Polayvi®
UcraniaContenido
INSTRUCCIONES para uso médico del medicamento Polayvī® (Polivy®)
Composición:
Principio activo: polatuzumab vedotin;
1 frasco contiene 140 mg de polatuzumab vedotin;
1 frasco contiene 30 mg de polatuzumab vedotin;
1 ml de la solución reconstituida contiene 20 mg/ml de polatuzumab vedotin;
Excipientes: ácido succínico, hidróxido de sodio, sacarosa, polisorbato 20.
Forma farmacéutica. Polvo para concentrado para solución para perfusión.
Características físicas y químicas principales: masa liofilizada de color blanco a blanco grisáceo.
Grupo farmacoterapéutico. Agentes antineoplásicos e inmunomoduladores. Agentes antineoplásicos. Anticuerpos monoclonales y conjugados de anticuerpos con fármaco. Otros anticuerpos monoclonales y conjugados de anticuerpos con fármaco.
Código ATC L01FX14.
Propiedades farmacodinámicas.
Farmacodinámica.
Mecanismo de acción
Polatuzumab vedotin es un conjugado anticuerpo-fármaco que contiene el agente antimítico monometil auristatina E (MMAE), unido covalentemente a un anticuerpo monoclonal (inmunoglobulina G1 [IgG1] recombinante humanizada) dirigido contra CD79b. Los anticuerpos monoclonales se producen mediante tecnología de ADN recombinante en células de ovario de hámster chino. El anticuerpo monoclonal se une con alta afinidad y selectividad a CD79b, un componente del receptor de células B en la superficie celular. La expresión de CD79b está limitada a células normales de la línea B diferenciadas (excepto células plasmáticas) y células B malignas. CD79b se expresa en > 95 % de los linfomas difusos de grandes células B. Tras unirse a CD79b, polatuzumab vedotin se internaliza rápidamente y el enlazador se escinde mediante proteasas lisosomales, lo que permite la liberación intracelular de MMAE. MMAE se une a los microtúbulos y mata las células en división mediante la inhibición de la división celular e inducción de apoptosis.
Efectos farmacodinámicos
Electrofisiología cardíaca
Según datos electrocardiográficos (ECG) obtenidos en dos estudios abiertos en pacientes con neoplasias malignas B previamente tratadas, polatuzumab vedotin en la dosis recomendada no prolongó de forma clínicamente significativa el intervalo medio QTc.
Eficacia y seguridad clínicas
Linfaoma difuso de grandes células B (LDGCB) previamente no tratado
La eficacia de Polivy® se evaluó en un estudio internacional, multicéntrico, aleatorizado, doble ciego, controlado con placebo (POLARIX, GO39942) con participación de 879 pacientes con LDGCB previamente no tratado.
Los criterios de inclusión incluían pacientes de 18 a 80 años de edad, con un índice pronóstico internacional (IPI) de 2 a 5 y un estado general de 0 a 2 según la escala del Grupo Cooperativo de Oncología del Este (ECOG). Los tipos histológicos de tumor incluían LDGCB (no especificado [NSE], de células B activadas [ABC], de centro germinal [GCB]), linfoma B de alto grado (NSE, "double-hit", "triple-hit") y otros subtipos de linfomas de grandes células B (positivos para el virus de Epstein-Barr, enriquecidos en células T/histiocitos). Los pacientes no tenían linfoma del SNC conocido ni neuropatía periférica > grado 1.
Los pacientes fueron aleatorizados en una proporción 1:1 para recibir Polivy® más R-CHP o R-CHOP durante seis ciclos de 21 días, seguidos por dos ciclos adicionales de monoterapia con rituximab en ambos grupos. Los pacientes se estratificaron según el índice IPI (2 frente a 3–5), presencia o ausencia de enfermedad voluminosa (lesión ≥ 7,5 cm) y región geográfica.
Polivy® se administró por vía intravenosa a una dosis de 1,8 mg/kg en el día 1 de los ciclos 1–6. R-CHP o R-CHOP se administraron desde el día 1 de los ciclos 1–6, seguido de monoterapia con rituximab en el día 1 de los ciclos 7–8. La dosificación en cada grupo de tratamiento se realizó según los siguientes esquemas:
- Grupo Polivy® + R-CHP: Polivy® 1,8 mg/kg, rituximab 375 mg/m², ciclofosfamida 750 mg/m², doxorrubicina 50 mg/m² y prednisona 100 mg/día por vía oral en los días 1–5 de cada ciclo.
- Grupo R-CHOP: rituximab 375 mg/m², ciclofosfamida 750 mg/m², doxorrubicina 50 mg/m², vincristina 1,4 mg/m² y prednisona 100 mg/día por vía oral en los días 1–5 de cada ciclo.
Ambos grupos de tratamiento fueron generalmente equilibrados en cuanto a características demográficas basales y características de la enfermedad. La edad media de los pacientes fue de 65 años (rango de 19 a 80 años), el 53,6 % eran de raza caucásica, el 53,8 % eran hombres, el 43,8 % tenían enfermedad voluminosa, el 38 % tenían un índice IPI de 2, el 62 % tenían un índice IPI de 3–5 y el 88,7 % tenían estadio III o IV de la enfermedad. La mayoría de los pacientes (84,2 %) tenían LDGCB (incluyendo NSE, ABC y GCB). En 211 pacientes no se informaron resultados sobre el origen celular (COO). Entre la población analizada respecto al COO (n = 668), según el perfil de expresión génica, el 33,1 % de los pacientes tenían LDGCB tipo ABC y el 52,7 % tenían LDGCB tipo GCB.
El punto final primario del estudio fue la supervivencia libre de progresión evaluada por el investigador. La mediana de duración del seguimiento posterior fue de 28,2 meses.
Tabla 1
Resumen de eficacia en pacientes con LDGCB previamente no tratado en el estudio GO39942 (POLARIX)
| Polayvі® + R-CHP N = 440 |
R-CHOP N = 439 |
|
| Punto final primario |
||
| Sobrevida libre de progresión1,* |
||
| Número (%) de pacientes con eventos |
107 (24,3 %) |
134 (30,5 %) |
| HR (95 % IC) |
0,73 [0,57, 0,95] |
|
| valor p3,** |
0,0177 |
|
| Tasa de SLP a 2 años (%) |
76,7 |
70,2 |
| [95 % IC] |
[72,65, 80,76] |
[65,80, 74,61] |
| Puntos finales secundarios clave |
||
| Sobrevida libre de eventos (SLCeef)1 |
||
| Número (%) de pacientes con evento |
112 (25,5 %) |
138 (31,4 %) |
| HR [95 % IC] |
0,75 [0,58, 0,96] |
|
| valor p3,** |
0,0244 |
|
| Tasa de respuesta objetiva (TRO) al final del tratamiento2 |
||
| Pacientes que respondieron al tratamiento (%) (respuesta completa, respuesta parcial) |
376 (85,5%) |
368 (83,8%) |
| Diferencia en la tasa de respuesta (%) [95 % IC] |
1,63 [-3,32, 6,57] |
|
| Tasa de respuesta completa (%) (RC)2,* |
||
| Pacientes que respondieron al tratamiento (%) |
343 (78%) |
325 (74%) |
| Diferencia en la tasa de respuesta (%) [95 % IC] |
3,92 [-1,89, 9,70] |
|
| Respuesta parcial (%) (RP) |
33 (7,5%) |
43 (9,8%) |
| 95 % IC por método de Clopper-Pearson |
[5,22, 10,37] |
[7,18, 12,97] |
IC: intervalo de confianza; HR: razón de riesgos; EFS: supervivencia libre de progresión; EFSeff: índice de supervivencia sin eventos debidos a la eficacia: se utiliza para reflejar la supervivencia sin eventos relacionados con la eficacia, definidos como el tiempo desde la fecha de aleatorización hasta la aparición más temprana de cualquiera de los siguientes eventos: progresión/recidiva de la enfermedad, muerte por cualquier causa, causa primaria de eficacia determinada por el investigador distinta de la progresión/recidiva de la enfermedad que condujo al inicio de cualquier tratamiento no previsto en el protocolo para linfoma (NALT), si se obtuvo una biopsia tras finalizar el tratamiento y esta fue positiva para enfermedad residual, independientemente de si se inició el tratamiento NALT o no; CMH: Cochran-Mantel-Haenszel.
1 Evaluación del investigador.
2 Evaluación mediante BICR (análisis centralizado independiente enmascarado).
3 Prueba de rango logarítmico, estratificada.
* Según los criterios de evaluación de respuesta de Lugano del año 2014.
** Estratificado según IPI (2 frente a 3-5), presencia o ausencia de enfermedad voluminosa y región geográfica.
Durante el análisis intermedio, el punto final secundario clave de supervivencia global fue inmaduro y no mostró diferencias estadísticamente significativas [razón de riesgos estratificada de 0,94 (IC del 95 %: 0,65, 1,37); p = 0,7524].
| Probabilidad de supervivencia sin progresión (%) |
|
|||||||
| Tiempo, meses |
||||||||
| Pacientes en riesgo |
||||||||
| R-CHOP |
439 |
389 |
330 |
296 |
220 |
78 |
3 |
NE |
| Polivy® + R-CHP |
440 |
404 |
353 |
327 |
246 |
78 |
NE |
NE |
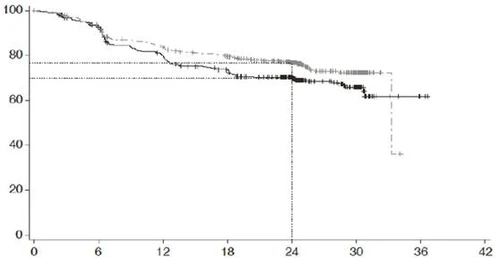
NE – no evaluable
Grupos de tratamiento: R-CHOP (n = 439); Polivy® + R-CHP; censurado
Valor p (prueba log-rank) = 0,0177; razón de riesgo (IC 95 %) = 0,73 (0,57 – 0,95)
Fig. 1. Curva de Kaplan-Meier de supervivencia libre de progresión (SLP) según evaluación del investigador en el estudio GO39942 (POLARIX).
LBGC difusa recidivante o refractaria
La eficacia de Polivy® se evaluó en un estudio internacional, multicéntrico y abierto (GO29365), que incluyó una cohorte aleatorizada de 80 pacientes con linfoma B de grandes células difuso (LBGC-D) previamente tratado. Los pacientes fueron aleatorizados en una proporción 1:1 para recibir Polivy® + bendamustina y rituximab (BR) o solo BR durante seis ciclos de 21 días. Los pacientes fueron estratificados según la duración de la respuesta al último tratamiento previo: ≤ 12 meses o > 12 meses.
Los pacientes no eran candidatos a trasplante autólogo de células madre hematopoyéticas (TCMH) y presentaban enfermedad recidivante o refractaria tras al menos un régimen previo de quimioterapia sistémica. No se incluyeron en este estudio pacientes con trasplante alogénico previo de CTHM, linfoma del sistema nervioso central, linfoma indolente transformado, linfoma folicular estadio 3b, enfermedad cardiovascular o pulmonar significativa, infecciones activas, niveles de aspartato aminotransferasa (AST) o alanina aminotransferasa (ALT) > 2,5 × límite superior normal (LSN), niveles de bilirrubina total ≥ 1,5 × LSN, niveles de creatinina > 1,5 × LSN (o aclaramiento de creatinina < 40 ml/min), salvo que el aumento se debiera directamente al linfoma en estudio.
Polivy® se administró por vía intravenosa a una dosis de 1,8 mg/kg el día 2 del ciclo 1 y el día 1 de los ciclos 2–6. La bendamustina se administró a una dosis de 90 mg/m²/día por vía intravenosa en los días 2 y 3 del ciclo 1 y en los días 1 y 2 de los ciclos 2–6. El rituximab se administró a una dosis de 375 mg/m² el día 1 de los ciclos 1–6.
De los 80 pacientes aleatorizados para recibir Polivy® + BR (n = 40) o solo BR (n = 40), la mayoría eran de raza caucásica (71 %) y del sexo masculino (66 %). La edad media fue de 69 años (rango: 30–86 años). 64 de 80 pacientes (80 %) tenían un estado general ECOG de 0–1 y 14 de 80 pacientes (18 %) tenían un estado general ECOG de 2. La mayoría de los pacientes (98 %) tenían LBGC-D sin especificación adicional. En total, el 48 % de los pacientes tenían LBGC-D de células B activadas y el 40 % tenían LBGC-D de células B del centro germinal. Las principales razones por las que los pacientes no eran candidatos a TCMH fueron la edad (40 %), respuesta insuficiente a la terapia de rescate (26 %) y fracaso del trasplante previo (20 %). La mediana del número de terapias previas fue de 2 (rango: 1–7), siendo que el 29 % (n = 23) recibió una terapia previa, el 25 % (n = 20) dos terapias previas y el 46 % (n = 37) tres o más terapias previas. Todos los pacientes, excepto uno en el grupo Polivy® + BR del estudio de fase II aleatorizado, no habían recibido previamente tratamiento con bendamustina. El 80 % de los pacientes tenían enfermedad refractaria. En los pacientes que recibieron polatuzumab vedotin + BR y en los que se determinó el recuento de linfocitos CD3+, el recuento absoluto de linfocitos CD3+ fue > 200 células/µl en el 95 %, 79 % y 83 % de los individuos en quienes la determinación se realizó antes del inicio del tratamiento (n = 134), al final del tratamiento (n = 72) y a los 6 meses tras la finalización del tratamiento (n = 18), respectivamente.
El punto final primario de este estudio fue la tasa de respuesta completa (RC) al final del tratamiento (6–8 semanas después del día 1 del ciclo 6 o de la última dosis del tratamiento en estudio), evaluada por tomografía por emisión de positrones (PET-TC) mediante un comité de revisión independiente.
Tabla 2
Resumen de la eficacia en pacientes con LBGC-D previamente tratado (estudio GO29365)
| Indicador |
Polayvi® + bendamustina + rituximab N = 40 |
Bendamustina + rituximab N = 40 |
| Mediana de duración del seguimiento: 22 meses |
||
| Punto final primario |
||
| Frecuencia de respuesta completa* (según evaluación del comité de vigilancia independiente) al final del tratamiento** |
||
| Pacientes que respondieron al tratamiento (%) |
16 (40,0) |
7 (17,5) |
| Diferencia en la frecuencia de respuesta (%) [IC del 95 %] |
22,5 [2,6; 40,2] |
|
| Valor p (prueba de chi-cuadrado, prueba de Cochran-Mantel-Haenszel***) |
0,0261 |
|
| Puntos finales secundarios y exploratorios |
||
| Duración de la respuesta (según evaluación del investigador) |
||
| Número de pacientes incluidos en el análisis Número (%) de pacientes con evento |
28 17 (60,7) |
13 11 (84,6) |
| Mediana de duración de la respuesta (IC del 95 %), meses HR [IC del 95 %] |
10,3 (5,6; LIM) |
4,1 (2,6; 12,7) |
| 0,44 [0,20; 0,95] |
||
| Valor p (prueba log-rank, estratificada***) |
0,0321 |
|
| Frecuencia de respuesta global* (según evaluación del investigador) al final del tratamiento** |
||
| Pacientes que respondieron al tratamiento (%) (RP, CR) |
19 (47,5) |
7 (17,5) |
| Diferencia en la frecuencia de respuesta (%) [IC del 95 %] |
30,0 [9,5; 47,4] |
|
| Valor p (prueba de chi-cuadrado, prueba de Cochran-Mantel-Haenszel***) |
0,0036 |
|
| Respuesta completa (%) (CR) |
17 (42,5) |
6 (15,0) |
| Diferencia en la frecuencia de respuesta (%) [IC del 95 %] |
27,5 [7,7; 44,7] |
|
| Valor p (prueba de chi-cuadrado, prueba de Cochran-Mantel-Haenszel***) |
0,0061 |
|
| Respuesta parcial (%) (RP) IC del 95 % mediante el método de Clopper-Pearson |
2 (5,0) [0,6; 16,9] |
1 (2,5) [0,06; 13,2] |
| Frecuencia de mejor respuesta global* (según evaluación del investigador) |
||
| Pacientes que respondieron al tratamiento (%) (RP, CR) |
28 (70,0) |
13 (32,5) |
| Diferencia en la frecuencia de respuesta (%) [IC del 95 %] |
37,5 [15,6; 54,7] |
|
| Respuesta completa (%) (CR) |
23 (57,5) |
8 (20,0) |
| IC del 95 % mediante el método de Clopper-Pearson |
[40,9; 73,0] |
[9,1; 35,7] |
| Respuesta parcial (%) (RP) IC del 95 % mediante el método de Clopper-Pearson |
5 (12,5) [4,2; 26,8] |
5 (12,5) [4,2; 26,8] |
DI – intervalo de confianza; VR – razón de riesgo; EV – estimación no disponible; RV – respuesta completa; PR – respuesta parcial.
* Según criterios modificados de Lugano 2014: se requiere confirmación de la respuesta completa mediante TEP-TAC del médula ósea. La PR confirmada mediante tomografía por emisión de positrones-tomografía computarizada (TEP-TAC) debe cumplir los criterios de TEP-TAC y tomografía computarizada (TAC).
** 6–8 semanas después del día 1 del ciclo 6 o de la última dosis del tratamiento en estudio.
*** Estratificación según duración de la respuesta al tratamiento previo (≤ 12 meses frente a > 12 meses).
La supervivencia global (SG) fue un punto final exploratorio no controlado respecto al error de tipo I. La mediana de SG en el grupo Polivy® + BR fue de 12,4 meses (95 % DI: 9,0, EV) frente a 4,7 meses (95 % DI: 3,7, 8,3) en el grupo control. La estimación no ajustada del VR respecto a la SG fue de 0,42. Tras el ajuste por el efecto de covariables basales, el VR para la SG se ajustó a 0,59. Las covariables incluyeron el estado de refractariedad primaria, número de líneas previas de tratamiento, índice pronóstico internacional y trasplante previo de células madre.
La supervivencia libre de progresión (SLP) según evaluación del investigador fue un punto final exploratorio no controlado respecto al error de tipo I. La mediana de SLP en el grupo Polivy® + BR fue de 7,6 meses (95 % DI: 6,0, 17,0) frente a 2,0 meses (95 % DI: 1,5, 3,7) en el grupo control. La estimación no ajustada del VR respecto a la SLP fue de 0,34.
Inmunogenicidad
Como todas las proteínas con efecto terapéutico, el polatuzumab vedotin tiene potencial para inducir una respuesta inmune. En los estudios GO39442 (POLARIX) y GO29365, se detectaron anticuerpos frente al polatuzumab vedotin en el 1,4 % (6/427) y 5,2 % (12/233) de los pacientes, respectivamente, sin que en ninguno de ellos se detectaran anticuerpos neutralizantes. Debido al número limitado de pacientes en los que se detectaron anticuerpos frente al polatuzumab vedotin, no es posible concluir sobre el posible impacto de la inmunogenicidad en la eficacia o seguridad.
Los resultados de la evaluación de inmunogenicidad dependen significativamente de varios factores, incluyendo la sensibilidad y especificidad del método analítico, la metodología de análisis, el manejo de las muestras, el momento de recogida de las muestras, el tratamiento concomitante y la enfermedad de base. Debido a estas razones, la comparación de la frecuencia de detección de anticuerpos frente al polatuzumab vedotin con la frecuencia de detección de anticuerpos frente a otros medicamentos podría llevar a interpretaciones erróneas de los datos.
Farmacocinética.
La exposición al MMAE conjugado al anticuerpo (acMMAE) en plasma aumentó proporcionalmente con la dosis en el rango de dosis de polatuzumab vedotin de 0,1 a 2,4 mg/kg. Tras la administración de la primera dosis de polatuzumab vedotin a 1,8 mg/kg, la concentración máxima media (Cmáx) de acMMAE fue de 803 (± 233) ng/ml y el área bajo la curva concentración-tiempo de 0 a infinito (AUCinf) fue de 1860 (± 966) día•ng/ml. Según el análisis farmacocinético poblacional, la AUC de acMMAE en el ciclo 3 aumentó aproximadamente un 30 % en comparación con la AUC en el ciclo 1 y alcanzó más del 90 % de la AUC en el ciclo 6. El período de semivida terminal de acMMAE en el ciclo 6 fue de aproximadamente 12 días (95 % DI: 8,1–19,5 días). Según el análisis farmacocinético poblacional, la concentración prevista de acMMAE al final del ciclo 6 es aproximadamente el 80 % del valor teórico en estado de equilibrio. La exposición al MMAE no conjugado, componente citotóxico del polatuzumab vedotin, aumentó proporcionalmente con la dosis en el rango de dosis de polatuzumab vedotin de 0,1 a 2,4 mg/kg. Las concentraciones plasmáticas de MMAE siguieron una cinética limitada por la velocidad de formación del MMAE.
Tras la administración de la primera dosis de polatuzumab vedotin a 1,8 mg/kg, la Cmáx fue de 6,82 (± 4,73) ng/ml, el tiempo hasta alcanzar la concentración máxima en plasma fue de aproximadamente 2,5 días y el período de semivida terminal fue de aproximadamente 4 días. La exposición al MMAE no conjugado en plasma representa < 3 % de la exposición al acMMAE. Según el análisis farmacocinético poblacional, se observa una disminución de la exposición (AUC) al MMAE no conjugado en plasma tras la administración repetida cada tres semanas.
Según la modelización farmacocinética poblacional basada en un análisis post-hoc, se prevé que la exposición al MMAE no conjugado en pacientes con peso corporal superior a 100 kg aumente no más del 55 %.
Absorción
Polivy® se administra mediante perfusión intravenosa. No se han realizado estudios con otras vías de administración.
Distribución
La estimación del volumen central de distribución del acMMAE en la población fue de 3,15 l, lo que equivale aproximadamente al volumen de plasma sanguíneo. In vitro, el MMAE se une moderadamente (71–77 %) a las proteínas plasmáticas humanas. El MMAE no penetra significativamente en los eritrocitos humanos in vitro; la relación entre la concentración en sangre total y plasma oscila entre 0,79 y 0,98.
Los datos in vitro indican que el MMAE es sustrato de P-gp, pero no inhibe la P-gp en concentraciones clínicamente relevantes.
Biotransformación
Se espera que en pacientes el polatuzumab vedotin sufra catabolismo, produciendo péptidos pequeños, aminoácidos, MMAE no conjugado y metabolitos derivados del MMAE no conjugado. Los niveles de metabolitos del MMAE no se han determinado en plasma humano.
Los estudios in vitro indican que el MMAE es sustrato de CYP3A4/5, pero no estimula las principales enzimas CYP. El MMAE es un inhibidor débil y dependiente del tiempo de CYP3A4/5, pero no inhibe competitivamente a CYP3A4/5 en concentraciones clínicamente relevantes.
El MMAE no inhibe CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ni CYP2D6.
Eliminación
Según los datos del análisis farmacocinético poblacional, el conjugado (acMMAE) se elimina principalmente mediante un aclaramiento lineal no específico de 0,9 l/día. Los estudios in vivo en ratas a las que se administró polatuzumab vedotin (marcado con radioisótopo en el MMAE) demostraron que la mayor parte de la radioactividad se excreta por heces y una fracción menor por orina.
Grupos especiales de pacientes
Pacientes pediátricos
No se han realizado estudios de farmacocinética del polatuzumab vedotin en niños (menores de 18 años).
Pacientes de edad avanzada
Según el análisis farmacocinético poblacional en pacientes de 19 a 89 años, la edad no influye en la farmacocinética del acMMAE ni del MMAE no conjugado. Según el análisis farmacocinético poblacional, no se observaron diferencias clínicamente relevantes en la farmacocinética del acMMAE ni del MMAE no conjugado entre pacientes menores de 65 años (n = 394) y pacientes de 65 años o más (n = 495).
Alteración de la función renal
En pacientes con alteración renal leve (aclaramiento de creatinina de 60–89 ml/min, n = 361) o moderada (aclaramiento de creatinina de 30–59 ml/min, n = 163), la exposición al acMMAE y al MMAE no conjugado fue similar a la observada en pacientes con función renal normal (aclaramiento de creatinina ≥ 90 ml/min, n = 356), según el análisis farmacocinético poblacional. No hay suficientes datos para evaluar el impacto de la alteración renal grave (aclaramiento de creatinina de 15–29 ml/min, n = 4) sobre la farmacocinética. No existen datos en pacientes con insuficiencia renal en fase terminal ni en pacientes sometidos a diálisis.
Alteración de la función hepática
Según el análisis farmacocinético poblacional, en pacientes con alteración hepática leve (TGP o TGO > 1,0–2,5 × LSN o bilirrubina total > 1,0–1,5 × LSN, n = 133), la exposición al acMMAE es similar a la de pacientes con función hepática normal (n = 737), mientras que el valor de AUC del MMAE no conjugado es hasta un 40 % mayor.
No hay suficientes datos para evaluar el impacto de la alteración hepática moderada (bilirrubina total > 1,5–3 × LSN, n = 11) sobre la farmacocinética. Existen datos limitados en pacientes con alteración hepática grave o tras trasplante hepático.
Características clínicas.
Indicaciones.
El medicamento Polivy® en combinación con rituximab, ciclofosfamida, doxorrubicina y prednisona (R-CHP) está indicado para el tratamiento de pacientes adultos con linfoma difuso de células B grandes (LDCBG) previamente no tratado.
El medicamento Polivy® en combin游戏副本ión con bendamustina y rituximab está indicado para el tratamiento de pacientes adultos con linfoma difuso de células B grandes recidivante/refractario que no sean candidatos al trasplante de células madre hematopoyéticas.
Contraindicaciones.
Hipersensibilidad al polatuzumab vedotin o a cualquier excipiente del medicamento.
Infecciones activas graves (véase la sección «Precauciones de uso»).
Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios clínicos relevantes sobre la interacción del polatuzumab vedotin con otros medicamentos en humanos.
Interacción al administrarse simultáneamente con medicamentos que son inhibidores, sustratos o inductores del CYP3A4, así como al administrarse simultáneamente con medicamentos que son inhibidores de la glucoproteína P (P-gp)
Según los resultados del modelo de simulación farmacocinética basado en fisiología sobre la liberación de MMAE a partir del polatuzumab vedotin, los inhibidores potentes del CYP3A4 y de la P-gp (por ejemplo, ketoconazol) podrían aumentar el área bajo la curva concentración-tiempo (AUC) del MMAE no conjugado en un 48 %. Se recomienda precaución al administrar simultáneamente un inhibidor del CYP3A4. Debe vigilarse cuidadosamente a los pacientes que reciben simultáneamente inhibidores potentes del CYP3A4 (por ejemplo, boceprevir, claritromicina, cobicistat, indinavir, itraconazol, nefazodona, nelfinavir, posaconazol, ritonavir, saquinavir, telaprevir, telitromicina, voriconazol) para detectar signos de toxicidad.
No se prevé que el MMAE no conjugado modifique la AUC de otros medicamentos administrados simultáneamente que sean sustratos del CYP3A4 (como el midazolam).
Los inductores potentes del CYP3A4 (por ejemplo, rifampicina, carbamazepina, fenobarbital, fenitoína, hipérico [Hypericum perforatum]) podrían reducir la exposición al MMAE no conjugado.
Interacción al administrar simultáneamente los medicamentos rituximab, bendamustina, ciclofosfamida y doxorrubicina junto con polatuzumab vedotin
La farmacocinética de rituximab, bendamustina, ciclofosfamida y doxorrubicina no se ve afectada por la administración concomitante de polatuzumab vedotin. La administración concomitante de rituximab se asocia con un aumento del 24 % en la AUC del anticuerpo conjugado con MMAE (acMMAE) en plasma y una reducción del 37 % en la AUC del MMAE no conjugado en plasma, según el análisis farmacocinético poblacional. Los valores de AUC de acMMAE y del MMAE no conjugado en plasma para el medicamento Polivy® en combinación con R-CHP son coherentes con los datos obtenidos en otros estudios con Polivy®. No se requiere ajuste de dosis.
La bendamustina no afecta la AUC de acMMAE ni del MMAE no conjugado en plasma.
Características de uso.
Seguimiento
Con el fin de mejorar la trazabilidad de los medicamentos biológicos, en la documentación médica del paciente debe indicarse clara y explícitamente el nombre comercial y el número de lote del medicamento administrado.
Mielosupresión
Se han notificado casos graves y severos de neutropenia y neutropenia febril en pacientes que recibieron Polivy®, ya durante el primer ciclo de tratamiento. Se debe considerar la administración de factores estimulantes de colonias de granulocitos (G-CSF), cuya administración profiláctica fue requerida en el programa de desarrollo clínico. Asimismo, durante el tratamiento con Polivy® puede presentarse trombocitopenia o anemia de grado 3 o 4. Se debe realizar un análisis sanguíneo completo antes de la administración de cada dosis de Polivy®. En pacientes con neutropenia y/o trombocitopenia de grado 3 o 4, se debe considerar un control de laboratorio más frecuente y/o la demora o suspensión del tratamiento con Polivy® (ver sección «Instrucciones de uso y dosis»).
Neuropatía periférica (NP)
Se ha notificado NP en pacientes ya durante el primer ciclo de tratamiento con Polivy®, y el riesgo aumenta con la administración de dosis posteriores. En pacientes con NP preexistente, esta condición puede empeorar. La NP notificada durante el tratamiento con Polivy® es principalmente sensorial. Sin embargo, también se han notificado casos de NP motora y sensoriomotora. Se debe vigilar a los pacientes en busca de síntomas de NP, tales como hipestesia, hiperestesia, parestesia, disestesia, dolor neuropático, sensación de ardor, debilidad muscular o alteraciones en la marcha. A los pacientes en quienes aparezca por primera vez o empeore la NP, puede ser necesario demorar la administración, reducir la dosis o suspender el tratamiento con Polivy® (ver sección «Instrucciones de uso y dosis»).
Infecciones
En pacientes que recibieron tratamiento con Polivy® se han notificado infecciones graves, potencialmente mortales o letales, incluyendo infecciones oportunistas, tales como neumonía (incluyendo la causada por Pneumocystis jirovecii y otras neumonías micóticas), bacteriemia, sepsis, infección herpética e infección por citomegalovirus (ver sección «Reacciones adversas»). Se han notificado reactivaciones de infecciones latentes. Se debe vigilar cuidadosamente a los pacientes durante el tratamiento en busca de signos de infecciones bacterianas, fúngicas o virales, y ante la aparición de tales signos, se debe consultar al médico. Durante el tratamiento con Polivy® se debe considerar la profilaxis de infecciones. No se debe administrar Polivy® en caso de infección activa grave. En pacientes que desarrollen una infección grave, se debe suspender el tratamiento con Polivy® y cualquier quimioterapia concomitante.
Virus de la inmunodeficiencia humana (VIH)
El medicamento Polivy® no ha sido estudiado en pacientes con VIH. Para información sobre la administración concomitante con inhibidores del CYP3A, ver sección «Interacción con otros medicamentos y otras formas de interacción».
Inmunización
No se deben administrar vacunas vivas ni vivas atenuadas simultáneamente con el tratamiento. Los pacientes que recientemente hayan recibido vacunas vivas no participaron en los estudios clínicos.
Leucoencefalopatía multifocal progresiva (LMP)
Durante el tratamiento con Polivy® se ha notificado leucoencefalopatía multifocal progresiva (LMP) (ver sección «Reacciones adversas»). Se debe vigilar cuidadosamente a los pacientes en busca de aparición de nuevos síntomas neurológicos, cognitivos o conductuales, o de empeoramiento de los mismos, que puedan indicar LMP. El tratamiento con Polivy® y cualquier quimioterapia concomitante debe suspenderse ante sospecha de LMP y debe descontinuarse permanentemente si se confirma el diagnóstico.
Síndrome de lisis tumoral (SLT)
En pacientes con alta carga tumoral y tumores con proliferación activa, puede aumentar el riesgo de SLT. Antes de iniciar el tratamiento con Polivy®, se deben tomar las medidas profilácticas adecuadas según las recomendaciones locales. Durante el tratamiento con Polivy® se debe vigilar cuidadosamente a los pacientes en busca de desarrollo de SLT.
Reacciones de infusión
Polivy® puede provocar reacciones de infusión, incluyendo reacciones graves. Se han presentado reacciones de infusión retardadas hasta 24 horas después de la administración del medicamento. Antes de la administración de Polivy® se debe administrar un antihistamínico y un antipirético, y se debe vigilar estrechamente al paciente durante toda la infusión. Si ocurren reacciones de infusión, se debe interrumpir la infusión y comenzar el tratamiento médico adecuado (ver sección «Instrucciones de uso y dosis»).
Toxicidad embrionofetal
Debido al mecanismo de acción y a los resultados de estudios preclínicos, Polivy® puede causar daño al feto si se administra durante el embarazo. Se debe informar a las mujeres embarazadas sobre el riesgo para el feto.
Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con Polivy® y al menos durante 9 meses después de la última dosis (ver sección «Uso durante el embarazo o la lactancia»). A los pacientes de sexo masculino con parejas femeninas en edad fértil se les debe aconsejar el uso de métodos anticonceptivos eficaces durante el tratamiento con Polivy® y al menos durante 6 meses después de la última dosis (ver sección «Uso durante el embarazo o la lactancia»).
Fertilidad
En estudios preclínicos, la administración de polatuzumab vedotin estuvo asociada con toxicidad testicular, lo que podría resultar en alteración de la función reproductiva y fertilidad en hombres. Por lo tanto, se recomienda a los hombres que recibirán tratamiento con Polivy® que antes del inicio del tratamiento conserven y almacenen muestras de esperma (ver sección «Uso durante el embarazo o la lactancia»).
Pacientes de edad avanzada
De los 435 pacientes previamente no tratados con LBDCD que recibieron tratamiento con Polivy® en combinación con el régimen R-CHP en el estudio GO39942, 227 (52,2 %) tenían ≥ 65 años de edad. En pacientes con ≥ 65 años, la frecuencia de reacciones adversas graves fue del 39,2 %, mientras que en pacientes con < 65 años fue del 28,4 %. Una frecuencia similar de reacciones adversas graves se observó en pacientes de edad avanzada en el grupo de tratamiento R-CHOP.
De los 151 pacientes previamente tratados con LBDCD que recibieron tratamiento con Polivy® en combinación con bendamustina y rituximab en el estudio GO29365, 103 (68 %) tenían ≥ 65 años de edad. En pacientes con ≥ 65 años, la frecuencia de reacciones adversas graves (55 %) fue similar a la de pacientes con < 65 años (56 %). Los estudios clínicos con Polivy® no incluyeron un número suficiente de pacientes con ≥ 65 años para establecer diferencias en la respuesta en comparación con pacientes más jóvenes.
Hepatotoxicidad
En pacientes que recibieron tratamiento con Polivy® se han presentado casos graves de hepatotoxicidad, indicativos de daño hepatocelular, incluyendo elevación de transaminasas y/o bilirrubina (ver sección «Reacciones adversas»). Enfermedades hepáticas preexistentes, niveles iniciales elevados de enzimas hepáticas y el uso concomitante de otros medicamentos pueden aumentar el riesgo de hepatotoxicidad. Se debe controlar los niveles de enzimas hepáticas y bilirrubina (ver sección «Instrucciones de uso y dosis»).
Excipientes
Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, por lo tanto, es prácticamente libre de sodio.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil/anticoncepción en hombres y mujeres
Mujeres
Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces durante el tratamiento con polatuzumab vedotin y al menos durante 9 meses después de la última dosis.
Hombres
A los pacientes de sexo masculino con parejas femeninas en edad fértil se les debe aconsejar el uso de métodos anticonceptivos eficaces durante el tratamiento con polatuzumab vedotin y al menos durante 6 meses después de la última dosis.
Embarazo
No existen datos sobre el uso de Polivy® en mujeres embarazadas. Los estudios en animales mostraron toxicidad reproductiva. Debido al mecanismo de acción y a los resultados de estudios preclínicos, el polatuzumab vedotin puede causar daño al feto si se administra durante el embarazo. Se debe realizar una prueba de embarazo a las mujeres en edad fértil antes del inicio del tratamiento. No se recomienda el uso de Polivy® durante el embarazo ni en mujeres en edad fértil que no utilicen anticoncepción, a menos que el beneficio potencial para la mujer supere el riesgo potencial para el feto.
Lactancia
No se sabe si el polatuzumab vedotin o sus metabolitos se excretan en la leche materna humana. No puede descartarse el riesgo para los lactantes. Durante el tratamiento con Polivy® y al menos durante 3 meses después de la última dosis, las mujeres deben interrumpir la lactancia.
Fertilidad
En estudios preclínicos, la administración de polatuzumab vedotin estuvo asociada con toxicidad testicular y podría provocar alteración de la función reproductiva y fertilidad en hombres.
Por lo tanto, se recomienda a los hombres que recibirán tratamiento con este medicamento que antes del tratamiento conserven y almacenen muestras de esperma. No se recomienda la concepción durante el tratamiento con Polivy® ni durante los 6 meses posteriores a la última dosis.
Capacidad para conducir y utilizar máquinas.
Polivy® tiene un efecto insignificante sobre la capacidad para conducir vehículos o manejar maquinaria. Durante el tratamiento con Polivy® pueden presentarse reacciones de infusión, neuropatía periférica, fatiga y mareo (ver secciones «Características de uso» y «Reacciones adversas»).
Vía de administración y dosis.
Polivy® debe administrarse bajo la estrecha supervisión de un médico con experiencia en el diagnóstico y tratamiento de pacientes con enfermedades oncológicas.
Dosificación
Linfoma B difuso de grandes células
Pacientes previamente no tratados
La dosis recomendada de Polivy® es de 1,8 mg/kg en forma de infusión intravenosa cada 21 días, en combinación con rituximab, ciclofosfamida, doxorubicina y prednisona (R-CHP), durante 6 ciclos. Polivy®, rituximab, ciclofosfamida y doxorubicina pueden administrarse en cualquier orden el día 1, tras la administración de prednisona. La prednisona se administra del día 1 al 5 de cada ciclo. Los ciclos 7 y 8 incluyen la administración de rituximab como monoterapia.
Véase el prospecto de los medicamentos quimioterápicos utilizados en combinación con Polivy® en pacientes con linfoma B difuso de grandes células previamente no tratado.
Pacientes con recidiva o refractarios
La dosis recomendada de Polivy® es de 1,8 mg/kg en forma de infusión intravenosa, en combinación con bendamustina y rituximab, cada 21 días durante 6 ciclos. Polivy®, bendamustina y rituximab pueden administrarse en cualquier orden el día 1 de cada ciclo. La dosis recomendada de bendamustina cuando se utiliza junto con Polivy® es de 90 mg/m²/día el día 1 y el día 2 de cada ciclo; la dosis recomendada de rituximab es de 375 mg/m² el día 1 de cada ciclo. Debido a la experiencia clínica limitada en pacientes que han recibido Polivy® a una dosis de 1,8 mg/kg con una dosis acumulada > 240 mg, no se recomienda exceder la dosis de 240 mg por ciclo.
Pacientes previamente no tratados y pacientes con recidiva o refractarios
Si no se ha realizado una premedicación previa, antes de la administración de Polivy® debe administrarse premedicación con antihistamínicos y medicamentos antipiréticos.
Retraso o omisión de la administración
Si no se ha administrado la dosis programada de Polivy®, debe administrarse tan pronto como sea posible, ajustando el calendario para mantener un intervalo de 21 días entre administraciones.
Modificación de la dosis
La velocidad de infusión de Polivy® debe reducirse o la infusión debe interrumpirse si el paciente presenta una reacción relacionada con la infusión. La administración de Polivy® debe suspenderse inmediatamente y el medicamento debe retirarse definitivamente si el paciente experimenta una reacción que ponga en peligro su vida.
Las opciones de modificación de la dosis de Polivy® difieren entre los pacientes con linfoma B difuso de grandes células previamente no tratado y los pacientes con recidiva o refractarios.
Para información sobre la modificación de la dosis en caso de neuropatía periférica (véase la sección «Propiedades farmacodinámicas»), véase la tabla 3.
Tabla 3
Modificación de la dosis del medicamento Polivy® en caso de aparición de neuropatía periférica (NP)
| Indicaciones |
Gravedad de la PN en el día 1 de cualquier ciclo |
Modificación de la dosis |
| LBCL no tratado previamente |
Grado 2a |
Neuropatía sensorial:
Neuropatía motora:
En caso de que ocurran simultáneamente neuropatía sensorial y motora, se deben seguir las recomendaciones con las restricciones más estrictas indicadas anteriormente. |
| Grado 3a |
Neuropatía sensorial:
Neuropatía motora:
En caso de que ocurran simultáneamente neuropatía sensorial y motora, se deben seguir las recomendaciones con las restricciones más estrictas indicadas anteriormente. |
|
| Grado 4 |
Interrumpir el uso del medicamento Polivy®. |
|
| LBCL recidivante/refractaria |
Grado 2–3 |
Interrumpir el uso del medicamento Polivy® hasta mejoría hasta grado ≤ 1. Si la recuperación alcanza grado ≤ 1 en el día 14 o antes, continuar el tratamiento con Polivy® a una dosis permanentemente reducida de 1,4 mg/kg. Si previamente ya se había reducido la dosis a 1,4 mg/kg, se debe interrumpir el tratamiento con Polivy®. Si no hay recuperación hasta grado ≤ 1 en el día 14 o antes, se debe interrumpir el tratamiento con Polivy®. |
| Grado 4 |
Interrumpir el uso del medicamento Polivy®. |
a Se puede continuar con la administración de R-CHP.
Para obtener información sobre la modificación de la dosis en caso de aparición de mielosupresión (véase la sección «Instrucciones de uso»), consulte la tabla 4.
Tabla 4
Modificación de la dosis del medicamento Polayvī®, quimioterapia y rituximab en caso de aparición de mielosupresión
| Indicaciones |
Gravedad de la mielosupresión en el día 1 de cualquier ciclo |
Modificación de la dosis |
| DLBCL no tratada previamente |
Neutropenia grados 3-4 |
Interrumpir todos los medicamentos hasta la recuperación del recuento absoluto de neutrófilos (RAN)* a > 1000/μl. Si el RAN se recupera a > 1000/μl en el día 7 o antes, continuar todos los medicamentos sin reducción de dosis. Si el RAN se recupera a > 1000/μl después del día 7:
|
| Trombocitopenia grados 3-4 |
Interrumpir todos los medicamentos hasta la recuperación del recuento de plaquetas a > 75 000/μl. Si el recuento de plaquetas se recupera a > 75 000/μl en el día 7 o antes, continuar todos los medicamentos sin reducción de dosis. Si el recuento de plaquetas se recupera a > 75 000/μl después del día 7:
|
|
| DLBCL recidivante/refractaria |
Neutropenia grados 3-41 |
Interrumpir todos los medicamentos hasta la recuperación del RAN a > 1000/μl. Si el RAN se recupera a > 1000/μl en el día 7 o antes, continuar todos los medicamentos sin reducción de dosis. Si el RAN se recupera a > 1000/μl después del día 7:
|
| Trombocitopenia grados 3-41 |
Interrumpir todos los medicamentos hasta la recuperación del recuento de plaquetas a > 75 000/μl. Si el recuento de plaquetas se recupera a > 75 000/μl en el día 7 o antes, continuar todos los medicamentos sin reducción de dosis. Si el recuento de plaquetas se recupera a > 75 000/μl después del día 7:
|
1 Si la causa principal es linfoma, posiblemente no sea necesario reducir la dosis de bendamustina.
Para información sobre la modificación de la dosis en caso de reacciones de infusión (véase la sección «Instrucciones de uso»), véase la tabla 5.
Tabla 5
Modificación de la dosis del medicamento Polivy® ante la aparición de reacciones de infusión (RI)
| Indicaciones |
Gravedad de la RI en el día 1 de cualquier ciclo |
Modificación de la dosis |
| Linfoma difuso de grandes células B previamente no tratado y recidivante/refractario |
RI grados 1-3 |
Interrumpir la infusión de Polivy® y proporcionar tratamiento de apoyo. En caso de presentación por primera vez de sibilancias, broncoespasmo o urticaria generalizada de grado 3, se debe interrumpir Polivy® de forma permanente. En caso de recurrencia de sibilancias o urticaria de grado 2, o recurrencia de cualquier síntoma de grado 3, se debe interrumpir Polivy® de forma permanente. En otros casos, tras la resolución completa de los síntomas, la infusión puede reiniciarse a una velocidad del 50 % de la velocidad alcanzada antes de la interrupción. En ausencia de reacciones por infusión, la velocidad de infusión puede aumentarse en incrementos de 50 mg/hora cada 30 minutos. En el ciclo siguiente, la infusión de Polivy® debe administrarse durante 90 minutos. Si no se presentan reacciones por infusión, las infusiones siguientes pueden administrarse en 30 minutos. La premedicación debe administrarse en todos los ciclos. |
| RI grado 4 |
Interrumpir inmediatamente la infusión de Polivy®. Proporcionar tratamiento de apoyo. Interrumpir Polivy® de forma permanente. |
Grupos de pacientes especiales
Pacientes de edad avanzada
No se requiere ajuste de la dosis del medicamento Polivy® en pacientes de 65 años o más (ver sección «Farmacocinética»).
Alteraciones de la función renal
No se requiere ajuste de la dosis del medicamento Polivy® en pacientes con aclaramiento de creatinina ≥ 30 ml/min. La dosis recomendada en pacientes con aclaramiento de creatinina < 30 ml/min no ha sido establecida debido a datos limitados.
Alteraciones de la función hepática
Debe evitarse el uso de Polivy® en pacientes con alteración hepática moderada o grave (niveles de bilirrubina superiores a 1,5 veces el límite superior normal [LSN]).
No se requiere ajuste de la dosis inicial de Polivy® en pacientes con alteración hepática leve (niveles de bilirrubina superiores al LSN pero menores o iguales a 1,5 veces el LSN o niveles de AST superiores al LSN).
En la población estudiada con alteración hepática leve (definida como niveles de AST o ALT > 1,0–2,5 veces el LSN o niveles de bilirrubina total > 1,0–1,5 veces el LSN), se observó un aumento en la exposición al MMAE no conjugado de hasta un 40 %, lo cual no se consideró clínicamente relevante.
Pacientes pediátricos
La seguridad y eficacia del medicamento en niños (menores de 18 años) no han sido establecidas. No existen datos disponibles.
Vía de administración
El medicamento Polivy® está indicado para administración por vía intravenosa.
La dosis inicial de Polivy® debe administrarse como una infusión intravenosa durante 90 minutos. Durante la infusión y al menos durante 90 minutos tras la administración de la dosis inicial, se debe observar al paciente en busca de reacciones por infusión o reacciones de hipersensibilidad.
Si la infusión previa fue bien tolerada, la siguiente dosis de Polivy® puede administrarse durante 30 minutos, y al paciente se le debe observar durante la infusión y al menos durante 30 minutos tras la finalización de la infusión.
El medicamento Polivy® debe reconstituirse y diluirse siguiendo técnicas asépticas bajo la supervisión de personal sanitario. El medicamento debe administrarse mediante infusión intravenosa a través de un sistema de infusión independiente, equipado con un filtro estéril, apirógeno, incorporado o adicional, con baja unión proteica (de poro 0,2 o 0,22 μm) y un catéter. No se debe administrar Polivy® mediante inyección intravenosa en bolo o por vía intravenosa rápida.
Precauciones que deben tomarse antes de la preparación o administración del medicamento
Polivy® contiene un componente citotóxico unido covalentemente a un anticuerpo monoclonal. Se deben seguir las normas adecuadas para la manipulación y eliminación del medicamento.
Precauciones generales
Polivy® contiene un componente citotóxico. El medicamento debe administrarse bajo supervisión médica de un profesional con experiencia en el uso de agentes citotóxicos. Se deben seguir las normas adecuadas para la manipulación y eliminación de medicamentos antineoplásicos y citotóxicos.
El medicamento reconstituido no contiene conservantes y está destinado únicamente para uso único. Se deben seguir las normas adecuadas de asepsia durante la manipulación de este medicamento.
Polivy® debe reconstituirse con agua para inyección estéril y diluirse antes de la administración en una bolsa para infusión intravenosa que contenga solución inyectable de cloruro de sodio 9 mg/ml (0,9 %) o solución inyectable de cloruro de sodio 4,5 mg/ml (0,45 %), o solución glucosada al 5 %.
No se debe congelar ni exponer la solución reconstituida ni la solución para infusión a la luz solar directa.
Instrucciones para la reconstitución
- Polivy®, 30 mg: utilizando una jeringa estéril, inyectar lentamente 1,8 ml de agua para inyección estéril en el vial de 30 mg de Polivy® para obtener una solución monodosis que contenga 20 mg/ml de polatuzumab vedotin. Dirigir el chorro contra la pared del vial, no directamente sobre la masa liofilizada.
- Polivy®, 140 mg: utilizando una jeringa estéril, inyectar lentamente 7,2 ml de agua para inyección estéril en el vial de 140 mg de Polivy® para obtener una solución monodosis que contenga 20 mg/ml de polatuzumab vedotin. Dirigir el chorro contra la pared del vial, no directamente sobre la masa liofilizada.
- Agitar suavemente el vial hasta lograr la disolución completa. No agitar.
- Examinar visualmente la solución reconstituida en busca de partículas sólidas y cambios de color. La solución reconstituida debe ser incolora o ligeramente marrón, transparente o ligeramente opalescente y no debe contener partículas visibles. No utilizar la solución reconstituida si presenta un color alterado, si está turbia o si contiene partículas visibles.
Instrucciones para la dilución
- Diluir Polivy® hasta una concentración final de 0,72–2,7 mg/ml en una bolsa para infusión intravenosa con un volumen mínimo de 50 ml que contenga solución inyectable de cloruro de sodio 9 mg/ml, solución inyectable de cloruro de sodio 4,5 mg/ml o solución glucosada al 5 %.
- Determinar el volumen de la solución reconstituida de 20 mg/ml necesario para obtener la dosis prescrita (ver a continuación):
| Dosis total del medicamento Polayvi® (ml) para posterior dilución |
= |
dosificación del medicamento Polayvi® (mg/kg) × peso corporal del paciente (kg) |
| concentración de la solución reconstituida en el vial (20 mg/ml) |
-
Con un jeringa estéril, extraer del frasco el volumen necesario de la solución reconstituida del medicamento Polivy® y diluirlo en una bolsa para perfusión intravenosa. Desechar cualquier parte no utilizada del medicamento que quede en el frasco.
-
Mezclar suavemente el contenido de la bolsa de perfusión girándola lentamente. No agitar.
-
Examinar el contenido de la bolsa de perfusión en busca de partículas y, si se observan, desechar el contenido.
Se debe evitar el transporte de la solución para perfusión preparada, ya que el estrés provocado por el movimiento puede provocar agregación. Si es necesario transportar la solución preparada, se debe extraer el aire de la bolsa de perfusión y limitar el transporte a un máximo de 30 minutos a temperatura ambiente (9–25 °C) o 24 horas en refrigeración (2–8 °C). Si se extrae el aire, debe utilizarse un conjunto de perfusión con aguja metálica para la conexión, a fin de garantizar una administración precisa durante la perfusión. El tiempo total de almacenamiento más el tiempo de transporte de la solución diluida no debe exceder el período de almacenamiento indicado en la tabla 6.
Polivy® debe administrarse mediante un sistema de perfusión independiente, equipado con un filtro incorporado o adicional, estéril y apirógeno, con baja unión a proteínas (con tamaño de poro de 0,2 o 0,22 μm) y un catéter.
Polivy® es compatible con bolsas para perfusión intravenosa de cloruro de polivinilo (PVC) o poliolefinas, como polietileno (PE) y polipropileno. Asimismo, no se ha observado incompatibilidad de los conjuntos de perfusión o dispositivos con productos que contienen PVC, PE, poliuretano, polibutadieno, acrilonitrilo-butadieno-estireno, policarbonato, polieteruretano, etileno-propileno fluorado o politetrafluoroetileno, ni con membranas filtrantes compuestas de poliéter sulfona o polisulfona.
Período de validez
Solución reconstituida
Desde el punto de vista microbiológico, la solución reconstituida debe utilizarse inmediatamente. Si el medicamento no se utiliza inmediatamente, el período y las condiciones de almacenamiento de la solución reconstituida son responsabilidad del usuario. El período de almacenamiento en refrigeración (2–8 °C) no debe superar generalmente las 24 horas, siempre que la reconstitución se haya llevado a cabo en condiciones asépticas controladas y validadas. La estabilidad química y física de la solución reconstituida se ha demostrado durante un período de hasta 72 horas cuando se almacenó en refrigeración (2–8 °C) y hasta 24 horas a temperatura ambiente (9–25 °C).
Solución diluida
Desde el punto de vista microbiológico, la solución diluida para perfusión debe utilizarse inmediatamente. Si el medicamento no se utiliza inmediatamente, el período y las condiciones de almacenamiento de la solución diluida son responsabilidad del usuario. El período de almacenamiento en refrigeración (2–8 °C) no debe superar generalmente las 24 horas, siempre que la dilución se haya llevado a cabo en condiciones asépticas controladas y validadas. La estabilidad química y física de la solución diluida para perfusión se ha demostrado durante los períodos indicados en la tabla 6. La solución diluida debe desecharse si el tiempo de almacenamiento excede los períodos indicados en la tabla 6.
Tabla 6
Períodos durante los cuales se ha demostrado la estabilidad química y física de la solución para perfusión preparada
| Disolvente utilizado para la preparación de la solución para perfusión |
Condiciones de conservación de la solución para perfusión1 |
| Cloruro de sodio 9 mg/ml (0,9 %) |
Hasta 72 horas en nevera (2–8 °C) o hasta 4 horas a temperatura ambiente (9–25 °C) |
| Cloruro de sodio 4,5 mg/ml (0,45 %) |
Hasta 72 horas en nevera (2–8 °C) o hasta 8 horas a temperatura ambiente (9–25 °C) |
| Solución de glucosa al 5 % |
Hasta 72 horas en nevera (2–8 °C) o hasta 8 horas a temperatura ambiente (9–25 °C) |
1Con el fin de garantizar la estabilidad del medicamento, no se deben superar los períodos de conservación indicados.
El medicamento Polayvi® está indicado únicamente para uso único.
Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos normativos locales.
Niños
La seguridad y eficacia del medicamento en niños (menores de 18 años) no han sido establecidas. No existen datos disponibles.
Sobredosis.
No existe experiencia de sobredosis en seres humanos en estudios clínicos. La dosis más alta estudiada hasta la fecha es de 2,4 mg/kg administrada mediante infusión intravenosa; esta dosis se asoció con mayor frecuencia y gravedad de casos de neuropatía periférica. En caso de sobredosis, se debe interrumpir inmediatamente la infusión y observar cuidadosamente al paciente.
Reacciones adversas.
Resumen del perfil de seguridad
La seguridad del medicamento Polivy® se evaluó en 435 pacientes en el estudio GO39942 (POLARIX). Se observaron las siguientes reacciones adversas descritas en esta sección:
- durante el tratamiento y el período de seguimiento posterior en pacientes no tratados previamente con LBCL-DHIT, que participaron en el estudio clínico principal GO39942 (POLARIX) y recibieron Polivy® más R-CHP (n = 435) o R-CHOP (n = 438). En el grupo de tratamiento con Polivy® más R-CHP, el 91,7 % recibió 6 ciclos del medicamento Polivy® en comparación con el 88,5 % de los pacientes que recibieron 6 ciclos de vincristina en el grupo R-CHOP.
En pacientes no tratados previamente con LBCL-DHIT que recibieron tratamiento con Polivy® más R-CHP:
- Las reacciones adversas más frecuentemente notificadas (≥ 30 %) en pacientes que recibieron tratamiento con Polivy® más R-CHP por LBCL-DHIT no tratada previamente fueron neuropatía periférica (52,9 %), náuseas (41,6 %), neutropenia (38,4 %) y diarrea (30,8 %).
- Se notificaron reacciones adversas graves en el 24,1 % de los pacientes que recibieron tratamiento con Polivy® más R-CHP.
- Las reacciones adversas graves más frecuentes, notificadas en ≥ 5 % de los pacientes, fueron neutropenia febril (10,6 %) y neumonía (5,3 %).
- La reacción adversa que condujo a la interrupción prematura del régimen de tratamiento en > 1 % de los pacientes que recibieron tratamiento con Polivy® más R-CHP fue neumonía (1,1 %).
La seguridad del uso del medicamento Polivy® se evaluó en 151 pacientes en el estudio GO29365. Se observaron las siguientes reacciones adversas descritas en esta sección:
- durante el tratamiento y el período de observación en pacientes previamente tratados con LBCL-DHIT (n = 151) en el estudio clínico principal GO29365. Esto incluyó pacientes de la fase de entrada (n = 6), pacientes aleatorizados (n = 39) y pacientes de la cohorte de expansión (n = 106), que recibieron Polivy® + BR, en comparación con pacientes aleatorizados (n = 39) que recibieron solo BR. Los pacientes en los grupos de tratamiento recibieron un promedio de 5 ciclos de tratamiento, mientras que los pacientes aleatorizados en el grupo de comparación recibieron un promedio de 3 ciclos de tratamiento.
En pacientes previamente tratados con LBCL-DHIT que recibieron tratamiento con Polivy® más bendamustina y rituximab:
- Las reacciones adversas notificadas con mayor frecuencia (≥ 30 %) (todos los grados) en pacientes que recibieron tratamiento con Polivy® más bendamustina y rituximab por LBCL-DHIT previamente tratada fueron neutropenia (45,7 %), diarrea (35,8 %), náuseas (33,1 %), trombocitopenia (32,5 %), anemia (31,8 %) y neuropatía periférica (30,5 %).
- Se notificaron reacciones adversas graves en el 41,7 % de los pacientes que recibieron tratamiento con Polivy® más bendamustina y rituximab.
- Las reacciones adversas graves más frecuentes, notificadas en ≥ 5 % de los pacientes, fueron: neutropenia febril (10,6 %), sepsis (9,9 %), neumonía (8,6 %) y fiebre (7,9 %).
- La reacción adversa que condujo a la interrupción prematura del régimen de tratamiento en > 5 % de los pacientes que recibieron tratamiento con Polivy® más bendamustina y rituximab fue trombocitopenia (7,9 %).
A continuación se indican las reacciones adversas que ocurrieron en 586 pacientes que recibieron tratamiento con el medicamento Polivy® en estudios clínicos.
Las reacciones adversas se enumeran a continuación por clases de sistemas orgánicos MedDRA y categoría de frecuencia. La categoría de frecuencia correspondiente para cada reacción adversa se basa en lo siguiente: muy frecuente (≥ 1/10), frecuente (≥ 1/100 a < 1/10), poco frecuente (≥ 1/1000 a < 1/100), raro (≥ 1/10 000 a < 1/1000), muy raro (< 1/10 000). Dentro de cada grupo por frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Infecciones e infestaciones: muy frecuente – neumoníaa, infección de las vías respiratorias superiores; frecuente – sepsisa, infección por herpesvirusa, infección por citomegalovirus, infección del tracto urinarioc.
Alteraciones de la sangre y del sistema linfático: muy frecuente – neutropenia febril, neutropenia, trombocitopenia, anemia, leucopenia; frecuente – linfopenia, pancitopenia.
Alteraciones del metabolismo y de la nutrición: muy frecuente – hipokalemia, disminución del apetito; frecuente – hipocalcemia, hipoalbuminemia.
Alteraciones del sistema nervioso: muy frecuente – neuropatía periférica; frecuente – vértigo.
Alteraciones del ojo: poco frecuente – visión borrosab.
Alteraciones del aparato respiratorio, del tórax y del mediastino: muy frecuente – tos; frecuente – neumonitis, disnea.
Alteraciones gastrointestinales: muy frecuente – diarrea, náuseas, estreñimiento, vómitos, mucositis, dolor abdominal.
Alteraciones de la piel y del tejido subcutáneo: muy frecuente – alopeciac; frecuente – prurito, infecciones de la piel, erupciones cutáneasc, sequedad de la piel.
Alteraciones del sistema musculoesquelético y del tejido conectivo: frecuente – artralgia, mialgias.
Alteraciones sistémicas y condiciones en el sitio de administración: muy frecuente – fatiga, fiebre, astenia; frecuente – edema periféricoc, escalofríos.
Alteraciones en los resultados de laboratorio: muy frecuente – pérdida de peso; frecuente – aumento de transaminasas, aumento de lipasab, hipofosfatemia.
Lesiones, intoxicaciones y complicaciones de procedimientos: muy frecuente – reacción de infusión.
a Reacciones adversas asociadas con desenlace letal.
b Reacciones adversas observadas únicamente en LBCL-DHIT recurrente o refractaria.
c Reacciones adversas observadas únicamente en LBCL-DHIT no tratada previamente.
Las reacciones adversas indicadas se observaron tanto en LBCL-DHIT no tratada previamente como en LBCL-DHIT recurrente o refractaria, excepto las indicadas en las notas.
Reacciones adversas que ocurrieron rara y muy raramente: ausentes.
Descripción de reacciones adversas individuales observadas en estudios clínicos
Mielosupresión
En el estudio controlado con placebo GO39942 (POLARIX), el 0,5 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP interrumpieron prematuramente el tratamiento investigacional debido a neutropenia. Ningún paciente interrumpió el tratamiento investigacional en el grupo R-CHOP por neutropenia. La trombocitopenia provocó la interrupción prematura del tratamiento investigacional en el 0,2 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP en comparación con ausencia de tales pacientes en el grupo R-CHOP. El tratamiento de ningún paciente se interrumpió prematuramente por anemia, ni en el grupo Polivy® más R-CHP ni en el grupo R-CHOP.
En el estudio abierto GO29365, el medicamento Polivy® fue interrumpido prematuramente por neutropenia en el 4 % de los pacientes en los grupos de tratamiento con Polivy® más bendamustina y rituximab en comparación con el 2,6 % de los pacientes en el grupo de tratamiento con bendamustina y rituximab, en quienes el tratamiento fue interrumpido por neutropenia. La trombocitopenia provocó la interrupción del tratamiento en el 7,9 % de los pacientes en los grupos de tratamiento con Polivy® + BR y en el 5,1 % de los pacientes en el grupo BR. En ningún paciente de ambos grupos (grupos de tratamiento con Polivy® + BR o solo grupo BR) se interrumpió el tratamiento por anemia. En los grupos de tratamiento con Polivy® más bendamustina y rituximab, se notificaron neutropenia, trombocitopenia y anemia de grado 3 o superior en el 40,4 %, 25,8 % y 12,6 % de los pacientes, respectivamente.
Neuropatía periférica (NP)
En el estudio controlado con placebo GO39942 (POLARIX), en el grupo de tratamiento con Polivy® más R-CHP, se notificó NP de grado 1, 2 y 3 en el 39,1 %, 12,2 % y 1,6 % de los pacientes, respectivamente. En el grupo R-CHOP, se notificó NP de grado 1, 2 y 3 en el 37,2 %, 15,5 % y 1,1 % de los pacientes, respectivamente. No se notificaron eventos de NP de grado 4–5 ni en el grupo de tratamiento con Polivy® más R-CHP ni en el grupo R-CHOP. En el grupo Polivy® más R-CHP, el 0,7 % de los pacientes interrumpieron prematuramente el tratamiento investigacional por NP en comparación con el 2,3 % en el grupo R-CHOP. En el grupo Polivy® más R-CHP, el 4,6 % de los pacientes tuvieron reducción de la dosis del tratamiento investigacional por NP en comparación con el 8,2 % en el grupo R-CHOP. En el grupo de tratamiento con Polivy® más R-CHP, la mediana del tiempo hasta la aparición del primer evento de NP fue de 2,27 meses en comparación con 1,87 meses en el grupo R-CHOP. En la fecha de cierre de recopilación de datos clínicos, los eventos de NP desaparecieron en el 57,8 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP en comparación con el 66,9 % en el grupo R-CHOP. La mediana del tiempo hasta la resolución de la neuropatía periférica fue de 4,04 meses en el grupo de tratamiento con Polivy® más R-CHP en comparación con 4,6 meses en el grupo R-CHOP.
En el estudio abierto GO29365, en los grupos de tratamiento con Polivy® más bendamustina y rituximab, se notificó NP de grado 1 y 2 en el 15,9 % y 12,6 % de los pacientes, respectivamente. En el grupo de tratamiento con bendamustina y rituximab, los eventos de NP de grado 1 y 2 se registraron en el 2,6 % y 5,1 % de los pacientes, respectivamente. Se notificó un caso de NP de grado 3 en los grupos Polivy® + BR y en ningún paciente del grupo BR. No se notificó NP de grado 4–5 en ningún grupo (grupos de tratamiento Polivy® + BR o solo grupo BR). En el 2,6 % de los pacientes, el tratamiento con el medicamento Polivy® se interrumpió prematuramente por NP y en el 2 % de los pacientes la dosis del medicamento Polivy® se redujo por NP. En ningún paciente del grupo BR se interrumpió prematuramente el tratamiento ni se redujo la dosis por NP. En los grupos Polivy® + BR, la mediana del tiempo hasta la aparición del primer evento de NP fue de 1,6 meses y en el 39,1 % de los pacientes con NP se notificó la desaparición de la NP.
Infecciones
En el estudio controlado con placebo GO39942 (POLARIX), se notificaron infecciones, incluyendo neumonía y otros tipos de infecciones, en el 49,7 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP y en el 42,7 % de los pacientes en el grupo R-CHOP. Las infecciones de grado 3–4 ocurrieron en el 14 % de los pacientes en el grupo Polivy® más R-CHP y en el 11,2 % de los pacientes en el grupo R-CHOP. En el grupo de tratamiento con Polivy® más R-CHP, se notificaron infecciones graves en el 14 % de los pacientes y se registraron infecciones con desenlace letal en el 1,1 % de los pacientes. En el grupo R-CHOP, se notificaron infecciones graves en el 10,3 % de los pacientes y se registraron infecciones con desenlace letal en el 1,4 % de los pacientes. El tratamiento se interrumpió por infección en 7 pacientes (1,6 %) en el grupo de tratamiento con Polivy® más R-CHP en comparación con 10 pacientes (2,3 %) en el grupo R-CHOP.
En el estudio abierto GO29365, se notificaron infecciones, incluyendo neumonía y otros tipos de infecciones, en el 48,3 % de los pacientes en los grupos Polivy® + BR y en el 51,3 % de los pacientes en el grupo BR. En los grupos Polivy® + BR, se notificaron infecciones graves en el 27,2 % de los pacientes y se notificaron infecciones con desenlace letal en el 6,6 % de los pacientes. En el grupo BR, se notificaron infecciones graves en el 30,8 % de los pacientes y se notificaron infecciones con desenlace letal en el 10,3 % de los pacientes. En 4 pacientes (2,6 %) en los grupos Polivy® + BR, el tratamiento se interrumpió prematuramente por infección en comparación con 2 pacientes (5,1 %) en el grupo BR.
Leucoencefalopatía multifocal progresiva (LMP)
En el estudio controlado con placebo GO39942 (POLARIX), no se notificaron casos de leucoencefalopatía multifocal progresiva.
En el estudio abierto GO29365, se observó un caso de LMP con desenlace letal en un paciente que recibía tratamiento con Polivy® + bendamustina y obinutuzumab. Este paciente había recibido previamente tres líneas de terapia, que incluyeron anticuerpos anti-CD20.
Hepatotoxicidad
En el estudio controlado con placebo GO39942 (POLARIX), se notificó hepatotoxicidad en el 10,6 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP y en el 7,3 % de los pacientes en el grupo R-CHOP. En el grupo de tratamiento con Polivy® más R-CHP, la mayoría de los eventos fueron de grado 1–2 (8,7 %); se notificaron eventos de grado 3 en el 1,8 % de los pacientes. No se observaron eventos de grado 4 o 5. En 1 paciente (0,2 %) se registró hepatotoxicidad grave que fue reversible.
En otro estudio, se notificaron dos casos de hepatotoxicidad grave (lesión hepatocelular y esteatosis hepática), que fueron reversibles.
Toxicidad gastrointestinal
En el estudio controlado con placebo GO39942 (POLARIX), los eventos de toxicidad gastrointestinal se registraron en el 76,1 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP en comparación con el 71,9 % de los pacientes en el grupo R-CHOP. La mayoría de los eventos fueron de grado 1–2, y los eventos de grado ≥ 3 se registraron en el 9,7 % de los pacientes en el grupo de tratamiento con Polivy® más R-CHP en comparación con el 8,2 % de los pacientes en el grupo R-CHOP. Los eventos más comunes de toxicidad gastrointestinal fueron náuseas y diarrea.
En el estudio abierto GO29365, se notificaron eventos de toxicidad gastrointestinal en el 72,8 % de los pacientes en el grupo Polivy® + BR en comparación con el 66,7 % de los pacientes en el grupo BR. La mayoría de los eventos fueron de grado 1–2. Se notificaron eventos de grado 3–4 en el 16,5 % de los pacientes en el grupo Polivy® + BR en comparación con el 12,9 % de los pacientes en el grupo BR. Los eventos más comunes de toxicidad gastrointestinal fueron diarrea y náuseas.
Período de validez.
30 meses.
Condiciones de almacenamiento.
Conservar a una temperatura de entre 2 y 8 ºC en el envase original para protegerlo de la luz. No congelar. Mantener en un lugar fuera del alcance de los niños.
Incompatibilidades.
Este medicamento no debe mezclarse ni diluirse con otros medicamentos, excepto los indicados en la sección «Precauciones que deben tomarse antes de la reconstitución o administración del medicamento».
Envase.
Frasco de 6 ml de vidrio incoloro (vidrio borosilicato, clase I), tapado con un tapón de caucho de butilo (revestido con película de fluorocaucho) y sellado con una cápsula de aluminio con disco plástico tipo «flip-off». 30 mg por frasco. 1 frasco por caja de cartón.
Frasco de 20 ml de vidrio incoloro (vidrio borosilicato, clase I), tapado con un tapón de caucho de butilo (revestido con película de fluorocaucho) y sellado con una cápsula de aluminio con disco plástico tipo «flip-off». 140 mg por frasco. 1 frasco por caja de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
F. Hoffmann-La Roche Ltd
Dirección del fabricante y lugar de actividad.
Wurmisweg, 4303 Kaiseraugst, Suiza