritardo o omissione della somministrazione
Se una dose programmata di Polayvi® non è stata somministrata, essa deve essere somministrata il prima possibile e il programma di somministrazione deve essere aggiornato per mantenere un intervallo di 21 giorni tra le infusioni.
Modificazione della posologia
La velocità di infusione di Polayvi® deve essere ridotta o l’infusione deve essere interrotta in caso di reazione da infusione. La somministrazione di Polayvi® deve essere immediatamente interrotta e il farmaco deve essere definitivamente sospeso in caso di reazione potenzialmente letale.
Le opzioni di modifica della dose di Polayvi® differiscono tra i pazienti con blastoma linfocitico B diffuso precedentemente non trattato e i pazienti con malattia recidivante o refrattaria.
Per informazioni sulla modifica della dose in caso di neuropatia periferica (vedere il paragrafo «Proprietà farmacologiche»), consultare la tabella 3.
Tabella 3
Modificazione della dose del farmaco Polayvi® in caso di neuropatia periferica (NP)
| Indicazione |
Gravità della NP nel giorno 1 di qualsiasi ciclo |
Modifica della dose |
| DLBCL precedentemente non trattata |
Grado 2a |
Neuropatia sensoriale:
Neuropatia motoria:
In caso di neuropatia sensoriale e motoria concomitante, seguire le raccomandazioni relative alle restrizioni più severe indicate sopra. |
| Grado 3a |
Neuropatia sensoriale:
Neuropatia motoria:
In caso di neuropatia sensoriale e motoria concomitante, seguire le raccomandazioni relative alle restrizioni più severe indicate sopra. |
|
| Grado 4 |
Interrompere l'uso del farmaco Polayvi®. |
|
| DLBCL recidivante/refrattaria |
Grado 2–3 |
Interrompere l'uso del farmaco Polayvi® fino al miglioramento dello stato a grado ≤ 1. Se lo stato si recupera a grado ≤ 1 entro il giorno 14 o prima, continuare il trattamento con Polayvi® alla dose ridotta costante di 1,4 mg/kg. Se è già stata effettuata una precedente riduzione della dose a 1,4 mg/kg, il trattamento con Polayvi® deve essere interrotto. Se lo stato non si recupera a grado ≤ 1 entro il giorno 14 o prima, il trattamento con Polayvi® deve essere interrotto. |
| Grado 4 |
Interrompere l'uso del farmaco Polayvi®. |
a Il trattamento con R-CHP può essere proseguito.
Per informazioni sulla modifica della dose in caso di mielosoppressione (vedere il paragrafo «Informazioni importanti sull’uso del medicinale»), vedere la tabella 4.
Tabella 4
Modifica della dose di Polayvi®, chemioterapia e rituximab in caso di mielosoppressione
| Indicazioni |
Gravità della mielosoppressione nel giorno 1 di qualsiasi ciclo |
Modifica della dose |
| DLBCL precedentemente non trattata |
Neutropenia di grado 3-4 |
Sospendere l'uso di tutti i farmaci fino al recupero del conteggio assoluto dei neutrofili (ANC)* a > 1000/µL. Se l'ANC si ripristina a > 1000/µL entro il giorno 7 o prima, continuare l'uso di tutti i farmaci senza alcuna riduzione della dose. Se l'ANC si ripristina a > 1000/µL dopo il giorno 7:
|
| Trombocitopenia di grado 3-4 |
Sospendere l'uso di tutti i farmaci fino al recupero del conteggio delle piastrine a > 75 000/µL. Se il conteggio delle piastrine si ripristina a > 75 000/µL entro il giorno 7 o prima, continuare l'uso di tutti i farmaci senza alcuna riduzione della dose. Se il conteggio delle piastrine si ripristina a > 75 000/µL dopo il giorno 7:
|
|
| DLBCL recidivante/refrattaria |
Neutropenia di grado 3-41 |
Sospendere l'uso di tutti i farmaci fino al recupero dell'ANC a > 1000/µL. Se l'ANC si ripristina a > 1000/µL entro il giorno 7 o prima, continuare l'uso di tutti i farmaci senza alcuna riduzione della dose. Se l'ANC si ripristina a > 1000/µL dopo il giorno 7:
|
| Trombocitopenia di grado 3-41 |
Sospendere l'uso di tutti i farmaci fino al recupero del conteggio delle piastrine a > 75 000/µL. Se il conteggio delle piastrine si ripristina a > 75 000/µL entro il giorno 7 o prima, continuare l'uso di tutti i farmaci senza alcuna riduzione della dose. Se il conteggio delle piastrine si ripristina a > 75 000/µL dopo il giorno 7:
|
1Se la causa principale è il linfoma, potrebbe non essere necessario ridurre la dose di bendamustina.
Per informazioni sulla modifica della dose in caso di reazioni da infusione (vedere il paragrafo «Istruzioni per l'uso»), vedere la tabella 5.
Tabella 5
Modifica della dose di Polayvi® in caso di reazioni da infusione (IR)
| Indicazioni |
Gravità della RI nel giorno 1 di qualsiasi ciclo |
Modifica della dose |
| DLBCL non precedentemente trattata e recidivante/refrattaria |
RI di grado 1-3 |
Interrompere l'infusione di Polayvi® e fornire un trattamento di supporto. In caso di primo verificarsi di sibili, broncospasmo o orticaria generalizzata di grado 3, Polayvi® deve essere definitivamente interrotto. In caso di recidiva di sibili o orticaria di grado 2 o di recidiva di qualsiasi sintomo di grado 3, Polayvi® deve essere definitivamente interrotto. Altrimenti, dopo la completa scomparsa dei sintomi, l'infusione può essere ripresa alla metà della velocità raggiunta prima dell'interruzione. In assenza di reazioni da infusione, la velocità di infusione può essere aumentata di 50 mg/ora ogni 30 minuti. Nel ciclo successivo, l'infusione di Polayvi® deve essere effettuata in 90 minuti. In assenza di reazioni da infusione, le infusioni successive possono essere somministrate in 30 minuti. La premedicazione deve essere effettuata in tutti i cicli. |
| RI di grado 4 |
Interrompere immediatamente l'infusione di Polayvi®. Fornire un trattamento di supporto. Interrompere definitivamente Polayvi®. |
Gruppi particolari di pazienti
Pazienti anziani
Nei pazienti di età ≥ 65 anni non è necessaria alcuna modifica della dose del medicinale Polayvi® (vedere il paragrafo «Farmacocinetica»).
Alterazioni della funzionalità renale
Nei pazienti con clearance della creatinina ≥ 30 ml/min non è necessaria alcuna modifica della dose del medicinale Polayvi®. La dose raccomandata nei pazienti con clearance della creatinina < 30 ml/min non è stata stabilita a causa della limitatezza dei dati disponibili.
Alterazioni della funzionalità epatica
L’uso di Polayvi® deve essere evitato nei pazienti con compromissione epatica moderata o grave (livelli di bilirubina > 1,5 × limite superiore della norma [LSN]).
Non è necessaria alcuna modifica della dose iniziale di Polayvi® nei pazienti con compromissione epatica lieve (livelli di bilirubina > LSN – ≤ 1,5 × LSN oppure livelli di AST > LSN).
Nella popolazione studiata con compromissione epatica lieve (definita come livelli di AST o ALT > 1,0–2,5 × LSN oppure livelli di bilirubina totale > 1,0–1,5 × LSN) è stata osservata un’aumentata esposizione al MMAE non coniugato fino al 40%, che non è stata considerata clinicamente rilevante.
Popolazione pediatrica
La sicurezza e l’efficacia del medicinale nei bambini (età inferiore a 18 anni) non sono state stabilite. I dati non sono disponibili.
Modalità di somministrazione
Polayvi® è destinato alla somministrazione endovenosa.
La dose iniziale di Polayvi® deve essere somministrata come infusione endovenosa della durata di 90 minuti. Durante l’infusione e per almeno 90 minuti dopo la somministrazione della dose iniziale, i pazienti devono essere monitorati per la comparsa di reazioni da infusione/reazioni di ipersensibilità.
Se l’infusione precedente è stata ben tollerata, la successiva dose di Polayvi® può essere somministrata in 30 minuti e i pazienti devono essere monitorati durante l’infusione e per almeno 30 minuti dopo il completamento dell’infusione.
Polayvi® deve essere ricostituito e diluito seguendo le procedure asettiche sotto la supervisione di un operatore sanitario. Il medicinale deve essere somministrato mediante infusione endovenosa attraverso un sistema di infusione separato, dotato di un filtro sterile, apiretico, integrato o aggiuntivo, con basso legame proteico (dimensione dei pori di 0,2 o 0,22 μm) e un catetere. Polayvi® non deve essere somministrato per via endovenosa diretta (in bolo) o come iniezione intravenosa rapida.
Precauzioni da adottare prima della preparazione o della somministrazione del medicinale
Polayvi® contiene un componente citotossico legato covalentemente a un anticorpo monoclonale. Devono essere seguite le opportune precauzioni per la manipolazione e lo smaltimento del medicinale.
Precauzioni generali
Polayvi® contiene un componente citotossico. Il medicinale deve essere somministrato sotto la supervisione di un medico esperto nell’uso di agenti citotossici. Devono essere seguite le opportune precauzioni per la manipolazione e lo smaltimento di farmaci antineoplastici e citotossici.
Il prodotto ricostituito non contiene conservanti ed è destinato all’uso monouso. Devono essere seguite le opportune procedure asettiche durante la manipolazione di questo medicinale.
Polayvi® deve essere ricostituito con acqua per preparazioni iniettabili sterile e successivamente diluito in un sacchetto per infusione endovenosa contenente soluzione di sodio cloruro per iniezione 9 mg/ml (0,9 %) o soluzione di sodio cloruro per iniezione 4,5 mg/ml (0,45 %), oppure soluzione glucosio al 5 %.
La soluzione ricostituita e la soluzione per infusione non devono essere congelate né esposte alla luce solare diretta.
Istruzioni per la ricostituzione
- Polayvi®, 30 mg: utilizzando una siringa sterile, iniettare lentamente 1,8 ml di acqua sterile per preparazioni iniettabili nel flaconcino contenente 30 mg di Polayvi® per ottenere una soluzione monodose contenente 20 mg/ml di polatuzumab vedotin. Dirigere il getto contro la parete del flaconcino e non direttamente sul residuo liofilizzato.
- Polayvi®, 140 mg: utilizzando una siringa sterile, iniettare lentamente 7,2 ml di acqua sterile per preparazioni iniettabili nel flaconcino contenente 140 mg di Polayvi® per ottenere una soluzione monodose contenente 20 mg/ml di polatuzumab vedotin. Dirigere il getto contro la parete del flaconcino e non direttamente sul residuo liofilizzato.
- Ruotare delicatamente il flaconcino fino a completo scioglimento. Non agitare.
- Esaminare visivamente la soluzione ricostituita per verificare la presenza di particelle solide e variazioni di colore. La soluzione ricostituita deve essere incolore o leggermente brunastra, trasparente o leggermente opalescente e non deve contenere particelle visibili. Non utilizzare la soluzione ricostituita se il colore è alterato, se è torbida o se contiene particelle visibili.
Istruzioni per la diluizione
- Diluire Polayvi® fino a una concentrazione finale di 0,72–2,7 mg/ml in un sacchetto per infusione endovenosa con un volume minimo di 50 ml contenente soluzione di sodio cloruro per iniezione 9 mg/ml o soluzione di sodio cloruro per iniezione 4,5 mg/ml, oppure soluzione glucosio al 5 %.
- Determinare il volume della soluzione ricostituita a 20 mg/ml necessario per ottenere la dose prescritta (vedere di seguito):
| Dose totale del medicinale Polayvi® (ml) da diluire ulteriormente |
= |
dose del medicinale Polayvi® (mg/kg) × peso del paziente (kg) |
| concentrazione della soluzione ricostituita nel flaconcino (20 mg/ml) |
- Prelevare il volume necessario della soluzione ricostituita del medicinale Polayvi® dal flaconcino mediante una siringa sterile e diluire nel sacchetto per infusione endovenosa. Eliminare qualsiasi parte non utilizzata del medicinale rimasta nel flaconcino.
- Mescolare delicatamente il contenuto del sacchetto per infusione, ruotando lentamente il sacchetto. Non agitare.
- Ispezionare il contenuto del sacchetto per infusione alla ricerca di particelle e, in caso di rilevamento, eliminare il contenuto.
Si raccomanda di evitare il trasporto della soluzione per infusione preparata, poiché lo stress meccanico derivante dal movimento potrebbe causare aggregazione. Se il trasporto della soluzione preparata è inevitabile, rimuovere l'aria dal sacchetto per infusione e limitare il trasporto a un massimo di 30 minuti a temperatura ambiente (9–25 °C) oppure a 24 ore in frigorifero (2–8 °C). Se l'aria viene rimossa, è necessario utilizzare un sistema per infusione con ago metallico per il collegamento, al fine di garantire un dosaggio preciso durante l'infusione. Il tempo totale di conservazione più il tempo di trasporto della soluzione diluita non deve superare il periodo di conservazione indicato nella Tabella 6.
Polayvi® deve essere somministrato attraverso un sistema per infusione separato, dotato di un filtro sterile, apirogeno, integrato o aggiuntivo con scarsa capacità di legame alle proteine (con dimensione dei pori di 0,2 o 0,22 µm) e di un catetere.
Polayvi® è compatibile con sacchetti per infusione endovenosa in cloruro di polivinile (PVC) o poliolefine, come polietilene (PE) e polipropilene. Inoltre, non sono state osservate incompatibilità con sistemi per infusione o dispositivi contenenti PVC, PE, poliuretano, polibutadiene, acrilonitrile-butadiene-stirene, policarbonato, polieteruretano, fluoruro di etilene-propilene o politetrafluoroetilene, né con membrane filtranti costituite da polietersolfone o polisolfone.
Periodo di validità
Soluzione ricostituita
Dal punto di vista microbiologico, la soluzione ricostituita deve essere utilizzata immediatamente. Se il medicinale non viene utilizzato immediatamente, il periodo e le condizioni di conservazione della soluzione ricostituita sono di responsabilità dell'utilizzatore. Il periodo di conservazione in frigorifero (2–8 °C) generalmente non deve superare le 24 ore, a condizione che la ricostituzione sia avvenuta in condizioni asettiche controllate e validate. La stabilità chimica e fisica della soluzione ricostituita è stata dimostrata fino a 72 ore di conservazione in frigorifero (2–8 °C) e fino a 24 ore a temperatura ambiente (9–25 °C).
Soluzione diluita
Dal punto di vista microbiologico, la soluzione diluita per infusione deve essere utilizzata immediatamente. Se il medicinale non viene utilizzato immediatamente, il periodo e le condizioni di conservazione della soluzione diluita sono di responsabilità dell'utilizzatore. Il periodo di conservazione in frigorifero (2–8 °C) generalmente non deve superare le 24 ore, a condizione che la diluizione sia avvenuta in condizioni asettiche controllate e validate. La stabilità chimica e fisica della soluzione diluita per infusione è stata dimostrata per i periodi indicati nella Tabella 6. La soluzione diluita deve essere eliminata se il tempo di conservazione supera i periodi indicati nella Tabella 6.
Tabella 6
Periodi durante i quali è stata dimostrata la stabilità chimica e fisica della soluzione per infusione preparata
| Solvente utilizzato per la preparazione della soluzione per infusione |
Condizioni di conservazione della soluzione per infusione1 |
| Cloruro di sodio 9 mg/ml (0,9 %) |
Fino a 72 ore in frigorifero (2–8 °C) o fino a 4 ore a temperatura ambiente (9–25 °C) |
| Cloruro di sodio 4,5 mg/ml (0,45 %) |
Fino a 72 ore in frigorifero (2–8 °C) o fino a 8 ore a temperatura ambiente (9–25 °C) |
| Soluzione glucosio 5 % |
Fino a 72 ore in frigorifero (2–8 °C) o fino a 8 ore a temperatura ambiente (9–25 °C) |
1Per garantire la stabilità del medicinale, non si devono superare i termini di conservazione indicati.
Il medicinale Polayvi® è destinato solo per uso monouso.
Qualsiasi medicinale non utilizzato o rifiuti derivati devono essere smaltiti in conformità con i requisiti normativi locali.
Popolazione pediatrica
L'efficacia e la sicurezza del medicinale nei bambini (di età inferiore ai 18 anni) non sono state stabilite. I dati non sono disponibili.
Sovradosaggio
Non vi è esperienza di sovradosaggio nell'uomo negli studi clinici. La dose più elevata studiata finora è di 2,4 mg/kg somministrata per infusione endovenosa; tale dose è stata associata a una maggiore frequenza e gravità di casi di neuropatia periferica. In caso di sovradosaggio, l'infusione deve essere immediatamente interrotta e il paziente deve essere attentamente monitorato.
Effetti indesiderati.
Riepilogo del profilo di sicurezza
La sicurezza del medicinale Polayvi® è stata valutata in 435 pazienti nello studio GO39942 (POLARIX). Sono state osservate le seguenti reazioni avverse descritte in questa sezione:
- durante il trattamento e nel periodo di follow-up successivo in pazienti precedentemente non trattati con LBCL-DH/TH, che partecipavano allo studio clinico principale GO39942 (POLARIX) e ricevevano Polayvi® più R-CHP (n = 435) o R-CHOP (n = 438). Nel gruppo di trattamento con Polayvi® più R-CHP, il 91,7% ha ricevuto 6 cicli di Polayvi® rispetto all’88,5% dei pazienti che hanno ricevuto 6 cicli di vincristina nel gruppo R-CHOP.
Nei pazienti precedentemente non trattati con LBCL-DH/TH che ricevevano Polayvi® più R-CHP:
- Le reazioni avverse più frequentemente riportate (≥ 30%) nei pazienti trattati con Polayvi® più R-CHP per LBCL-DH/TH precedentemente non trattata sono state neuropatia periferica (52,9%), nausea (41,6%), neutropenia (38,4%) e diarrea (30,8%).
- Reazioni avverse gravi sono state riportate nel 24,1% dei pazienti trattati con Polayvi® più R-CHP.
- Le reazioni avverse gravi più comuni riportate in ≥ 5% dei pazienti sono state neutropenia febbrile (10,6%) e polmonite (5,3%).
- La polmonite (1,1%) è stata l’unica reazione avversa che ha portato alla sospensione prematura del regime terapeutico in > 1% dei pazienti trattati con Polayvi® più R-CHP.
La sicurezza dell’uso di Polayvi® è stata valutata in 151 pazienti nello studio GO29365. Sono state osservate le seguenti reazioni avverse descritte in questa sezione:
- durante il trattamento e nel periodo di osservazione in pazienti precedentemente trattati con LBCL-DH/TH (n = 151) nello studio clinico principale GO29365. Questo includeva pazienti dello studio iniziale (n = 6), pazienti randomizzati (n = 39) e pazienti del braccio di espansione (n = 106) che ricevevano Polayvi® + BR, rispetto ai pazienti randomizzati (n = 39) che ricevevano solo BR. I pazienti nei gruppi di trattamento hanno ricevuto in media 5 cicli di trattamento, mentre i pazienti randomizzati nel gruppo di confronto hanno ricevuto in media 3 cicli di trattamento.
Nei pazienti precedentemente trattati con LBCL-DH/TH che ricevevano Polayvi® più bendamustina e rituximab:
- Le reazioni avverse riportate più frequentemente (≥ 30%) (tutti i gradi) nei pazienti trattati con Polayvi® più bendamustina e rituximab per LBCL-DH/TH precedentemente trattata sono state neutropenia (45,7%), diarrea (35,8%), nausea (33,1%), trombocitopenia (32,5%), anemia (31,8%) e neuropatia periferica (30,5%).
- Reazioni avverse gravi sono state riportate nel 41,7% dei pazienti trattati con Polayvi® più bendamustina e rituximab.
- Le reazioni avverse gravi più comuni riportate in ≥ 5% dei pazienti sono state: neutropenia febbrile (10,6%), sepsi (9,9%), polmonite (8,6%) e aumento della temperatura corporea (7,9%).
- La trombocitopenia (7,9%) è stata l’unica reazione avversa che ha portato alla sospensione prematura del regime terapeutico in > 5% dei pazienti trattati con Polayvi® più bendamustina e rituximab.
Di seguito sono riportate le reazioni avverse osservate nei 586 pazienti trattati con Polayvi® negli studi clinici.
Le reazioni avverse sono elencate in base alle classi di organi del sistema MedDRA e alla frequenza di insorgenza. La categoria di frequenza corrispondente per ciascuna reazione avversa si basa su: molto frequente (≥ 1/10), frequente (≥ 1/100 a < 1/10), non frequente (≥ 1/1.000 a < 1/100), raro (≥ 1/10.000 a < 1/1.000), molto raro (< 1/10.000). All’interno di ogni categoria di frequenza, le reazioni avverse sono elencate in ordine decrescente di gravità.
Infezioni e infestazioni: molto frequente – polmonitea, infezione delle vie respiratorie superiori; frequente – sepsia, infezione da herpesvirusa, infezione da citomegalovirus, infezione delle vie urinariec.
Disturbi del sangue e del sistema linfatico: molto frequente – neutropenia febbrile, neutropenia, trombocitopenia, anemia, leucopenia; frequente – linfopenia, pancitopenia.
Disturbi del metabolismo e della nutrizione: molto frequente – ipokaliemia, riduzione dell’appetito; frequente – ipocalcemia, ipoalbuminemia.
Disturbi del sistema nervoso: molto frequente – neuropatia periferica; frequente – vertigini.
Disturbi della vista: non frequente – offuscamento della vista b.
Disturbi del sistema respiratorio, toracico e mediastinico: molto frequente – tosse; frequente – pneumonite, dispnea c.
Disturbi gastrointestinali: molto frequente – diarrea, nausea, costipazione, vomito, mucosite c, dolore addominale.
Disturbi della cute e del tessuto sottocutaneo: molto frequente – alopeciac; frequente – prurito, infezioni cutanee c, eruzioni cutanee c, secchezza della pelle c.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo: frequente – artralgia, mialgia c.
Disturbi sistemici e condizioni in sede di somministrazione: molto frequente – affaticamento, aumento della temperatura corporea, astenia; frequente – edema periferico c, brividi.
Alterazioni degli esami di laboratorio: molto frequente – perdita di peso; frequente – aumento dei livelli delle transaminasi, aumento del livello della lipasi b, ipofosfatemia.
Lesioni, avvelenamenti e complicazioni da procedure: molto frequente – reazione da infusione.
a Reazioni avverse associate a esito fatale.
b Reazioni avverse osservate solo in LBCL-DH/TH recidivante o refrattaria.
c Reazioni avverse osservate solo in LBCL-DH/TH precedentemente non trattata.
Le reazioni avverse indicate sono state osservate sia in LBCL-DH/TH precedentemente non trattata che in LBCL-DH/TH recidivante o refrattaria, salvo diversamente specificato nelle note.
Reazioni avverse che si sono verificate raramente e molto raramente: assenti.
Descrizione delle singole reazioni avverse osservate negli studi clinici
Mielosoppressione
Nello studio controllato con placebo GO39942 (POLARIX), lo 0,5% dei pazienti nel gruppo di trattamento con Polayvi® più R-CHP ha interrotto prematuramente il trattamento sperimentale a causa di neutropenia. Nessun paziente ha interrotto il trattamento sperimentale nel gruppo R-CHOP a causa di neutropenia. La trombocitopenia ha causato l’interruzione prematura del trattamento sperimentale nello 0,2% dei pazienti nel gruppo Polayvi® più R-CHP, rispetto a nessun paziente nel gruppo R-CHOP. Nessun paziente ha interrotto prematuramente il trattamento a causa di anemia né nel gruppo Polayvi® più R-CHP né nel gruppo R-CHOP.
Nello studio aperto GO29365, il trattamento con Polayvi® è stato interrotto prematuramente a causa di neutropenia nel 4% dei pazienti nei gruppi di trattamento con Polayvi® più bendamustina e rituximab, rispetto al 2,6% dei pazienti nel gruppo di trattamento con bendamustina e rituximab. La trombocitopenia ha causato l’interruzione del trattamento nel 7,9% dei pazienti nei gruppi con Polayvi® + BR e nel 5,1% dei pazienti nel gruppo BR. Nessun paziente in entrambi i gruppi (gruppi di trattamento Polayvi® + BR o solo gruppo BR) ha interrotto il trattamento a causa di anemia. Nei gruppi di trattamento con Polayvi® più bendamustina e rituximab, sono state riportate neutropenia, trombocitopenia e anemia di grado 3 o superiore rispettivamente nel 40,4%, 25,8% e 12,6% dei pazienti.
Neuropatia periferica (PN)
Nello studio controllato con placebo GO39942 (POLARIX), nel gruppo di trattamento con Polayvi® più R-CHP sono state riportate PN di grado 1, 2 e 3 rispettivamente nel 39,1%, 12,2% e 1,6% dei pazienti. Nel gruppo R-CHOP, sono state riportate PN di grado 1, 2 e 3 rispettivamente nel 37,2%, 15,5% e 1,1% dei pazienti. Non sono state riportate reazioni di PN di grado 4–5 né nel gruppo Polayvi® più R-CHP né nel gruppo R-CHOP. Nel gruppo Polayvi® più R-CHP, lo 0,7% dei pazienti ha interrotto prematuramente il trattamento sperimentale a causa di PN, rispetto al 2,3% nel gruppo R-CHOP. Nel gruppo Polayvi® più R-CHP, il 4,6% dei pazienti ha avuto una riduzione della dose a causa di PN, rispetto all’8,2% nel gruppo R-CHOP. Nel gruppo di trattamento con Polayvi® più R-CHP, la mediana del tempo all’insorgenza del primo evento di PN è stata di 2,27 mesi, rispetto a 1,87 mesi nel gruppo R-CHOP. Alla data di chiusura della raccolta dati clinici, i sintomi di PN erano scomparsi nel 57,8% dei pazienti nel gruppo Polayvi® più R-CHP, rispetto al 66,9% nel gruppo R-CHOP. La mediana del tempo di risoluzione della neuropatia periferica è stata di 4,04 mesi nel gruppo Polayvi® più R-CHP rispetto a 4,6 mesi nel gruppo R-CHOP.
Nello studio aperto GO29365, nei gruppi di trattamento con Polayvi® più bendamustina e rituximab sono state riportate PN di grado 1 e 2 rispettivamente nel 15,9% e 12,6% dei pazienti. Nel gruppo di trattamento con bendamustina e rituximab, sono stati registrati eventi di PN di grado 1 e 2 rispettivamente nel 2,6% e 5,1% dei pazienti. Un singolo caso di PN di grado 3 è stato riportato nei gruppi Polayvi® + BR e nessun caso nel gruppo BR. Non sono stati riportati casi di PN di grado 4–5 in nessun gruppo (gruppi di trattamento Polayvi® + BR o solo gruppo BR). Nel 2,6% dei pazienti il trattamento con Polayvi® è stato interrotto prematuramente a causa di PN e nel 2% dei pazienti la dose di Polayvi® è stata ridotta a causa di PN. Nessun paziente nel gruppo BR ha interrotto prematuramente il trattamento o ha avuto riduzione della dose a causa di PN. Nei gruppi Polayvi® + BR, la mediana del tempo all’insorgenza del primo evento di PN è stata di 1,6 mesi e nel 39,1% dei pazienti con PN è stato riportato il suo scomparire.
Infezioni
Nello studio controllato con placebo GO39942 (POLARIX), infezioni, comprese polmonite e altri tipi di infezioni, sono state riportate nel 49,7% dei pazienti nel gruppo di trattamento con Polayvi® più R-CHP e nel 42,7% dei pazienti nel gruppo R-CHOP. Infezioni di grado 3–4 si sono verificate nel 14% dei pazienti nel gruppo Polayvi® più R-CHP e nell’11,2% dei pazienti nel gruppo R-CHOP. Nel gruppo di trattamento con Polayvi® più R-CHP sono state riportate infezioni gravi nel 14% dei pazienti e infezioni con esito fatale nel 1,1% dei pazienti. Nel gruppo R-CHOP sono state riportate infezioni gravi nel 10,3% dei pazienti e infezioni con esito fatale nell’1,4% dei pazienti. Sette pazienti (1,6%) nel gruppo di trattamento con Polayvi® più R-CHP hanno interrotto il trattamento a causa di infezione, rispetto a 10 pazienti (2,3%) nel gruppo R-CHOP.
Nello studio aperto GO29365, infezioni, comprese polmonite e altri tipi di infezioni, sono state riportate nel 48,3% dei pazienti nei gruppi Polayvi® + BR e nel 51,3% dei pazienti nel gruppo BR. Nei gruppi Polayvi® + BR sono state riportate infezioni gravi nel 27,2% dei pazienti e infezioni con esito fatale nel 6,6% dei pazienti. Nel gruppo BR sono state riportate infezioni gravi nel 30,8% dei pazienti e infezioni con esito fatale nel 10,3% dei pazienti. Quattro pazienti (2,6%) nei gruppi Polayvi® + BR hanno interrotto prematuramente il trattamento a causa di infezione, rispetto a 2 pazienti (5,1%) nel gruppo BR.
Leucoencefalopatia multifocale progressiva (LMP)
Nello studio controllato con placebo GO39942 (POLARIX) non sono stati riportati casi di leucoencefalopatia multifocale progressiva.
Nello studio aperto GO29365, un caso fatale di LMP è stato osservato in un paziente che riceveva trattamento con Polayvi® + bendamustina e obinutuzumab. Questo paziente aveva precedentemente ricevuto tre linee di terapia, compresi anticorpi anti-CD20.
Epatotossicità
Nello studio controllato con placebo GO39942 (POLARIX), epatotossicità è stata riportata nel 10,6% dei pazienti nel gruppo di trattamento con Polayvi® più R-CHP e nel 7,3% dei pazienti nel gruppo R-CHOP. Nel gruppo di trattamento con Polayvi® più R-CHP, la maggior parte degli eventi era di grado 1–2 (8,7%); eventi di grado 3 sono stati riportati nell’1,8% dei pazienti. Non sono stati osservati eventi di grado 4 o 5. In un paziente (0,2%) è stata riportata epatotossicità grave, che si è rivelata reversibile.
In un altro studio sono stati riportati due casi di epatotossicità grave (danno epatocellulare e steatosi epatica), che si sono rivelati reversibili.
Tossicità gastrointestinale
Nello studio controllato con placebo GO39942 (POLARIX), eventi di tossicità gastrointestinale sono stati osservati nel 76,1% dei pazienti nel gruppo di trattamento con Polayvi® più R-CHP rispetto al 71,9% dei pazienti nel gruppo R-CHOP. La maggior parte degli eventi era di grado 1–2, mentre eventi di grado ≥ 3 sono stati osservati nel 9,7% dei pazienti nel gruppo Polayvi® più R-CHP rispetto all’8,2% nel gruppo R-CHOP. Gli eventi più comuni di tossicità gastrointestinale sono stati nausea e diarrea.
Nello studio aperto GO29365, eventi di tossicità gastrointestinale sono stati riportati nel 72,8% dei pazienti nel gruppo Polayvi® + BR rispetto al 66,7% dei pazienti nel gruppo BR. La maggior parte degli eventi era di grado 1–2. Eventi di grado 3–4 sono stati riportati nel 16,5% dei pazienti nel gruppo Polayvi® + BR rispetto al 12,9% dei pazienti nel gruppo BR. Gli eventi più comuni di tossicità gastrointestinale sono stati diarrea e nausea.
Periodo di validità.
30 mesi.
Condizioni di conservazione.
Conservare a una temperatura compresa tra 2 e 8 ºC, nella confezione originale per proteggere dallo splendore della luce. Non congelare. Conservare fuori dalla portata dei bambini.
Incompatibilità.
Questo medicinale non deve essere mescolato o diluito con altri medicinali, ad eccezione di quelli indicati nella sezione «Precauzioni da adottare prima della ricostituzione o della somministrazione del medicinale».
Confezione.
Fiala da 6 ml in vetro incolore (vetro borosilicato, classe I), chiusa con tappo in butil gomma (rivestito con pellicola fluorogomma) e sigillata con capsula in alluminio con disco plastico di tipo «flip-off». Contiene 30 mg per fiala. Una fiala per confezione in cartone.
Fiala da 20 ml in vetro incolore (vetro borosilicato, classe I), chiusa con tappo in butil gomma (rivestito con pellicola fluorogomma) e sigillata con capsula in alluminio con disco plastico di tipo «flip-off». Contiene 140 mg per fiala. Una fiala per confezione in cartone.
Categoria di prescrizione.
Sotto prescrizione medica.
Produttore.
F. Hoffmann-La Roche Ltd
Sede del produttore e indirizzo del luogo di esercizio dell’attività.
Wurmisweg, 4303 Kaiseraugst, Svizzera