Polyvi
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT Polivy® (Polivy®)
Composition:
Active substance: polatuzumab vedotin;
1 vial contains 140 mg of polatuzumab vedotin;
1 vial contains 30 mg of polatuzumab vedotin;
1 ml of prepared (reconstituted) solution contains 20 mg/ml of polatuzumab vedotin;
Excipients: succinic acid, sodium hydroxide, sucrose, polysorbate 20.
Pharmaceutical form. Powder for concentrate for solution for infusion.
Main physicochemical properties: lyophilized powder of white to greyish-white color.
Pharmaco-therapeutic group. Antineoplastic and immunomodulating agents. Antineoplastic agents. Monoclonal antibodies and antibody-drug conjugates. Other monoclonal antibodies and antibody-drug conjugates.
ATC code L01F X14.
Pharmacological Properties
Pharmacodynamics
Mechanism of action
Polatuzumab vedotin is an antibody-drug conjugate composed of the antimitotic agent monomethyl auristatin E (MMAE) covalently linked to a monoclonal antibody (recombinant humanized immunoglobulin G1 [IgG1]) targeting CD79b. The monoclonal antibody is produced using recombinant DNA technology in Chinese hamster ovary cells. The monoclonal antibody binds with high affinity and selectivity to CD79b, a component of the B-cell receptor complex on the cell surface. CD79b expression is restricted to normal B-cell lineage cells (except plasma cells) and malignant B-cells. CD79b is expressed in > 95% of diffuse large B-cell lymphomas (DLBCL). After binding to CD79b, polatuzumab vedotin is rapidly internalized, and the linker is cleaved by lysosomal proteases, enabling intracellular release of MMAE. MMAE binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis.
Pharmacodynamic effects
Cardiac electrophysiology
Based on electrocardiography (ECG) data from two open-label studies in patients with previously treated B-cell malignancies, polatuzumab vedotin at the recommended clinical dose did not cause clinically meaningful prolongation of the mean QTc interval.
Clinical efficacy and safety
Previously untreated DLBCL
The efficacy of Polivy® was evaluated in an international, multicenter, randomized, double-blind, placebo-controlled study (POLARIX, GO39942) involving 879 patients with previously untreated DLBCL.
Eligible patients were aged 18–80 years, had an International Prognostic Index (IPI) score of 2–5, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2. Tumor histological subtypes included diffuse large B-cell lymphoma (DLBCL) not otherwise specified (NOS), activated B-cell (ABC) type, germinal center B-cell (GCB) type, high-grade B-cell lymphoma (NOS, double-hit, triple-hit), and other subtypes of large B-cell lymphomas (Epstein-Barr virus-positive, T-cell/histiocyte-rich). Patients with known CNS lymphoma or peripheral neuropathy > grade 1 were excluded.
Patients were randomized in a 1:1 ratio to receive either Polivy® plus R-CHP or R-CHOP for six 21-day cycles, followed by two additional cycles of rituximab monotherapy in both groups. Patients were stratified by IPI score (2 vs. 3–5), presence or absence of bulky disease (≥ 7.5 cm), and geographic region.
Polivy® was administered intravenously at 1.8 mg/kg on day 1 of cycles 1–6. R-CHP or R-CHOP was administered on day 1 of cycles 1–6, followed by rituximab monotherapy on day 1 of cycles 7–8. Dosing in each treatment group followed these regimens:
- Polivy® + R-CHP group: Polivy® 1.8 mg/kg, rituximab 375 mg/m², cyclophosphamide 750 mg/m², doxorubicin 50 mg/m², and prednisone 100 mg/day orally on days 1–5 of each cycle.
- R-CHOP group: rituximab 375 mg/m², cyclophosphamide 750 mg/m², doxorubicin 50 mg/m², vincristine 1.4 mg/m², and prednisone 100 mg/day orally on days 1–5 of each cycle.
The two treatment groups were generally balanced with respect to baseline demographic and disease characteristics. The median patient age was 65 years (range: 19 to 80 years); 53.6% were of Caucasian race, 53.8% were male, 43.8% had bulky disease, 38% had an IPI score of 2, 62% had an IPI score of 3–5, and 88.7% had stage III or IV disease. The majority of patients (84.2%) had DLBCL (including NOS, ABC, and GCB subtypes). Cell-of-origin (COO) status was not reported for 211 patients. Among the population analyzed for COO (n = 668), gene expression profiling showed that 33.1% of patients had ABC-type DLBCL and 52.7% had GCB-type DLBCL.
The primary endpoint of the study was investigator-assessed progression-free survival. The median duration of follow-up was 28.2 months.
Table 1
Summary of efficacy in patients with previously untreated DLBCL in study GO39942 (POLARIX)
| Polivy® + R-CHP N = 440 |
R-CHOP N = 439 |
|
| Primary endpoint |
||
| Progression-free survival1,* |
||
| Number (%) of patients with events |
107 (24.3%) |
134 (30.5%) |
| HR (95% CI) |
0.73 [0.57, 0.95] |
|
| p-value3,** |
0.0177 |
|
| 2-year PFS rate (%) |
76.7 |
70.2 |
| [95% CI] |
[72.65, 80.76] |
[65.80, 74.61] |
| Key secondary endpoints |
||
| Event-free survival (EFSeff)1 |
||
| Number (%) of patients with event |
112 (25.5%) |
138 (31.4%) |
| HR [95% CI] |
0.75 [0.58, 0.96] |
|
| p-value3,** |
0.0244 |
|
| Overall response rate (ORR) at end of treatment2 |
||
| Patients who responded to treatment (%) (complete response, partial response) |
376 (85.5%) |
368 (83.8%) |
| Difference in response rate (%) [95% CI] |
1.63 [-3.32, 6.57] |
|
| Complete response rate (%) (CR)2,* |
||
| Patients who responded to treatment (%) |
343 (78%) |
325 (74%) |
| Difference in response rate (%) [95% CI] |
3.92 [-1.89, 9.70] |
|
| Partial response (%) (PR) |
33 (7.5%) |
43 (9.8%) |
| 95% CI by Clopper-Pearson method |
[5.22, 10.37] |
[7.18, 12.97] |
CI – confidence interval; HR – hazard ratio; PFS – progression-free survival; EFS – event-free survival based on efficacy: used to reflect event-free survival due to efficacy and defined as the time from the date of randomization to the earliest occurrence of any of the following events: disease progression/relapse, death from any cause, primary efficacy reason established by the investigator other than disease progression/relapse leading to initiation of any non-protocol lymphoma therapy (NALT), if a biopsy was obtained after completion of therapy and was positive for residual disease regardless of whether NALT was initiated or not; CMH: Cochran–Mantel–Haenszel.
1 Assessed by investigator.
2 Assessed by BICR (blinded independent centralized review).
3 Stratified log-rank test.
* According to Lugano 2014 response assessment criteria.
** Stratified by IPI (2 vs. 3–5), presence or absence of bulky disease, and geographic region.
At the time of the interim analysis, the key secondary endpoint, overall survival, was immature and did not differ statistically [stratified hazard ratio 0.94 (95% CI, 0.65, 1.37); p = 0.7524].
| Progression-free survival probability (%) |
|
|||||||
| Time, months |
||||||||
| Patients at risk |
||||||||
| R-CHOP |
439 |
389 |
330 |
296 |
220 |
78 |
3 |
NE |
| Polivy® + R-CHP |
440 |
404 |
353 |
327 |
246 |
78 |
NE |
NE |
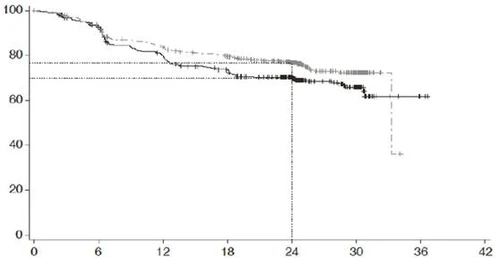
NE – not evaluable
Treatment groups: R-CHOP (n = 439); Polivy® + R-CHP; censored
P-value (log-rank test) = 0.0177; hazard ratio (95% CI) = 0.73 (0.57–0.95)
Fig. 1. Kaplan–Meier curve of progression-free survival (PFS) as assessed by investigator in study GO39942 (POLARIX).
Relapsed or refractory DLBCL
The efficacy of Polivy was evaluated in an international, multicenter, open-label study (GO29365), which included a randomized cohort of 80 patients with previously treated diffuse large B-cell lymphoma (DLBCL). Patients were randomized in a 1:1 ratio to receive either Polivy® + bendamustine and rituximab (BR) or BR alone for six 21-day cycles. Patients were stratified by duration of response to the last prior therapy: ≤12 months or >12 months.
Patients were not candidates for autologous hematopoietic stem cell transplantation (HSCT) and had relapsed or refractory disease after at least one prior systemic chemotherapy regimen. Patients with prior allogeneic HSCT, central nervous system lymphoma, transformed indolent lymphoma, stage 3b follicular lymphoma, significant cardiovascular or pulmonary disease, active infections, aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels >2.5 × upper limit of normal (ULN), total bilirubin ≥1.5 × ULN, creatinine >1.5 × ULN (or creatinine clearance <40 mL/min), unless elevated due to the lymphoma under study, were excluded from this trial.
Polivy was administered intravenously at a dose of 1.8 mg/kg on Day 2 of Cycle 1 and on Day 1 of Cycles 2–6. Bendamustine was administered at 90 mg/m²/day intravenously on Days 2 and 3 of Cycle 1 and on Days 1 and 2 of Cycles 2–6. Rituximab was administered at 375 mg/m² on Day 1 of Cycles 1–6.
Of the 80 patients randomized to receive Polivy + BR (n = 40) or BR alone (n = 40), the majority were of Caucasian race (71%) and male gender (66%). The median age was 69 years (range: 30–86 years). Sixty-four of 80 patients (80%) had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, and 14 of 80 patients (18%) had an ECOG performance status of 2. The majority of patients (98%) had DLBCL not otherwise specified. Overall, 48% of patients had activated B-cell-like DLBCL and 40% had germinal center B-cell-like DLBCL. The main reasons patients were not candidates for HSCT were age (40%), inadequate response to salvage therapy (26%), and failure of prior transplantation (20%). The median number of prior therapies was 2 (range: 1–7), with 29% (n = 23) having received one prior therapy, 25% (n = 20) two prior therapies, and 46% (n = 37) three or more prior therapies. All patients except one in the Polivy + BR arm of the Phase II randomized study had not previously received bendamustine. Eighty percent of patients had refractory disease. Among patients who received polatuzumab vedotin + BR and in whom CD3+ lymphocyte counts were assessed, the absolute CD3+ lymphocyte count was >200 cells/µL in 95%, 79%, and 83% of patients at assessments performed before treatment initiation (n = 134), at end of treatment (n = 72), and 6 months after treatment completion (n = 18), respectively.
The primary endpoint of this study was the complete response rate (CR) at the end of treatment (6–8 weeks after Day 1 of Cycle 6 or the last dose of investigational treatment), as assessed by an independent review committee using PET-CT.
Table 2
Summary of efficacy in patients with previously treated DLBCL (study GO29365)
| Parameter |
Polivy® + bendamustine + rituximab N = 40 |
Bendamustine + rituximab N = 40 |
| Median follow-up duration 22 months |
||
| Primary endpoint |
||
| Complete response rate* (by independent review committee assessment) at end of treatment** |
||
| Patients who responded to treatment (%) |
16 (40.0) |
7 (17.5) |
| Difference in response rate (%) [95% CI] |
22.5 [2.6, 40.2] |
|
| p-value (chi-square test, Cochran–Mantel–Haenszel***) |
0.0261 |
|
| Secondary and exploratory endpoints |
||
| Duration of response (by investigator assessment) |
||
| Number of patients included in analysis Number (%) of patients with event |
28 17 (60.7) |
13 11 (84.6) |
| Median duration of response (95% CI), months HR [95% CI] |
10.3 (5.6, NE) |
4.1 (2.6, 12.7) |
| 0.44 [0.20, 0.95] |
||
| p-value (log-rank test, stratified***) |
0.0321 |
|
| Overall response rate* (by investigator assessment) at end of treatment** |
||
| Patients who responded to treatment (%) (CR, PR) |
19 (47.5) |
7 (17.5) |
| Difference in response rate (%) [95% CI] |
30.0 [9.5, 47.4] |
|
| p-value (chi-square test, Cochran–Mantel–Haenszel***) |
0.0036 |
|
| Complete response (%) (CR) |
17 (42.5) |
6 (15.0) |
| Difference in response rate (%) [95% CI] |
27.5 [7.7, 44.7] |
|
| p-value (chi-square test, Cochran–Mantel–Haenszel***) |
0.0061 |
|
| Partial response (%) (PR) 95% CI by Clopper–Pearson method |
2 (5.0) [0.6, 16.9] |
1 (2.5) [0.06, 13.2] |
| Best overall response rate* (by investigator assessment) |
||
| Patients who responded to treatment (%) (CR, PR) |
28 (70.0) |
13 (32.5) |
| Difference in response rate (%) [95% CI] |
37.5 [15.6, 54.7] |
|
| Complete response (%) (CR) |
23 (57.5) |
8 (20.0) |
| 95% CI by Clopper–Pearson method |
[40.9, 73.0] |
[9.1, 35.7] |
| Partial response (%) (PR) 95% CI by Clopper–Pearson method |
5 (12.5) [4.2, 26.8] |
5 (12.5) [4.2, 26.8] |
CI – confidence interval; HR – hazard ratio; NE – not estimable; CR – complete response; PR – partial response.
* According to modified Lugano 2014 criteria: confirmation of complete response by bone marrow assessment using PET-CT is required. PR confirmed by positron emission tomography–computed tomography (PET-CT) must meet both PET-CT and computed tomography (CT) criteria.
** 6–8 weeks after Day 1 of Cycle 6 or last dose of investigational treatment.
*** Stratification by duration of response to prior therapy (≤ 12 months vs > 12 months).
Overall survival (OS) was an exploratory endpoint not controlled for type 1 error. Median OS in the Polivy® + BR group was 12.4 months (95% CI: 9.0, NE) versus 4.7 months (95% CI: 3.7, 8.3) in the control group. The unadjusted HR for OS was 0.42. Adjusting for the impact of baseline covariates, the HR for OS was adjusted to 0.59. Covariates included primary refractory status, number of prior lines of therapy, International Prognostic Index, and prior stem cell transplantation.
Progression-free survival (PFS) by investigator assessment was an exploratory endpoint not controlled for type 1 error. Median PFS in the Polivy® + BR group was 7.6 months (95% CI: 6.0, 17.0) versus 2.0 months (95% CI: 1.5, 3.7) in the control group. The unadjusted HR for PFS was 0.34.
Immunogenicity
Like all therapeutic proteins, polatuzumab vedotin has the potential to elicit an immune response. In studies GO39442 (POLARIX) and GO29365, anti-polatuzumab vedotin antibodies were detected in 1.4% (6/427) and 5.2% (12/233) of patients, respectively, with no neutralizing antibodies detected in any patient. Due to the limited number of patients in whom antibodies to polatuzumab vedotin were detected, it is not possible to draw conclusions regarding the potential impact of immunogenicity on efficacy or safety.
Immunogenicity assessment results are highly dependent on several factors, including the sensitivity and specificity of the assay method, analytical methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparisons of the incidence of anti-polatuzumab vedotin antibody detection with that of other medicinal products may lead to misinterpretation of the data.
Pharmacokinetics.
Exposure to MMAE conjugated to antibody (acMMAE) in plasma increased proportionally with dose over the polatuzumab vedotin dose range of 0.1 to 2.4 mg/kg. Following administration of the first dose of polatuzumab vedotin at 1.8 mg/kg, the mean maximum concentration (Cmax) of acMMAE was 803 (± 233) ng/mL, and the area under the concentration–time curve from zero to infinity (AUCinf) was 1860 (± 966) day•ng/mL. Based on population pharmacokinetic analysis, AUC of acMMAE in Cycle 3 increased by approximately 30% compared to Cycle 1 and reached more than 90% of AUC in Cycle 6. The terminal half-life of acMMAE in Cycle 6 was approximately 12 days (95% CI: 8.1–19.5 days). Based on population pharmacokinetic modeling, the predicted acMMAE concentration at the end of Cycle 6 is approximately 80% of the theoretical steady-state value. Exposure to unconjugated MMAE, the cytotoxic component of polatuzumab vedotin, increased proportionally with dose over the polatuzumab vedotin dose range of 0.1 to 2.4 mg/kg. Plasma concentrations of MMAE exhibited formation-rate-limited kinetics.
Following administration of the first dose of polatuzumab vedotin at 1.8 mg/kg, the Cmax of unconjugated MMAE was 6.82 (± 4.73) ng/mL, time to maximum plasma concentration was approximately 2.5 days, and terminal half-life was approximately 4 days. Plasma exposure to unconjugated MMAE accounted for < 3% of acMMAE exposure. Population pharmacokinetic analysis indicated a decrease in plasma exposure (AUC) of unconjugated MMAE with repeated administration every three weeks.
Based on population pharmacokinetic modeling and post-hoc analysis, predicted exposure to unconjugated MMAE in patients with body weight above 100 kg is expected to increase by no more than 55%.
Absorption
Polivy® is administered by intravenous infusion. Studies with other routes of administration have not been conducted.
Distribution
The population-estimated central volume of distribution of acMMAE was 3.15 L, approximately equivalent to plasma volume. In vitro, MMAE is moderately bound (71–77%) to human plasma proteins. In vitro, MMAE does not significantly penetrate human erythrocytes; the whole blood to plasma concentration ratio ranges from 0.79 to 0.98.
In vitro data indicate that MMAE is a substrate of P-gp, but does not inhibit P-gp at clinically relevant concentrations.
Metabolism
Polatuzumab vedotin is expected to undergo catabolism in patients, resulting in small peptides, amino acids, unconjugated MMAE, and metabolites related to unconjugated MMAE. Metabolite levels of MMAE were not detectable in human plasma.
In vitro studies indicate that MMAE is a substrate of CYP3A4/5, but does not induce major CYP enzymes. MMAE is a weak, time-dependent inhibitor of CYP3A4/5, but does not competitively inhibit CYP3A4/5 at clinically relevant concentrations.
MMAE does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6.
Elimination
Based on population pharmacokinetic analysis, the conjugate (acMMAE) is primarily eliminated via non-specific linear clearance at 0.9 L/day. In vivo studies in rats administered radiolabeled polatuzumab vedotin (with radiolabel on MMAE) demonstrated that the majority of radioactivity was excreted in feces and a smaller portion in urine.
Special patient populations
Paediatric population
Pharmacokinetic studies of polatuzumab vedotin in children (under 18 years of age) have not been conducted.
Older patients
Based on population pharmacokinetic analysis in patients aged 19–89 years, age does not affect the pharmacokinetics of acMMAE or unconjugated MMAE. Population pharmacokinetic analysis showed no clinically significant differences in the pharmacokinetics of acMMAE or unconjugated MMAE between patients aged < 65 years (n = 394) and those aged ≥ 65 years (n = 495).
Renal impairment
In patients with mild (creatinine clearance 60–89 mL/min, n = 361) or moderate (creatinine clearance 30–59 mL/min, n = 163) renal impairment, exposure to acMMAE and unconjugated MMAE was similar to that in patients with normal renal function (creatinine clearance ≥ 90 mL/min, n = 356), based on population pharmacokinetic analysis. Insufficient data are available to assess the impact of severe renal impairment (creatinine clearance 15–29 mL/min, n = 4) on pharmacokinetics. No data are available for patients with end-stage renal disease and/or patients on dialysis.
Hepatic impairment
Based on population pharmacokinetic analysis, in patients with mild hepatic impairment (AST or ALT > 1.0–2.5 × ULN or total bilirubin > 1.0–1.5 × ULN, n = 133), exposure to acMMAE is similar to that in patients with normal hepatic function (n = 737), while AUC of unconjugated MMAE is increased by up to 40%.
Insufficient data are available to assess the impact of moderate hepatic impairment (total bilirubin > 1.5–3 × ULN, n = 11) on pharmacokinetics. Limited data are available for patients with severe hepatic impairment or post-liver transplantation.
Clinical characteristics.
Indications.
The medicinal product Polivy® in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHP) is indicated for the treatment of adult patients with previously untreated diffuse large B-cell lymphoma (DLBCL).
The medicinal product Polivy® in combination with bendamustine and rituximab is indicated for the treatment of adult patients with relapsed/refractory diffuse large B-cell lymphoma who are not candidates for hematopoietic stem cell transplantation.
Contraindications.
Hypersensitivity to polatuzumab vedotin or to any excipient of the medicinal product.
Active severe infections (see section "Special warnings and precautions for use").
Interaction with other medicinal products and other forms of interaction.
Clinical studies on the interaction of polatuzumab vedotin with other medicinal products in humans have not been conducted.
Interaction when used concomitantly with medicinal products that are inhibitors, substrates, or inducers of CYP3A4, as well as when used concomitantly with medicinal products that are inhibitors of P-glycoprotein (P-gp)
Based on the results of a physiologically based pharmacokinetic simulation model for the release of MMAE from polatuzumab vedotin, strong inhibitors of CYP3A4 and P-gp (e.g., ketoconazole) may increase the area under the concentration–time curve (AUC) of unconjugated MMAE by 48%. Caution is recommended when administering CYP3A4 inhibitors concomitantly. Patients receiving strong CYP3A4 inhibitors (e.g., boceprevir, clarithromycin, cobicistat, indinavir, itraconazole, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telaprevir, telithromycin, voriconazole) should be closely monitored for signs of toxicity.
It is not expected that unconjugated MMAE will alter the AUC of concomitantly administered medicinal products that are substrates of CYP3A4 (such as midazolam).
Strong inducers of CYP3A4 (e.g., rifampicin, carbamazepine, phenobarbital, phenytoin, St. John’s wort [Hypericum perforatum]) may reduce the exposure of unconjugated MMAE.
Interaction when using medicinal products rituximab, bendamustine, cyclophosphamide, and doxorubicin in combination with polatuzumab vedotin
The pharmacokinetics of rituximab, bendamustine, cyclophosphamide, and doxorubicin are not affected by concomitant administration of polatuzumab vedotin. Concomitant administration of rituximab is associated with a 24% increase in the AUC of the MMAE-conjugated antibody (acMMAE) and a 37% decrease in the AUC of unconjugated MMAE in plasma, based on population pharmacokinetic analysis. The AUC values of acMMAE and unconjugated MMAE in plasma for Polivy® in combination with R-CHP are consistent with data obtained in other studies of Polivy®. Dose adjustment is not required.
Bendamustine does not affect the AUC of acMMAE and unconjugated MMAE in plasma.
Special precautions for use.
Traceability
To improve the traceability of biological medicinal products, the trade name and batch number of the administered product must be clearly and unambiguously recorded in the patient's medical documentation.
Myelosuppression
Severe and profound neutropenia, including febrile neutropenia, have been reported in patients receiving Polivy® as early as in the first treatment cycle. Consideration should be given to the use of granulocyte colony-stimulating factor (G-CSF), which was required in the clinical development program. Thrombocytopenia or anemia of grade 3 or 4 may also occur with Polivy®. A complete blood count should be monitored prior to administration of each dose of Polivy®. For patients with grade 3 or 4 neutropenia and/or thrombocytopenia, more frequent laboratory monitoring and/or dose delay or discontinuation of Polivy® should be considered (see section "Dosage and administration").
Peripheral neuropathy (PN)
PN has been reported in patients as early as the first cycle of Polivy® treatment, with risk increasing with subsequent doses. In patients with pre-existing PN, this condition may worsen. The PN reported during Polivy® treatment is predominantly sensory. However, motor and sensorimotor PN have also been reported. Patients should be monitored for the development of PN symptoms such as hypoesthesia, hyperesthesia, paresthesia, dysesthesia, neuropathic pain, burning sensations, muscle weakness, or gait disturbances. Patients who develop new or worsening PN may require dose delay, dose reduction, or discontinuation of Polivy® (see section "Dosage and administration").
Infections
Serious, life-threatening, or fatal infections, including opportunistic infections, have been reported in patients receiving Polivy® treatment. These include pneumonia (including that caused by Pneumocystis jirovecii and other fungal pneumonias), bacteremia, sepsis, herpes virus infections, and cytomegalovirus infection (see section "Adverse reactions"). Reactivation of latent infections has been reported. Patients should be closely monitored during treatment for signs of bacterial, fungal, or viral infections, and medical attention should be sought if such signs occur. Prophylaxis against infections should be considered during Polivy® treatment. Polivy® should not be administered in the presence of active severe infection. Treatment with Polivy® and any concomitant chemotherapy should be discontinued in patients who develop a serious infection.
Human immunodeficiency virus (HIV)
Polivy® has not been studied in HIV-positive patients. Information regarding concomitant use with CYP3A inhibitors is provided in the section "Interaction with other medicinal products and other forms of interaction".
Immunization
Live or live attenuated vaccines should not be administered concurrently with treatment. Patients who have recently received live vaccines were not included in clinical trials.
Progressive multifocal leukoencephalopathy (PML)
PML has been reported during Polivy® treatment (see section "Adverse reactions"). Patients should be closely monitored for the emergence of new or worsening neurological, cognitive, or behavioral changes that may indicate PML. Treatment with Polivy® and any concomitant chemotherapy should be discontinued upon suspicion of PML and permanently discontinued upon confirmation of diagnosis.
Tumour lysis syndrome (TLS)
Patients with high tumour burden and rapidly proliferating tumours may be at increased risk of TLS. Appropriate preventive measures should be taken prior to initiating Polivy® treatment, in accordance with local guidelines. Patients should be closely monitored for the development of TLS during Polivy® treatment.
Infusion-related reactions
Polivy® may cause infusion-related reactions, including severe reactions. Delayed infusion-related reactions have occurred up to 24 hours after Polivy® administration. Antihistamines and antipyretics should be administered prior to Polivy® administration, and patients should be closely monitored throughout the infusion. If infusion-related reactions occur, the infusion should be interrupted and appropriate medical treatment initiated (see section "Dosage and administration").
Embryo-fetal toxicity
Based on the mechanism of action and results from preclinical studies, Polivy® may cause harm to the foetus when administered to a pregnant woman. Pregnant women should be informed of the potential risk to the foetus.
Women of reproductive potential should be advised to use effective contraception during treatment with Polivy® and for at least 9 months after the last dose (see section "Pregnancy and breastfeeding"). Male patients with female partners of reproductive potential should be advised to use effective contraception during treatment with Polivy® and for at least 6 months after the last dose (see section "Pregnancy and breastfeeding").
Fertility
Preclinical studies of polatuzumab vedotin have shown toxic effects on the testes, which may result in impaired reproductive function and fertility in males. Therefore, males receiving Polivy® therapy are advised to consider sperm cryopreservation and storage prior to initiating treatment (see section "Pregnancy and breastfeeding").
Elderly patients
Among the 435 previously untreated diffuse large B-cell lymphoma (DLBCL) patients treated with Polivy® in combination with R-CHP in study GO39942, 227 (52.2%) were aged ≥65 years. The incidence of serious adverse reactions was 39.2% in patients aged ≥65 years and 28.4% in patients aged <65 years. A similar incidence of serious adverse reactions was observed in elderly patients in the R-CHOP treatment group.
Among the 151 previously treated DLBCL patients who received Polivy® in combination with bendamustine and rituximab in study GO29365, 103 (68%) were aged ≥65 years. The incidence of serious adverse reactions in patients aged ≥65 years (55%) was similar to that in patients aged <65 years (56%). Clinical trials of Polivy® did not include sufficient numbers of patients aged ≥65 years to determine whether they respond differently from younger patients.
Hepatotoxicity
Serious cases of hepatotoxicity, indicative of hepatocellular injury, including elevations in transaminases and/or bilirubin, have occurred in patients receiving Polivy® (see section "Adverse reactions"). Pre-existing liver disease, baseline elevated liver enzymes, and concomitant use of other medicinal products may increase the risk of hepatotoxicity. Liver enzymes and bilirubin levels should be monitored (see section "Dosage and administration").
Excipients
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, i.e. essentially "sodium-free".
Pregnancy and breastfeeding
Women of reproductive potential/contraception in men and women
Women
Women of reproductive potential should be advised to use effective contraception during treatment with polatuzumab vedotin and for at least 9 months after the last dose.
Men
Male patients with female partners of reproductive potential should be advised to use effective contraception during treatment with polatuzumab vedotin and for at least 6 months after the last dose.
Pregnancy
There are no data on the use of Polivy® in pregnant women. Animal studies have shown reproductive toxicity. Due to the mechanism of action and results from preclinical studies, polatuzumab vedotin may cause harm to the foetus when administered to a pregnant woman. Women of reproductive potential should be tested for pregnancy prior to initiating treatment. Polivy® is not recommended during pregnancy and in women of reproductive potential who are not using contraception, unless the potential benefit to the woman outweighs the potential risk to the foetus.
Breastfeeding
It is unknown whether polatuzumab vedotin or its metabolites are excreted in human milk. A risk to breastfed infants cannot be excluded. Women should discontinue breastfeeding during treatment with Polivy® and for at least 3 months after receiving the last dose.
Fertility
Preclinical studies of polatuzumab vedotin have shown toxic effects on the testes and may lead to impaired reproductive function and fertility in males.
Therefore, males receiving treatment with this medicinal product are advised to consider sperm cryopreservation and storage prior to treatment. Men receiving Polivy® should not father a child during treatment and for 6 months after the last dose.
Effect on ability to drive and use machines
Polivy® has a negligible influence on the ability to drive and use machines. Infusion-related reactions, peripheral neuropathy, fatigue, and dizziness may occur during Polivy® treatment (see sections "Special precautions for use" and "Adverse reactions").
Method of administration and dosage
Polivy® should be administered under the supervision of a physician experienced in the diagnosis and treatment of patients with oncological diseases.
Dosage
Diffuse large B-cell lymphoma (DLBCL)
Previously untreated patients
The recommended dose of Polivy® is 1.8 mg/kg as an intravenous infusion every 21 days in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (R-CHP) for 6 cycles. Polivy®, rituximab, cyclophosphamide, and doxorubicin can be administered in any order on day 1 after prednisone administration. Prednisone is administered on days 1–5 of each cycle. Cycles 7 and 8 consist of rituximab monotherapy.
Refer to the medical instructions for the use of chemotherapeutic agents used in combination with Polivy® in patients with previously untreated DLBCL.
Patients with relapsed or refractory disease
The recommended dose of Polivy® is 1.8 mg/kg as an intravenous infusion in combination with bendamustine and rituximab every 21 days for 6 cycles. Polivy®, bendamustine, and rituximab can be administered in any order on day 1 of each cycle. When used in combination with Polivy®, the recommended dose of bendamustine is 90 mg/m²/day on day 1 and day 2 of each cycle, and the recommended dose of rituximab is 375 mg/m² on day 1 of each cycle. Due to limited clinical experience, exceeding a dose of 240 mg per cycle is not recommended for patients receiving Polivy® at a dose of 1.8 mg/kg with a cumulative dose > 240 mg.
Previously untreated patients and patients with relapsed or refractory disease
If premedication has not been administered, premedication with antihistamines and antipyretic agents should be given prior to Polivy® infusion.
Delayed or missed administration
If a scheduled dose of Polivy® has not been administered, it should be given as soon as possible, and the administration schedule should be adjusted to maintain a 21-day interval between doses.
Dose modifications
The infusion rate of Polivy® should be reduced or the infusion interrupted if an infusion-related reaction occurs. Polivy® administration must be discontinued immediately and permanently if a life-threatening reaction occurs.
Dose modification options for Polivy® differ between patients with previously untreated DLBCL and patients with relapsed or refractory disease.
Information on dose modifications in the event of peripheral neuropathy (see section "Special precautions") is provided in Table 3.
Table 3
Dose modification of Polivy® in the event of peripheral neuropathy (PN)
| Indication |
Severity of PN on day 1 of any cycle |
Dose modification |
| Previously untreated DLBCL |
Grade 2a |
Sensory neuropathy:
Motor neuropathy:
If both sensory and motor neuropathy occur simultaneously, follow the most stringent recommendations outlined above. |
| Grade 3a |
Sensory neuropathy:
Motor neuropathy:
If both sensory and motor neuropathy occur simultaneously, follow the most stringent recommendations outlined above. |
|
| Grade 4 |
Discontinue Polivy®. |
|
| Relapsed/refractory DLBCL |
Grade 2–3 |
Discontinue Polivy® until improvement to grade ≤ 1. If recovery to grade ≤ 1 occurs by day 14 or earlier, resume Polivy® treatment at a permanently reduced dose of 1.4 mg/kg. If prior dose reduction to 1.4 mg/kg has already occurred, Polivy® treatment should be discontinued. If recovery to grade ≤ 1 does not occur by day 14 or earlier, Polivy® treatment should be discontinued. |
| Grade 4 |
Discontinue Polivy®. |
a R-CHP may be continued.
For information on dose modification in the event of myelosuppression (see section "Special Instructions"), refer to Table 4.
Table 4
Dose modification of Polivy®, chemotherapy, and rituximab in the event of myelosuppression
| Indications |
Severity of myelosuppression on day 1 of any cycle |
Dose modification |
| Previously untreated DLBCL |
Grade 3–4 neutropenia |
Discontinue all drugs until recovery of absolute neutrophil count (ANC)* to >1000/μL. If ANC recovers to >1000/μL on or before day 7, continue all drugs without any dose reduction. If ANC recovers to >1000/μL after day 7:
|
| Grade 3–4 thrombocytopenia |
Discontinue all drugs until platelet count recovers to >75,000/μL. If platelet count recovers to >75,000/μL on or before day 7, continue all drugs without any dose reduction. If platelet count recovers to >75,000/μL after day 7:
|
|
| Relapsed/refractory DLBCL |
Grade 3–4 neutropenia1 |
Discontinue all drugs until recovery of ANC to >1000/μL. If ANC recovers to >1000/μL on or before day 7, continue all drugs without any dose reduction. If ANC recovers to >1000/μL after day 7:
|
| Grade 3–4 thrombocytopenia1 |
Discontinue all drugs until platelet count recovers to >75,000/μL. If platelet count recovers to >75,000/μL on or before day 7, continue all drugs without any dose reduction. If platelet count recovers to >75,000/μL after day 7:
|
1If lymphoma is the main cause, the bendamustine dose may not need to be reduced.
For information on dose modification in the event of infusion reactions (see section "Special precautions for use"), refer to Table 5.
Table 5
Dose modification of Polivy® in the event of infusion reactions (IR)
| Indications |
IR severity on day 1 of any cycle |
Dose modification |
| Previously untreated and relapsed/refractory DLBCL |
Grade 1–3 IR |
Interrupt Polivy® infusion and provide supportive care. In case of first occurrence of grade 3 wheezing, bronchospasm, or generalized urticaria, Polivy® should be permanently discontinued. For recurrent wheezing or grade 2 urticaria, or for recurrence of any grade 3 symptoms, Polivy® should be permanently discontinued. Otherwise, after complete resolution of symptoms, the infusion may be resumed at 50% of the rate achieved prior to interruption. In the absence of infusion reaction symptoms, the infusion rate may be increased by 50 mg/hour every 30 minutes. In the next cycle, Polivy® infusion should be administered over 90 minutes. If no infusion reactions occur, subsequent infusions may be given over 30 minutes. Premedication should be administered in all cycles. |
| Grade 4 IR |
Immediately discontinue Polivy® infusion. Provide supportive care. Permanently discontinue Polivy®. |
Special patient populations
Elderly patients
Dose adjustment of Polivy® is not required in patients aged ≥ 65 years (see section "Pharmacokinetics").
Renal impairment
Dose adjustment of Polivy® is not required in patients with creatinine clearance ≥ 30 mL/min. The recommended dose in patients with creatinine clearance < 30 mL/min has not been established due to limited data.
Hepatic impairment
Polivy® should be avoided in patients with moderate or severe hepatic impairment (bilirubin level greater than 1.5 × upper limit of normal [ULN]).
No initial dose adjustment of Polivy® is required in patients with mild hepatic impairment (bilirubin level greater than ULN to less than or equal to 1.5 × ULN or AST level greater than ULN).
In the studied population with mild hepatic impairment (defined as AST or ALT level > 1.0–2.5 × ULN or total bilirubin level > 1.0–1.5 × ULN), an increase in exposure to unconjugated MMAE of up to 40% was observed, which was not considered clinically significant.
Children
Safety and efficacy of the drug in children (under 18 years of age) have not been established. Data are lacking.
Method of administration
Polivy® is intended for intravenous infusion.
The initial dose of Polivy® must be administered as an intravenous infusion over 90 minutes. Patients should be monitored for infusion reactions/hypersensitivity reactions during the infusion and for at least 90 minutes after administration of the initial dose.
If the previous infusion was well tolerated, the next dose of Polivy® may be administered over 30 minutes, and patients should be monitored during the infusion and for at least 30 minutes after completion of the infusion.
Polivy® must be reconstituted and diluted under aseptic conditions by a healthcare professional. The drug should be administered via intravenous infusion using a separate infusion set equipped with a sterile, pyrogen-free in-line or add-on filter with low protein binding (pore size 0.2 or 0.22 µm) and a catheter. Polivy® must not be administered by intravenous push or bolus injection.
Precautions to be taken prior to preparation or administration of the drug
Polivy® contains a cytotoxic component covalently linked to a monoclonal antibody. Appropriate handling and disposal procedures must be followed.
General precautions
Polivy® contains a cytotoxic component. The drug should be administered under the supervision of a physician experienced in the use of cytotoxic agents. Appropriate handling and disposal procedures for antineoplastic and cytotoxic medicinal products must be followed.
The reconstituted drug contains no preservatives and is intended for single use only. Appropriate aseptic techniques must be followed when handling this medicinal product.
Polivy® should be reconstituted with sterile water for injection and further diluted in an intravenous infusion bag containing 9 mg/mL (0.9%) sodium chloride injection solution, 4.5 mg/mL (0.45%) sodium chloride injection solution, or 5% glucose solution.
The reconstituted solution and the infusion solution must not be frozen or exposed to direct sunlight.
Reconstitution instructions
- Polivy®, 30 mg: Using a sterile syringe, slowly inject 1.8 mL of sterile water for injection into the vial containing 30 mg of Polivy® to obtain a single-dose solution containing 20 mg/mL of polatuzumab vedotin. Direct the stream onto the vial wall, not directly onto the lyophilized powder.
- Polivy®, 140 mg: Using a sterile syringe, slowly inject 7.2 mL of sterile water for injection into the vial containing 140 mg of Polivy® to obtain a single-dose solution containing 20 mg/mL of polatuzumab vedotin. Direct the stream onto the vial wall, not directly onto the lyophilized powder.
- Gently rotate the vial until complete dissolution. Do not shake.
- Inspect the reconstituted solution for the presence of particulate matter and discoloration. The reconstituted solution should be colorless to slightly brown, clear to slightly opalescent, and free of visible particles. Do not use reconstituted solution that is discolored, cloudy, or contains visible particles.
Dilution instructions
- Dilute Polivy® to a final concentration of 0.72–2.7 mg/mL in an intravenous infusion bag with a minimum volume of 50 mL containing 9 mg/mL sodium chloride injection solution, 4.5 mg/mL sodium chloride injection solution, or 5% glucose solution.
- Determine the volume of the reconstituted 20 mg/mL solution required to achieve the prescribed dose (see below):
| Total volume of Polivy® solution (ml) to be diluted |
= |
dose of Polivy® (mg/kg) × patient's body weight (kg) |
| concentration of reconstituted solution in vial (20 mg/ml) |
- Withdraw the required volume of reconstituted Polyvii® solution from the vial using a sterile syringe and dilute in an intravenous infusion bag. Discard any unused portion remaining in the vial.
- Gently mix the contents of the infusion bag by slowly inverting it. Do not shake.
- Inspect the infusion bag for particles and, if any are observed, discard the solution.
Transportation of the prepared infusion solution should be avoided, as stress from agitation may lead to aggregation. If transportation of the prepared solution is necessary, remove air from the infusion bag and limit transportation to 30 minutes at room temperature (9–25 °C) or 24 hours under refrigeration (2–8 °C). If air is removed, an infusion set with a metal needle connector must be used to ensure accurate dosing during infusion. The total storage time plus transportation time of the diluted solution must not exceed the storage period specified in Table 6.
Polyvii® should be administered through a separate infusion system equipped with a sterile, pyrogen-free in-line or add-on filter with minimal protein binding (pore size 0.2 or 0.22 µm) and a catheter.
Polyvii® is compatible with intravenous infusion bags made of polyvinyl chloride (PVC) or polyolefins such as polyethylene (PE) and polypropylene. Additionally, no incompatibility has been observed with infusion sets or devices made of PVC, PE, polyurethane, polybutadiene, acrylonitrile-butadiene-styrene, polycarbonate, polyetherurethane, fluorinated ethylene propylene, or polytetrafluoroethylene, or with filter membranes composed of polyethersulfone or polysulfone.
Shelf life
Reconstituted solution
From a microbiological standpoint, the reconstituted solution should be used immediately. If not used immediately, the storage duration and conditions of the reconstituted solution are the responsibility of the user. Storage under refrigeration (2–8 °C) should generally not exceed 24 hours, provided reconstitution was performed under controlled and validated aseptic conditions. Chemical and physical stability of the reconstituted solution has been demonstrated for up to 72 hours when stored under refrigeration (2–8 °C) and up to 24 hours at room temperature (9–25 °C).
Diluted solution
From a microbiological standpoint, the diluted infusion solution should be used immediately. If not used immediately, the storage duration and conditions of the diluted solution are the responsibility of the user. Storage under refrigeration (2–8 °C) should generally not exceed 24 hours, provided dilution was performed under controlled and validated aseptic conditions. Chemical and physical stability of the diluted infusion solution has been demonstrated for the periods specified in Table 6. The diluted solution must be discarded if the storage time exceeds the periods specified in Table 6.
Table 6
Periods during which chemical and physical stability of the prepared infusion solution has been demonstrated
| Solvent used for preparation of infusion solution |
Storage conditions of infusion solution1 |
| Sodium chloride 9 mg/mL (0.9%) |
Up to 72 hours in a refrigerator (2–8 °C) or up to 4 hours at room temperature (9–25 °C) |
| Sodium chloride 4.5 mg/mL (0.45%) |
Up to 72 hours in a refrigerator (2–8 °C) or up to 8 hours at room temperature (9–25 °C) |
| 5% glucose solution |
Up to 72 hours in a refrigerator (2–8 °C) or up to 8 hours at room temperature (9–25 °C) |
1To ensure the stability of the medicinal product, the specified storage periods should not be exceeded.
Polayvi® is intended for single use only.
Any unused medicinal product or waste material should be disposed of in accordance with local regulatory requirements.
Children
The safety and efficacy of the medicinal product in children (under 18 years of age) have not been established. Data are lacking.
Overdose.
There is no experience of overdose in humans from clinical trials. The highest dose studied to date is 2.4 mg/kg administered by intravenous infusion; this dose was associated with increased frequency and severity of peripheral neuropathy. In case of overdose, the infusion should be immediately discontinued and the patient closely monitored.
Adverse Reactions
Summary of Safety Profile
The safety of Polivy® was evaluated in 435 patients in the GO39942 (POLARIX) study. The following adverse reactions were observed in this section:
- During treatment and during the subsequent follow-up period in previously untreated patients with DLBCL who participated in the main clinical trial GO39942 (POLARIX) and received Polivy® plus R-CHP (n = 435) or R-CHOP (n = 438). In the Polivy® plus R-CHP treatment group, 91.7% received 6 cycles of Polivy® compared to 88.5% of patients who received 6 cycles of vincristine in the R-CHOP group.
In previously untreated patients with DLBCL receiving Polivy® plus R-CHP:
- The most frequently reported (≥ 30%) adverse reactions were peripheral neuropathy (52.9%), nausea (41.6%), neutropenia (38.4%), and diarrhea (30.8%).
- Serious adverse reactions were reported in 24.1% of patients receiving Polivy® plus R-CHP.
- The most common serious adverse reactions occurring in ≥ 5% of patients were febrile neutropenia (10.6%) and pneumonia (5.3%).
- Pneumonia (1.1%) was the only adverse reaction leading to premature discontinuation of treatment in > 1% of patients receiving Polivy® plus R-CH0P.
The safety of Polivy® was evaluated in 151 patients in the GO29365 study. The following adverse reactions were observed in this section:
- During treatment and the observation period in previously treated patients with DLBCL (n = 151) in the main clinical trial GO29365. This included patients from the lead-in phase (n = 6), randomized patients (n = 39), and patients from the expansion cohort (n = 106), who received Polivy® + BR, compared to randomized patients (n = 39) who received BR alone. Patients in the treatment groups received a mean of 5 treatment cycles, while patients randomized to the control group received a mean of 3 treatment cycles.
In previously treated patients with DLBCL receiving Polivy® plus bendamustine and rituximab:
- The most frequently reported adverse reactions (≥ 30%) (all grades) were neutropenia (45.7%), diarrhea (35.8%), nausea (33.1%), thrombocytopenia (32.5%), anemia (31.8%), and peripheral neuropathy (30.5%).
- Serious adverse reactions were reported in 41.7% of patients receiving Polivy® plus bendamustine and rituximab.
- The most common serious adverse reactions reported in ≥ 5% of patients were febrile neutropenia (10.6%), sepsis (9.9%), pneumonia (8.6%), and pyrexia (7.9%).
- Thrombocytopenia (7.9%) was the only adverse reaction leading to premature discontinuation of treatment in > 5% of patients receiving Polivy® plus bendamustine and rituximab.
The adverse reactions listed below occurred in 586 patients treated with Polivy® in clinical studies.
Adverse reactions are listed below by MedDRA system organ class and frequency category. The corresponding frequency category for each adverse reaction is based on the following: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000). Within each frequency group, adverse reactions are listed in order of decreasing severity.
Infections and infestations: very common – pneumoniaa, upper respiratory tract infection; common – sepsisa, herpesvirus infectiona, cytomegalovirus infection, urinary tract infectionc.
Blood and lymphatic system disorders: very common – febrile neutropenia, neutropenia, thrombocytopenia, anemia, leukopenia; common – lymphopenia, pancytopenia.
Metabolism and nutrition disorders: very common – hypokalemia, decreased appetite; common – hypocalcemia, hypoalbuminemia.
Nervous system disorders: very common – peripheral neuropathy; common – dizziness.
Eye disorders: uncommon – blurred visionb.
Respiratory, thoracic and mediastinal disorders: very common – cough; common – pneumonitis, dyspneac.
Gastrointestinal disorders: very common – diarrhea, nausea, constipation, vomiting, mucositisc, abdominal pain.
Skin and subcutaneous tissue disorders: very common – alopeciac; common – pruritus, skin infectionsc, rashc, dry skinc.
Musculoskeletal and connective tissue disorders: common – arthralgia, myalgiac.
General disorders and administration site conditions: very common – fatigue, pyrexia, asthenia; common – peripheral edemac, chills.
Investigations: very common – weight decreased; common – increased transaminases, increased lipaseb, hypophosphatemia.
Injury, poisoning and procedural complications: very common – infusion reaction.
a Adverse reactions associated with fatal outcomes.
b Adverse reactions observed only in relapsed or refractory DLBCL.
c Adverse reactions observed only in previously untreated DLBCL.
The listed adverse reactions were observed in both previously untreated and relapsed/refractory DLBCL, except as noted in the footnotes.
Adverse reactions occurring rarely and very rarely: none reported.
Description of Selected Adverse Reactions Observed in Clinical Studies
Myelosuppression
In the placebo-controlled study GO39942 (POLARIX), 0.5% of patients in the Polivy® plus R-CHP treatment group discontinued study treatment prematurely due to neutropenia. No patients discontinued study treatment in the R-CHOP group due to neutropenia. Thrombocytopenia led to premature discontinuation of study treatment in 0.2% of patients in the Polivy® plus R-CHP group compared to none in the R-CHOP group. No patient discontinued treatment due to anemia in either the Polivy® plus R-CHP or R-CHOP group.
In the open-label study GO29365, Polivy® was prematurely discontinued due to neutropenia in 4% of patients in the Polivy® plus bendamustine and rituximab treatment groups compared to 2.6% of patients in the bendamustine and rituximab group who discontinued due to neutropenia. Thrombocytopenia led to treatment discontinuation in 7.9% of patients in the Polivy® + BR groups and in 5.1% of patients in the BR group. No patient in either group (Polivy® + BR treatment groups or BR only group) discontinued treatment due to anemia. In the Polivy® plus bendamustine and rituximab treatment groups, grade 3 or higher neutropenia, thrombocytopenia, and anemia were reported in 40.4%, 25.8%, and 12.6% of patients, respectively.
Peripheral Neuropathy (PN)
In the placebo-controlled study GO39942 (POLARIX), PN of grades 1, 2, and 3 was reported in 39.1%, 12.2%, and 1.6% of patients, respectively, in the Polivy® plus R-CHP group. In the R-CHOP group, PN of grades 1, 2, and 3 was reported in 37.2%, 15.5%, and 1.1% of patients, respectively. No grade 4–5 PN events were reported in either the Polivy® plus R-CHP or R-CHOP group. In the Polivy® plus R-CHP group, 0.7% of patients discontinued study treatment prematurely due to PN compared to 2.3% in the R-CHOP group. In the Polivy® plus R-CHP group, 4.6% of patients had dose reductions due to PN compared to 8.2% in the R-CHOP group. The median time to first PN event was 2.27 months in the Polivy® plus R-CHP group compared to 1.87 months in the R-CHOP group. At the time of clinical data cutoff, PN had resolved in 57.8% of patients in the Polivy® plus R-CHP group compared to 66.9% in the R-CHOP group. The median time to resolution of peripheral neuropathy was 4.04 months in the Polivy® plus R-CHP group compared to 4.6 months in the R-CHOP group.
In the open-label study GO29365, PN of grades 1 and 2 was reported in 15.9% and 12.6% of patients, respectively, in the Polivy® plus bendamustine and rituximab treatment groups. In the bendamustine and rituximab treatment group, PN of grades 1 and 2 was reported in 2.6% and 5.1% of patients, respectively. One case of grade 3 PN was reported in the Polivy® + BR groups and none in the BR group. No grade 4–5 PN events were reported in either group (Polivy® + BR treatment groups or BR only group). Polivy® treatment was discontinued prematurely due to PN in 2.6% of patients, and the dose of Polivy® was reduced due to PN in 2% of patients. No patients in the BR group discontinued treatment or had dose reductions due to PN. In the Polivy® + BR groups, the median time to first PN event was 1.6 months, and PN resolved in 39.1% of patients with PN.
Infections
In the placebo-controlled study GO39942 (POLARIX), infections including pneumonia and other types of infections were reported in 49.7% of patients in the Polivy® plus R-CHP group and in 42.7% of patients in the R-CHOP group. Grade 3–4 infections occurred in 14% of patients in the Polivy® plus R-CHP group and in 11.2% of patients in the R-CHOP group. Serious infections were reported in 14% of patients in the Polivy® plus R-CHP group, and fatal infections occurred in 1.1% of patients. In the R-CHOP group, serious infections were reported in 10.3% of patients, and fatal infections occurred in 1.4% of patients. Treatment was discontinued due to infection in 7 patients (1.6%) in the Polivy® plus R-CHP group compared to 10 patients (2.3%) in the R-CHOP group.
In the open-label study GO29365, infections including pneumonia and other types of infections were reported in 48.3% of patients in the Polivy® + BR groups and in 51.3% of patients in the BR group. Serious infections were reported in 27.2% of patients in the Polivy® + BR groups, and fatal infections occurred in 6.6% of patients. In the BR group, serious infections were reported in 30.8% of patients, and fatal infections occurred in 10.3% of patients. Treatment was discontinued prematurely due to infection in 4 patients (2.6%) in the Polivy® + BR groups compared to 2 patients (5.1%) in the BR group.
Progressive Multifocal Leukoencephalopathy (PML)
In the placebo-controlled study GO39942 (POLARIX), no cases of progressive multifocal leukoencephalopathy were reported.
In the open-label study GO29365, one fatal case of PML was observed in a patient receiving Polivy® + bendamustine and obinutuzumab. This patient had previously received three lines of therapy, including anti-CD20 antibodies.
Hepatotoxicity
In the placebo-controlled study GO39942 (POLARIX), hepatotoxicity was reported in 10.6% of patients in the Polivy® plus R-CHP group and in 7.3% of patients in the R-CHOP group. In the Polivy® plus R-CHP group, most events were grade 1–2 (8.7%); grade 3 events were reported in 1.8% of patients. No grade 4 or 5 events were observed. One patient (0.2%) experienced a serious hepatotoxicity event, which was reversible.
In another study, two cases of serious hepatotoxicity (hepatocellular injury and hepatic steatosis) were reported, both of which were reversible.
Gastrointestinal Toxicity
In the placebo-controlled study GO39942 (POLARIX), gastrointestinal toxicity events were reported in 76.1% of patients in the Polivy® plus R-CHP group compared to 71.9% of patients in the R-CHOP group. Most events were of grade 1–2 severity, and events ≥ grade 3 were reported in 9.7% of patients in the Polivy® plus R-CHP group compared to 8.2% in the R-CHOP group. The most common gastrointestinal toxicity events were nausea and diarrhea.
In the open-label study GO29365, gastrointestinal toxicity events were reported in 72.8% of patients in the Polivy® + BR group compared to 66.7% of patients in the BR group. Most events were of grade 1–2 severity. Grade 3–4 events were reported in 16.5% of patients in the Polivy® + BR group compared to 12.9% in the BR group. The most common gastrointestinal toxicity events were diarrhea and nausea.
Shelf life.
30 months.
Storage conditions.
Store at 2 to 8 °C in the original packaging to protect from light. Do not freeze. Keep out of the reach of children.
Incompatibilities.
This medicinal product must not be mixed with or diluted by other medicinal products except as specified in the section “Precautions to be taken before reconstitution or administration of the medicinal product”.
Packaging.
30 mg in a 6 mL vial made of colorless glass (borosilicate glass, Class I), stoppered with a butyl rubber stopper (fluoroelastomer laminated) and sealed with an aluminum cap with a plastic “flip-off” disc. One vial in a cardboard box.
140 mg in a 20 mL vial made of colorless glass (borosilicate glass, Class I), stoppered with a butyl rubber stopper (fluoroelastomer laminated) and sealed with an aluminum cap with a plastic “flip-off” disc. One vial in a cardboard box.
Prescription status.
Prescription only.
Manufacturer.
F. Hoffmann-La Roche Ltd
Manufacturer's address and location of activity.
Wurmisweg, 4303 Kaiseraugst, Switzerland