Ilaris
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL FARMACO ILARIS (ILARIS®)
Composizione:
principio attivo: canakinumab;
1 flaconcino contiene 150 mg di canakinumab;
eccipienti: mannitolo, L-istidina, cloridrato di L-istidina monoidrato, polisorbato 80, acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile.
Principali caratteristiche fisico-chimiche: soluzione da incolore a giallo pallido.
Gruppo farmacoterapeutico. Agenti antineoplastici e immunomodulatori. Immunosoppressori. Inibitori dell'interleuchina. Codice ATC L04A C08.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Canakinumab è un anticorpo monoclonale completamente umano di isotype IgG1/κ diretto contro l'interleuchina-1 beta (IL-1 beta). Canakinumab si lega specificamente con alta affinità all'IL-1 beta umano e neutralizza l'attività biologica dell'IL-1 beta umano, bloccandone l'interazione con i recettori dell'IL-1, prevenendo così l'attivazione del gene indotto dall'IL-1 beta e la produzione di mediatori infiammatori.
Effetti farmacodinamici
Sindromi periodiche associate alla criopirina (CAP-sindrome), sindrome periodica associata al recettore del fattore di necrosi tumorale (TRAP-sindrome), sindrome da iperimmunoglobulinemia D (HID-sindrome)/deficit di mevalonato chinasi (MKD), febbre mediterranea familiare (FMF)
Negli studi clinici condotti su pazienti affetti da sindromi periodiche associate alla criopirina (CAP-sindrome), sindrome periodica associata al recettore del fattore di necrosi tumorale (TRAP-sindrome), sindrome da iperimmunoglobulinemia D (HID-sindrome)/deficit di mevalonato chinasi (MKD), febbre mediterranea familiare (FMF), caratterizzate da un rilascio incontrollato di IL-1 beta, si è osservata una rapida risposta alla terapia con canakinumab: parametri di laboratorio come livelli elevati di proteina C-reattiva (PCR), amiloyde sierico A (SAA), neutrofili, piastrine e leucocitosi si normalizzano rapidamente.
Malattia di Still (compresa la malattia di Still dell'adulto (AOSD) e l'artrite idiopatica giovanile sistemica (SJIA))
La malattia di Still dell'adulto e l'artrite idiopatica giovanile sistemica sono gravi malattie autoinfiammatorie innate causate da citochine pro-infiammatorie, tra cui l'IL-1 beta svolge un ruolo chiave.
I principali segni dell'artrite idiopatica giovanile sistemica comprendono febbre, eruzione cutanea, epatosplenomegalia, linfadenopatia, polisierosite e artrite.
Il trattamento con canakinumab determina una rapida e sostenuta riduzione sia delle manifestazioni articolari che di quelle sistemiche dell'artrite idiopatica giovanile sistemica, con una significativa riduzione del numero di articolazioni infiammate, attenuazione della febbre e riduzione della reattività acuta in gran parte dei pazienti.
Artrite gotta
L'artrite gotta è causata dalla presenza di cristalli di urato (urato monosodico monoidrato) nelle articolazioni e nei tessuti circostanti, che attivano i macrofagi residenti alla produzione di IL-1 beta attraverso il complesso infiammasoma NALP3. L'attivazione dei macrofagi e il conseguente aumento del rilascio di IL-1 beta portano a una reazione infiammatoria acuta e dolorosa. Altri attivatori del sistema immunitario innato, come agonisti endogeni dei recettori simili ai Toll (TLR), possono contribuire all'attivazione trascrizionale del gene dell'IL-1 beta, innescando l'attacco di artrite gotta. Dopo il trattamento con canakinumab, i marcatori infiammatori come la proteina C-reattiva (PCR) e l'amiloyde sierico A (SAA), insieme ai segni di infiammazione acuta (ad esempio dolore, gonfiore, arrossamento) nell'articolazione colpita, scompaiono rapidamente.
Efficacia clinica e sicurezza
Sindromi periodiche associate alla criopirina (CAP-sindrome)
L'efficacia e la sicurezza del medicinale Ilaris sono state dimostrate in pazienti con diversi gradi di gravità della malattia e diversi fenotipi (inclusi i casi gravi di sindrome autoinfiammatoria familiare da freddo/orticaria familiare da freddo, sindrome di Muckle-Wells e malattia infiammatoria multisistemica neonatale/sindrome neurologica cutaneo-articolare cronica infantile). Solo pazienti con mutazione NLRP3 confermata sono stati inclusi nello studio principale.
Nella fase I/II dello studio, Ilaris ha mostrato un rapido inizio d'azione: i sintomi sono scomparsi o si sono notevolmente attenuati entro un giorno. I parametri di laboratorio, come i livelli di PCR e SAA, nonché i livelli di neutrofili e piastrine, si sono normalizzati rapidamente entro pochi giorni dall'amministrazione di Ilaris.
Lo studio principale di 48 settimane, multicentrico, è stato suddiviso in tre parti: un periodo aperto di 8 settimane (parte I), un periodo randomizzato, in doppio cieco, controllato con placebo di 24 settimane di sospensione (parte II) e un periodo aperto di 16 settimane (parte III). L'obiettivo dello studio era valutare l'efficacia, la sicurezza e la tollerabilità di Ilaris (150 mg oppure 2 mg/kg ogni 8 settimane) in pazienti affetti da sindromi periodiche associate alla criopirina.
- Parte I: una risposta clinica completa e una risposta dei biomarcatori a Ilaris (definita come valutazione globale del medico sulla malattia autoinfiammatoria e sulla malattia cutanea ≤ al minimo e valori di PCR o SAA < 10 mg/l) è stata osservata nel 97% dei pazienti, manifestandosi entro 7 giorni dall'inizio del trattamento. È stato osservato un notevole miglioramento nella valutazione clinica del medico sull'attività dell'infiammazione: valutazione globale dell'attività della malattia autoinfiammatoria, valutazione della malattia cutanea (orticaria, eruzione cutanea), dolore articolare, mialgia, cefalea/migrazione, congiuntivite, affaticamento/malessere, valutazione di altri sintomi correlati, valutazione dei sintomi da parte del paziente.
- Parte II: nel periodo di sospensione dello studio principale, il punto finale primario è stato definito come la frazione di pazienti con recidiva della malattia/riacutizzazione: 0% di pazienti con recidiva nel gruppo trattato con Ilaris rispetto all'81% di pazienti randomizzati al gruppo placebo.
- Parte III: i pazienti che avevano ricevuto placebo nella parte II e nei quali si era verificata una recidiva, e che mantenevano risposta clinica e sierologica dopo l'ingresso nella parte aperta di estensione dello studio con Ilaris.
Tabella 1. Efficacia nella fase III dello studio principale controllato con placebo durante il periodo di sospensione (parte II)
| Indice |
Canakinumab N = 15 n (%) |
Placebo N = 16 n (%) |
p-valore |
| Endpoint primario (ricaduta) Numero di pazienti con recidiva della malattia nella parte II |
0 (0 %) |
13 (81 %) |
< 0,001 |
| Marker di infiammazione* Proteina C reattiva, mg/l Amiloidina sierica A, mg/l |
1,10 (0,40) 2,27 (-0,20) |
19,93 (10,50) 71,09 (14,35) |
< 0,001 0,002 |
| *Variazione media rispetto all'inizio della parte II |
|||
Sono stati condotti due studi di fase III aperti e non controllati a lungo termine. In uno di questi è stata valutata la sicurezza, la tollerabilità e l'efficacia di canakinumab in pazienti con sindromi periodiche associate alla criopirina. La durata complessiva del trattamento è stata da 6 mesi fino a 2 anni. L'altro studio era uno studio aperto di canakinumab per valutare efficacia e sicurezza in pazienti giapponesi con sindromi periodiche associate alla criopirina, della durata di 24 settimane con una fase di estensione fino a 48 settimane. L'obiettivo primario era valutare la percentuale di pazienti senza recidiva alla 24^ settimana, inclusi i pazienti a cui era stata aumentata la dose.
Nell'analisi combinata di efficacia di questi due studi, il 65,6% dei pazienti precedentemente non trattati con canakinumab ha raggiunto una risposta completa alla dose di 150 mg o 2 mg/kg, mentre l'85,2% dei pazienti ha raggiunto una remissione completa a qualsiasi dose. Tra i pazienti trattati con 600 mg o 8 mg/kg (o anche dosi superiori), il 43,8% ha raggiunto una risposta completa. La percentuale di pazienti di età compresa tra 2 e 4 anni che hanno raggiunto una risposta completa (57,1%) è risultata inferiore rispetto ai pazienti pediatrici più grandi e agli adulti. Tra i pazienti che hanno raggiunto una risposta completa, l'89,3% ha mantenuto la risposta senza recidive.
L'esperienza clinica in singoli pazienti che hanno raggiunto una risposta completa dopo un aumento della dose a 600 mg (8 mg/kg) ogni 8 settimane indica che una dose più elevata può essere utile per i pazienti che non hanno raggiunto una risposta completa o che non mantengono una risposta completa alle dosi raccomandate (150 mg o 2 mg/kg per pazienti con peso corporeo ≥ 15 kg e ≤ 40 kg). La dose aumentata è stata somministrata più frequentemente a pazienti di età compresa tra 2 e 4 anni e a pazienti con sintomi di NOMID/CINCA rispetto a FCAS o MWS.
È stato condotto uno studio osservazionale di registro della durata di 6 anni per ottenere dati sulla sicurezza e l'efficacia a lungo termine del trattamento con canakinumab in pazienti pediatrici e adulti con sindromi periodiche associate alla criopirina nella pratica clinica abituale. Lo studio ha incluso 243 pazienti con sindromi periodiche associate alla criopirina (tra cui 85 pazienti di età inferiore a 18 anni). L'attività della malattia è stata valutata come assente o lieve/moderata in oltre il 90% dei pazienti in tutti i momenti successivi al periodo iniziale dello studio, e i marcatori sierologici medi dell'infiammazione (PCR e SAA) sono risultati normali (< 10 mg/litro) in tutti i momenti successivi al basale. Sebbene circa il 22% dei pazienti trattati con canakinumab abbia richiesto un aggiustamento della dose, solo una piccola percentuale di pazienti (1,2%) ha interrotto il trattamento con canakinumab per mancanza di effetto terapeutico.
Popolazione pediatrica
Negli studi sulle sindromi periodiche associate alla criopirina hanno partecipato complessivamente 80 pazienti pediatrici di età compresa tra 2 e 17 anni (circa la metà dei quali ha ricevuto una dose basata sul peso corporeo). Nel complesso, nei pazienti pediatrici non sono state osservate differenze clinicamente significative in termini di efficacia, sicurezza e profilo di tollerabilità di Ilaris rispetto alla popolazione generale. La maggior parte dei pazienti pediatrici ha mostrato un miglioramento dei sintomi clinici e dei marcatori oggettivi di infiammazione (ad esempio SAA e PCR).
L'efficacia, la sicurezza e la tollerabilità del medicinale Ilaris sono state valutate in uno studio aperto della durata di 56 settimane in pazienti di età pediatrica (≤ 4 anni) con sindromi periodiche associate alla criopirina. Sono stati valutati diciassette pazienti (inclusi 6 pazienti di età inferiore a 2 anni), con dosi iniziali calcolate in base al peso corporeo, pari a 2–8 mg/kg. Nello studio è stata inoltre valutata l'influenza di canakinumab sulla formazione di anticorpi contro i vaccini pediatrici standard. Non sono state osservate differenze in termini di sicurezza o efficacia nei pazienti di età inferiore a 2 anni rispetto ai pazienti di età pari o superiore a 2 anni. Tutti i pazienti che hanno ricevuto vaccini pediatrici standard inattivati (N = 7) hanno mostrato livelli protettivi di anticorpi.
Sindrome periodica associata al recettore del fattore di necrosi tumorale (TRAPS), sindrome da iper-IgD (HIDS) / deficit di mevalonato chinasi (MKD), febbre familiare mediterranea (FMF)
L'efficacia e la sicurezza di canakinumab nel trattamento della sindrome periodica associata al recettore del fattore di necrosi tumorale (TRAPS), della sindrome da iper-IgD (HIDS) / deficit di mevalonato chinasi (MKD) e della febbre familiare mediterranea (FMF) sono state dimostrate in uno studio chiave di fase III, composto da quattro parti (N2301) e comprendente tre coorti distinte di malattia.
- Parte I: I partecipanti di ogni gruppo di pazienti di età ≥ 2 anni sono stati inclusi in un periodo di screening di 12 settimane, durante il quale è stata valutata la presenza di un'esacerbazione della malattia.
- Parte II: I pazienti con esacerbazione della malattia sono stati randomizzati in un periodo di trattamento in doppio cieco controllato con placebo della durata di 16 settimane, durante il quale hanno ricevuto 150 mg di canakinumab (2 mg/kg per pazienti con peso corporeo ≤ 40 kg) sottocute o placebo ogni 4 settimane. Ai pazienti di età compresa tra 28 giorni e < 2 anni è stato consentito partecipare direttamente alla parte aperta della parte II come pazienti non randomizzati (sono stati esclusi dall'analisi primaria di efficacia).
- Parte III: I pazienti che hanno completato il trattamento di 16 settimane e sono stati classificati come responder (rispondenti al trattamento) sono stati nuovamente randomizzati in un periodo in doppio cieco di sospensione della durata di 24 settimane, durante il quale hanno ricevuto 150 mg di canakinumab (2 mg/kg per pazienti con peso corporeo ≤ 40 kg) sottocute o placebo ogni 8 settimane.
- Parte IV: Tutti i pazienti della parte III che hanno ricevuto canakinumab hanno avuto diritto a partecipare a un periodo di trattamento aperto di estensione della durata di 72 settimane.
In totale sono stati arruolati 185 pazienti di età compresa tra 28 giorni e 181 pazienti di età ≥ 2 anni sono stati randomizzati nella parte II dello studio.
Il punto finale primario di efficacia del periodo di trattamento randomizzato (parte II) è stata la percentuale di responder in ciascun gruppo che ha mostrato una riduzione dell'indice di esacerbazione della malattia al giorno 15 e non ha avuto nuove esacerbazioni durante il resto del periodo di trattamento di 16 settimane (definita come risposta completa). Una riduzione dell'indice di esacerbazione della malattia è stata definita come un punteggio di valutazione globale del medico (PGA) per l'attività della malattia < 2 ("malattia minima o assente") e un livello di PCR entro i limiti normali (≤ 10 mg/l) o una riduzione ≥ 70% rispetto al basale. Una nuova esacerbazione è stata definita come un punteggio PGA ≥ 2 ("malattia lieve, moderata o grave") e un livello di PCR ≥ 30 mg/l. I punti finali secondari, basati sui risultati alla settimana 16 (fine della parte II), includevano la percentuale di pazienti che hanno raggiunto un PGA < 2, la percentuale di pazienti con remissione sierologica (definita come PCR ≤ 10 mg/l) e la percentuale di pazienti con normalizzazione del livello di SAA (definita come SAA ≤ 10 mg/l).
Per quanto riguarda il punto finale primario di efficacia, canakinumab si è dimostrato superiore al placebo in tutti e tre i gruppi di malattia. Canakinumab ha mostrato anche un'efficacia superiore al placebo nei punti finali secondari PGA < 2 e PCR ≤ 10 mg/l in tutti e tre i gruppi. Una maggiore percentuale di pazienti ha normalizzato il livello di SAA (≤ 10 mg/l) alla settimana 16 con canakinumab rispetto al placebo in tutti e tre i gruppi, con una differenza statisticamente significativa osservata nei pazienti con TRAPS.
Tabella 2. Efficacia nella fase III dello studio chiave randomizzato controllato con placebo durante il periodo di trattamento (parte II)
| Indice |
Canakinumab, n/N (%) |
Placebo, n/N (%) |
p-valore |
| Endpoint primario (riacutizzazione) Numero di pazienti che al giorno 15 presentavano un indice di riacutizzazione della malattia e che non avevano sperimentato una nuova riacutizzazione durante il restante periodo di trattamento di 16 settimane |
|||
| FMF Sindrome HID/MKD Sindrome TRAP |
19/31 (61,29) 13/37 (35,14) 10/22 (45,45) |
2/32 (6,25) 2/35 (5,71) 2/24 (8,33) |
< 0,0001* 0,0020* 0,0050* |
| Endpoint secondario (malattia e marcatori dell'infiammazione) |
|||
| Valutazione globale del medico < 2 FMF Sindrome HID/MKD Sindrome TRAP |
20/31 (64,52) 17/37 (45,95) 10/22 (45,45) |
3/32 (9,38) 2/35 (5,71) 1/24 (4,17) |
<0,0001** 0,0006** 0,0028** |
| Proteina C reattiva ≤ 10 mg/l FMF Sindrome HID/MKD Sindrome TRAP |
21/31 (67,74) 15/37 (40,54) 8/22 (36,36) |
2/32 (6,25) 2/35 (5,71) 2/24 (8,33) |
<0,0001** 0,0010** 0,0149** |
| Serum Amyloid A ≤ 10 mg/l FMF Sindrome HID/MKD Sindrome TRAP |
8/31 (25,81) 5/37 (13,51) 6/22 (27,27) |
0/32 (0,00) 1/35 (2,86) 0/24 (0,00) |
0,0286 0,0778 0,0235* |
| n – numero di rispondenti; N – numero di pazienti valutati. *Indica significatività statistica (unilaterale) al livello 0,025 basata sul test esatto di Fisher. **Indica significatività statistica (unilaterale) al livello 0,025 basata su un modello di regressione logistica con il gruppo di trattamento e il valore basale di PGA, PCR o SAA rispettivamente come variabili esplicative per ciascun gruppo. |
|||
Titrazione della dose
Nella parte II dello studio, i pazienti trattati con canakinumab che presentavano un’attività persistente della malattia hanno ricevuto una dose aggiuntiva di 150 mg (o 2 mg/kg per i pazienti con peso corporeo ≤ 40 kg) entro il primo mese. Questa dose aggiuntiva può essere somministrata già a partire dal 7° giorno dopo la prima dose di trattamento. Tutti i pazienti sottoposti a titolazione sono rimasti nel gruppo con dose aumentata di 300 mg (o 4 mg/kg per i pazienti con peso corporeo ≤ 40 kg) ogni 4 settimane.
Durante l’analisi sperimentale del punto finale primario, si è osservato che nei pazienti con risposta inadeguata dopo la prima dose, l’aumento della dose entro il primo mese fino a 300 mg (o 4 mg/kg) ogni 4 settimane ha ulteriormente migliorato il controllo delle riacutizzazioni, la riduzione dell’attività della malattia e la normalizzazione dei livelli di PCR e SAA.
Pazienti pediatrici
Due pazienti non randomizzati con sindrome HID/MKD di età > 28 giorni – < 2 anni sono stati inclusi nello studio e hanno ricevuto canakinumab. Un paziente ha ottenuto la soppressione dell’indice di riacutizzazione entro il 15° giorno dopo una singola dose di canakinumab da 2 mg/kg, ma ha interrotto il trattamento dopo questa prima dose a causa di eventi avversi gravi (pancitopenia e insufficienza epatica). Prima dell’inclusione nello studio, questo paziente aveva anamnesi di purpura trombocitopenica immune e uno stato medico attivo con alterazioni della funzionalità epatica. Il secondo paziente ha ricevuto una dose iniziale di canakinumab da 2 mg/kg e una dose aggiuntiva da 2 mg/kg alla 3ª settimana, e alla 5ª settimana è stato titolato alla dose di 4 mg/kg, somministrata ogni 4 settimane fino alla fine della parte II dello studio. La remissione della riacutizzazione è stata raggiunta entro la 5ª settimana e il paziente non ha presentato nuove riacutizzazioni fino alla fine della parte II dello studio (16ª settimana).
Malattia di Still (malattia di Still dell'adulto (AOSD) e artrite idiopatica giovanile sistemica (SJIA))
Artrite idiopatica giovanile sistemica
L’efficacia di Ilaris nel trattamento dell’artrite idiopatica giovanile sistemica attiva è stata valutata in due studi principali (G2305 e G2301). I pazienti inclusi nello studio avevano un’età compresa tra 2 e 20 anni (età media 8,5 anni e durata media della malattia di 3,5 anni all’inizio dello studio) e presentavano una malattia attiva definita dalla presenza di almeno 2 articolazioni con artrite attiva, febbre e aumento della PCR.
Studio G2305
G2305 è uno studio randomizzato, in doppio cieco, controllato con placebo, della durata di 4 settimane. In questo studio è stata valutata l’efficacia a breve termine di Ilaris in 84 pazienti, randomizzati a ricevere una singola dose di 4 mg/kg (fino a un massimo di 300 mg) di Ilaris o placebo. L’obiettivo primario era determinare la percentuale di pazienti che al giorno 15 raggiungevano un miglioramento minimo del 30% secondo i criteri pediatrici dell’American College of Rheumatology (ACR). Questi criteri sono stati adattati per consentire l’inclusione di pazienti senza febbre. Il trattamento con Ilaris ha migliorato tutti gli indicatori ACR rispetto al placebo sia al giorno 15 che al giorno 29 (tabella 3).
Tabella 3. Indici pediatrici ACR e stato della malattia al giorno 15 e al giorno 29
| Indicatore |
Giorno 15 |
Giorno 29 |
||
| Ilaris N=43 |
Placebo N=41 |
Ilaris N=43 |
Placebo N=41 |
|
| ACR30 |
84 % |
10 % |
81 % |
10 % |
| ACR50 |
67 % |
5 % |
79 % |
5 % |
| ACR70 |
61 % |
2 % |
67 % |
2 % |
| ACR90 |
42 % |
0 % |
47 % |
2 % |
| ACR100 |
33 % |
0 % |
33 % |
2 % |
| Malattia inattiva |
33 % |
0 % |
30 % |
0 % |
| La differenza nel trattamento per tutti gli indicatori ACR è stata significativa (p ≤ 0,0001) |
||||
I risultati degli indici pediatrici adattati ACR, che includevano componenti sistemici e articolari, erano coerenti con i risultati complessivi degli indici ACR. Al giorno 15, la variazione media dal basale nel numero di articolazioni con artrite attiva e nell'ampiezza di movimento limitata era rispettivamente del -67% e -73% per Ilaris (N = 43), rispetto a una mediana di variazione dello 0% e 0% nel gruppo placebo (N = 41). La variazione media del punteggio del dolore nel paziente (0–100 mm della scala visiva analogica) al giorno 15 era di -50,0 mm per Ilaris (N = 43) rispetto a +4,5 mm per il placebo (N = 25). La variazione media del punteggio del dolore nei pazienti era coerente al giorno 29.
Studio G2301
G2301 è uno studio randomizzato, in doppio cieco, controllato con placebo, sulla prevenzione delle ricadute con la terapia con Ilaris. Lo studio era composto da due parti con due endpoint primari indipendenti (successo nella riduzione dei corticosteroidi e tempo fino all'infiammazione). Nella parte I (aperta), 177 pazienti sono stati arruolati e hanno ricevuto 4 mg/kg (fino a 300 mg) di Ilaris, somministrato ogni 4 settimane per un periodo fino a 32 settimane. I pazienti nella parte II (in doppio cieco) hanno ricevuto Ilaris 4 mg/kg o placebo ogni 4 settimane fino all'osservazione di 37 ricadute.
Riduzione della dose di corticosteroidi
Dei 128 pazienti che assumevano corticosteroidi e partecipavano alla parte I dello studio, 92 pazienti hanno tentato di ridurre la dose di corticosteroidi. Cinquantasette (62%) di questi sono riusciti a ridurre significativamente la dose di corticosteroidi, e 42 (46%) hanno interrotto completamente l'uso di corticosteroidi.
Tempo fino alla prima ricaduta
Nei pazienti trattati con Ilaris nella parte II dello studio, si è osservata una riduzione del rischio di ricaduta della malattia del 64% rispetto al gruppo placebo (rapporto di rischio 0,36; IC 95%: 0,17–0,75; p = 0,0032). In 63 dei 100 pazienti arruolati nella parte II dello studio nel gruppo placebo o canakinumab, non si è verificata alcuna ricaduta durante il periodo di osservazione (fino a un massimo di 80 settimane).
Risultati degli studi G2305 e G2301 relativi alla salute e alla qualità della vita
Durante il trattamento con Ilaris si è osservato un miglioramento clinicamente significativo della funzione fisica e della qualità della vita dei pazienti. Nello studio G2305, secondo il questionario di valutazione dello stato di salute pediatrico, il miglioramento medio corretto per i minimi quadrati era di 0,69 nel gruppo Ilaris rispetto al gruppo placebo, superando di 3,6 volte la differenza minima clinicamente significativa di 0,19 (p = 0,0002). Il miglioramento medio rispetto al basale alla fine della parte II dello studio G2301 era di 0,88 (79%). Miglioramenti statisticamente significativi nel questionario di valutazione dello stato di salute pediatrico PF50 sono stati osservati nel gruppo Ilaris rispetto al gruppo placebo nello studio G2305 (stato fisico p = 0,0012; benessere psicosociale p = 0,0017).
Analisi combinata dell'efficacia
I dati dei primi 12 settimane di trattamento con Ilaris ottenuti negli studi G2305, G2301 e in uno studio esteso sono stati combinati per valutare l'efficacia. Questi dati indicano miglioramenti simili rispetto al basale negli indici pediatrici adattati ACR e nei loro componenti già alla settimana 12, rispetto a quanto osservato nello studio controllato con placebo (G2305). Alla settimana 12, gli indici pediatrici adattati ACR30, 50, 70, 90 e 100 erano rispettivamente del 70%, 69%, 61%, 49% e 30%, mentre il 28% dei pazienti presentava una malattia inattiva (N = 178).
Sebbene i dati siano limitati, gli studi clinici indicano che i pazienti che non rispondono al trattamento con tocilizumab o anakinra possono rispondere al trattamento con canakinumab.
Studio G2306
Lo studio G2306 era uno studio aperto per valutare il mantenimento della risposta al trattamento con riduzione della dose di canakinumab (2 mg/kg ogni 4 settimane) o allungamento dell'intervallo (4 mg/kg ogni 8 settimane) in pazienti con artrite idiopatica giovanile sistemica trattati con canakinumab 4 mg/kg ogni 4 settimane. Settantacinque pazienti di età compresa tra 2 e 22 anni, che avevano mantenuto uno stato di malattia inattivo per almeno 6 mesi consecutivi (remissione clinica) con monoterapia canakinumab, inclusi pazienti che avevano mantenuto uno stato di malattia inattivo interrompendo il trattamento concomitante di corticosteroidi e/o metotrexato per almeno 4 settimane, sono stati randomizzati per ricevere canakinumab 2 mg/kg ogni 4 settimane (N = 38) o canakinumab 4 mg/kg ogni 8 settimane (N = 37). Dopo 24 settimane, il 71% (27/38) dei pazienti che ricevevano la dose ridotta (2 mg/kg ogni 4 settimane) e l'84% (31/37) dei pazienti con intervallo allungato (4 mg/kg ogni 8 settimane) sono riusciti a mantenere uno stato di malattia inattivo per 6 mesi. Tra i pazienti in remissione clinica che hanno proseguito con ulteriore riduzione della dose (1 mg/kg ogni 4 settimane) o allungamento dell'intervallo di somministrazione (4 mg/kg ogni 12 settimane), il 93% (26/28) e il 91% (30/33) dei pazienti rispettivamente hanno mantenuto uno stato di malattia inattivo per 6 mesi. Ai pazienti che hanno mantenuto uno stato di malattia inattivo per ulteriori 6 mesi a questa dose minima è stato permesso di interrompere il trattamento con canakinumab. Nel complesso, il 33% (25/75) dei pazienti randomizzati per ridurre la dose o allungare l'intervallo di somministrazione è riuscito a interrompere il trattamento con canakinumab e mantenere uno stato di malattia inattivo per 6 mesi. Il tasso di eventi avversi in entrambi i gruppi di trattamento era simile a quello dei pazienti che ricevevano canakinumab 4 mg/kg ogni 4 settimane.
Malattia di Still dell'adulto (AOSD)
L'efficacia di canakinumab alla dose di 4 mg/kg (massimo fino a 300 mg), somministrato ogni 4 settimane a pazienti con AOSD, in uno studio randomizzato, in doppio cieco, controllato con placebo con 36 pazienti (età compresa tra 22 e 70 anni) è risultata simile a quella osservata nei pazienti con SJIA. Nello studio GDE01T, una maggiore proporzione di pazienti (12/18, 66,7%) nel gruppo canakinumab rispetto al gruppo placebo (7/17, 41,2%) ha mostrato un miglioramento rispetto al basale nell'indice di attività della malattia DAS28-ESR > 1,2 alla settimana 12, risultato non statisticamente significativo (rapporto di odds 2,86, differenza nel trattamento [%] 25,49 [IC 95%: 9,43, 55,80]). Entro 4 settimane, 7 su 18 pazienti (38,9%) trattati con canakinumab avevano già raggiunto la remissione DAS28-ESR rispetto a 2 su 17 pazienti (11,8%) che ricevevano placebo.
Questi dati sono coerenti con i risultati dell'analisi combinata dell'efficacia di 418 pazienti con SJIA, che ha mostrato che l'efficacia di canakinumab nel sottogruppo di pazienti con SJIA di età compresa tra 16 e <20 anni (n = 34) corrispondeva all'efficacia osservata nei pazienti di età inferiore ai 16 anni (n = 384).
Artrite gotta
L'efficacia di Ilaris nel trattamento delle crisi acute di artrite gotta è stata dimostrata in due studi multicentrici, randomizzati, in doppio cieco, controllati attivamente, in pazienti con manifestazioni frequenti di artrite gotta (3 o più attacchi negli ultimi 12 mesi) in cui non era possibile utilizzare FANS o colchicina (a causa di controindicazioni, intolleranza o insufficiente efficacia). Gli studi hanno avuto una durata di 12 settimane con un'estensione in doppio cieco di ulteriori 12 settimane. In totale, 225 pazienti hanno ricevuto Ilaris sottocutaneo alla dose di 150 mg e 229 pazienti hanno ricevuto triamcinolone acetonide (TA) intramuscolare alla dose di 40 mg al momento dell'inizio dello studio e dopo una recidiva. Il numero medio di attacchi di artrite gotta negli ultimi 12 mesi era di 6,5. Oltre l'85% dei pazienti aveva comorbidità, tra cui ipertensione arteriosa (60%), diabete mellito (15%), malattia coronarica (12%) e malattia renale cronica di stadio ≥3 (25%). Circa un terzo dei pazienti inclusi negli studi (76 [33,8%] nel gruppo Ilaris e 84 [36,7%] nel gruppo triamcinolone) non poteva assumere FANS e colchicina (intolleranza, controindicazioni o mancata risposta). La terapia concomitante per la riduzione degli urati (ULT) era in uso nel 42% dei pazienti all'arruolamento.
Gli endpoint primari composti erano: (I) intensità del dolore nell'artrite gotta (su scala visiva analogica, VAS) a 72 ore dall'assunzione della dose e (II) tempo fino al primo nuovo attacco di artrite gotta.
Nella popolazione generale dello studio, l'intensità del dolore era statisticamente significativamente inferiore con Ilaris 150 mg rispetto al triamcinolone acetonide a 72 ore. Ilaris riduce anche il rischio di attacchi successivi (vedi tabella 4).
I risultati di efficacia nel sottogruppo di pazienti che non possono assumere FANS e colchicina e quelli che assumevano ULT, non avevano risposta a ULT o avevano controindicazioni a ULT (N = 101), erano coerenti con lo studio nella popolazione generale, con differenze statisticamente significative rispetto al triamcinolone acetonide per l'intensità del dolore a 72 ore (-10,2 mm, p = 0,0208) e riduzione del rischio di attacchi successivi (rapporto di rischio 0,39, p = 0,0047 alla settimana 24).
I risultati di efficacia per il sottogruppo ridotto, limitato ai pazienti che assumevano ULT (N = 62), sono riportati nella tabella 4. Il trattamento con Ilaris ha determinato una riduzione del dolore e del rischio di attacchi successivi nei pazienti che assumono ULT e che non possono assumere FANS e colchicina, anche se la differenza nel trattamento rispetto al triamcinolone acetonide era meno pronunciata rispetto alla popolazione generale dello studio.
Tabella 4. Efficacia nella popolazione generale dello studio e nel sottogruppo di pazienti che assumono ULT e che non possono assumere FANS o colchicina
| Endpoint di efficacia |
Popolazione totale dello studio N = 454 |
Pazienti che non possono assumere FANS e colchicina e che ricevono ULT N = 62 |
| Trattamento delle crisi di artrite gotta (intensità del dolore (VAS) a 72 ore) |
||
| Stima della differenza media con il metodo dei minimi quadrati per triamcinolone acetonide IC p-valore, unilaterale |
−10,7 (−15,4; −6,0) p < 0,0001* |
−3,8 (−16,7; 9,1) p = 0,2798 |
| Riduzione del rischio di successive crisi di artrite gotta, valutata in base al tempo alla prima riacutizzazione (24 settimane) |
||
| Rapporto di rischio per triamcinolone acetonide IC p-valore, unilaterale |
0,44 (0,32; 0,60) p < 0,0001* |
0,71 (0,29; 1,77) p = 0,2337 |
| *Indica un valore di p significativo ≤0,025. |
||
I risultati dello studio sulla sicurezza hanno mostrato un aumento del numero di eventi avversi dopo l'uso di canakinumab rispetto al triamcinolone acetonide: il 66 % contro il 53 % dei pazienti nei quali si sono verificati eventi negativi e il 20 % contro il 10 % dei pazienti nei quali si sono verificati casi di infezione, durante un periodo di 24 settimane.
Pazienti anziani
Complessivamente, l'efficacia, la sicurezza e il profilo di tollerabilità di Ilaris nei pazienti anziani (≥ 65 anni) sono risultati paragonabili a quelli dei pazienti di età inferiore ai 65 anni.
Pazienti in terapia per la riduzione degli urati (ULT)
Negli studi clinici, Ilaris è stato utilizzato in modo sicuro in associazione con ULT. Nella popolazione complessiva dello studio, i pazienti in terapia con ULT hanno mostrato una riduzione meno pronunciata del dolore e una riduzione del rischio di futuri attacchi di artrite gottaica rispetto ai pazienti non sottoposti a ULT.
Immunogenicità
Non sono state osservate reazioni anafilattiche nei pazienti trattati con Ilaris.
Anticorpi diretti contro il medicinale Ilaris sono stati osservati in circa l'1,5 %, il 3 % e il 2 % dei pazienti trattati con Ilaris per il trattamento dei sindromi periodici associati alla criopirina, artrite idiopatica giovanile sistemica (SJIA) e artrite gottaica, rispettivamente.
Non sono stati osservati anticorpi diretti contro canakinumab nei pazienti con sindrome TRAPS, sindrome HID/MKD e FMF che hanno ricevuto dosi di 150 mg e 300 mg per 16 settimane di trattamento.
Popolazione pediatrica
Il richiedente ha completato quattro piani di studio pediatrici riguardanti canakinumab (per il sindrome CAP, SJIA, FMF – sindrome HID/MKD e sindrome TRAPS, rispettivamente). Queste informazioni sul medicinale sono state aggiornate per includere i risultati degli studi sull'uso di canakinumab nella popolazione pediatrica.
L'Agenzia europea per i medicinali ha rinviato l'obbligo di presentare i risultati degli studi su Ilaris in tutti i sottogruppi della popolazione pediatrica per l'artrite gottaica.
Farmacocinetica.
Sindromi periodici associati alla criopirina (CAPS)
Assorbimento
La concentrazione plasmatica massima di canakinumab (Cmax) è stata osservata circa 7 giorni dopo una singola somministrazione sottocutanea di 150 mg in pazienti adulti con CAPS. L'emivita media è stata di 26 giorni. I valori medi di Cmax e AUCinf dopo una singola dose sottocutanea di 150 mg in un paziente adulto tipico con CAPS (70 kg) sono stati di 15,9 µg/ml e 708 µg*d/ml. La biodisponibilità assoluta dopo somministrazione sottocutanea di canakinumab è stimata pari al 66 %. I parametri di esposizione (ad esempio AUC e Cmax) sono aumentati in modo proporzionale alla dose nell'intervallo di dosi da 0,30 a 10,0 mg/kg somministrati come infusione endovenosa, o da 150 a 600 mg come iniezione sottocutanea. I valori previsti dell'esposizione a regime stazionario (Cmin,ss, Cmax,ss, AUC,ss,8w) dopo somministrazione sottocutanea di 150 mg (o 2 mg/kg) ogni 8 settimane sono risultati leggermente più elevati nei pazienti con peso corporeo compreso tra 40 e 70 kg (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) rispetto a quelli con peso corporeo < 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) e > 70 kg (4,6 µg/ml, 17,8 µg/ml, 545 µg*d/ml). Il coefficiente di accumulo previsto è stato di 1,3 volte dopo 6 mesi di somministrazione sottocutanea di 150 mg di canakinumab ogni 8 settimane.
Distribuzione
Canakinumab si lega all'IL-1 beta nel siero. Il volume di distribuzione (Vss) di canakinumab varia in base al peso corporeo. È stimato pari a 6,2 litri nei pazienti con sindromi periodici associati alla criopirina con peso corporeo di 70 kg.
Eliminazione
La clearance apparente (CL/F) di canakinumab aumenta con il peso corporeo. È stimata pari a 0,17 l/giorno nei pazienti con sindromi periodici associati alla criopirina con peso corporeo di 70 kg e a 0,11 l/giorno nei pazienti con artrite idiopatica giovanile sistemica con peso corporeo di 33 kg.
Non sono state osservate evidenze di clearance accelerata o variazioni farmacocinetiche dipendenti dal tempo dopo somministrazioni ripetute. Dopo correzione per il peso corporeo, non sono state osservate differenze farmacocinetiche in base al sesso o all'età del paziente.
Sindrome TRAPS, sindrome HID/MKD e FMF
La biodisponibilità nei pazienti con sindrome TRAPS, sindrome HID/MKD e FMF non è stata determinata separatamente. La clearance apparente (CL/F) nella popolazione con sindrome TRAPS, sindrome HID/MKD e FMF con peso corporeo di 55 kg (0,14 l/giorno) è risultata paragonabile a quella nella popolazione con sindrome CAP con peso corporeo di 70 kg (0,17 l/giorno). Il volume di distribuzione apparente (V/F) è stato di 4,96 l con un peso corporeo di 55 kg.
Dopo somministrazioni ripetute sottocutanee di 150 mg ogni 4 settimane, la concentrazione minima di canakinumab alla 16ª settimana (Cmin) è stata stimata pari a 15,4 ± 6,6 µg/ml. L'AUCtau a regime stazionario stimata è stata di 636,7 ± 260,2 µg*d/ml.
Malattia di Still (artrite idiopatica giovanile sistemica (SJIA) e malattia di Still dell'adulto (AOSD))
La biodisponibilità nei pazienti con artrite idiopatica giovanile sistemica non è stata determinata separatamente. La clearance apparente per chilogrammo di peso corporeo (CL/F per 1 kg) è stata confrontata nei gruppi di pazienti con artrite idiopatica giovanile sistemica e sindromi periodici associati alla criopirina (0,004 l/giorno/kg). Il volume di distribuzione apparente per chilogrammo di peso corporeo (V/F per kg) è stato di 0,14 l/kg.
Dopo somministrazione ripetuta di dosi di 4 mg/kg ogni 4 settimane, il coefficiente di accumulo di canakinumab è stato 1,6 volte maggiore nei pazienti con artrite idiopatica giovanile sistemica. Lo stato stazionario è stato raggiunto entro 110 giorni. I valori medi previsti complessivi (±DS) di Cmin,ss, Cmax,ss e AUC,ss4w sono stati rispettivamente di 14,7 ± 8,8 µg/ml, 36,5 ± 14,9 µg/ml e 696,1 ± 326,5 µg*giorno/ml.
Per fasce d'età, l'AUCss4w è stato di 692, 615, 707 e 742 µg*giorno/ml nei pazienti di età compresa tra 2-3, 4-5, 6-11 e 12-19 anni, rispettivamente. In base alla stratificazione per peso corporeo, si è osservata una mediana di esposizione inferiore (30-40 %) di Cmin,ss (11,4 rispetto a 19 µg/ml) e AUCss (594 rispetto a 880 µg*giorno/ml) nel gruppo di pazienti con peso corporeo più basso (≤ 40 kg) rispetto ai pazienti con peso corporeo più elevato (> 40 kg).
Sulla base dell'analisi di modellizzazione farmacocinetica di popolazione, la farmacocinetica di canakinumab in giovani adulti con SJIA di età compresa tra 16 e 20 anni è risultata simile a quella nei pazienti di età inferiore ai 16 anni. L'esposizione prevista a regime stazionario con una dose di 4 mg/kg (massimo 300 mg) nei pazienti di età superiore ai 20 anni è risultata paragonabile a quella nei pazienti con SJIA di età inferiore ai 20 anni.
Pazienti con artrite gottaica
La biodisponibilità nei pazienti con artrite gottaica non è stata determinata. La clearance apparente per chilogrammo di peso corporeo (CL/F per 1 kg) è stata confrontata nei gruppi di pazienti con artrite gottaica e CAPS (0,004 l/giorno/kg). L'esposizione media in un paziente tipico con artrite gottaica (93 kg) dopo una singola dose sottocutanea di 150 mg (Cmax: 10,8 µg/ml e AUCinf: 495 µg*d/ml) è risultata inferiore rispetto a quella nei pazienti tipici con CAPS con peso corporeo di 70 kg (15,9 µg/ml e 708 µg*d/ml). Questo è coerente con l'aumento osservato di CL/F in relazione al peso corporeo.
Il coefficiente di accumulo previsto è stato 1,1 volte maggiore dopo somministrazione sottocutanea di canakinumab alla dose di 150 mg ogni 12 settimane.
Bambini
La concentrazione massima di canakinumab è stata raggiunta tra 2 e 7 giorni dopo una singola somministrazione sottocutanea di canakinumab alla dose di 150 mg o 2 mg/kg nei pazienti pediatrici di età superiore ai 4 anni. L'emivita è variata tra 22,9 e 25,7 giorni, simile a quella negli adulti. Sulla base dell'analisi di modellizzazione farmacocinetica, la farmacocinetica di canakinumab nei bambini di età compresa tra 2 e 4 anni è risultata analoga a quella nei pazienti di età superiore ai 4 anni.
È stato determinato che, dopo somministrazione sottocutanea, il grado di assorbimento diminuisce con l'età e si accelera nei pazienti più giovani. Di conseguenza, il Tmax è risultato più breve (3,6 giorni) nei giovani pazienti con artrite idiopatica giovanile sistemica (2-3 anni) rispetto ai pazienti più anziani con artrite idiopatica giovanile sistemica (12-19 anni; Tmax: 6 giorni). Non è stato osservato alcun effetto negativo sulla biodisponibilità (AUCss).
Un'analisi farmacocinetica aggiuntiva ha mostrato che la farmacocinetica di canakinumab in 6 pazienti di età inferiore ai 2 anni con sindromi periodici associati alla criopirina era simile a quella nei bambini di età compresa tra 2 e 4 anni. La modellizzazione farmacocinetica di popolazione indica che i livelli previsti di esposizione dopo una dose di 2 mg/kg erano paragonabili nei pazienti pediatrici con sindromi periodici associati alla criopirina, ma erano del 40 % inferiori nei pazienti con peso corporeo molto basso, ad esempio 10 kg, rispetto ai pazienti adulti (dose di 150 mg). Questo è coerente con i livelli di esposizione più elevati osservati nei gruppi di pazienti con sindromi periodici associati alla criopirina con peso corporeo più elevato.
La farmacocinetica è la stessa nei bambini con sindromi periodici associati alla criopirina, sindrome TRAPS, sindrome HID/MKD, FMF e artrite idiopatica giovanile sistemica.
Pazienti anziani
Non sono state osservate variazioni dei parametri farmacocinetici basate su clearance o volume di distribuzione nei pazienti anziani rispetto ai pazienti adulti di età inferiore ai 65 anni.
Dati preclinici di sicurezza
I dati preclinici non hanno evidenziato rischi specifici per l'uomo sulla base di studi di reattività incrociata, somministrazione ripetuta di dosi, immunotossicità, tossicità riproduttiva e tossicità giovanile, condotti con canakinumab o con anticorpi murini anti-IL-1 beta murini.
Poiché canakinumab si lega all'IL-1 beta animale (C. jacchus) e umano con affinità simile, la sicurezza di canakinumab è stata studiata negli animali. Nessun effetto avverso di canakinumab è stato osservato dopo somministrazione agli animali due volte alla settimana per 26 settimane o nello studio di tossicità sullo sviluppo embrione-fetale in animali gravidi. Le concentrazioni nel plasma ben tollerate negli animali superano almeno di 42 volte (Cmax) e 78 volte (CAVG) le concentrazioni nel plasma nei pazienti pediatrici con CAPS (peso corporeo 10 kg) trattati con dosi cliniche di canakinumab fino a 8 mg/kg sottocutaneo ogni 8 settimane. Inoltre, gli anticorpi contro canakinumab non sono stati rilevati in questi studi. Nessuna reattività incrociata tissutale aspecifica è stata dimostrata dopo applicazione di canakinumab su tessuti umani sani.
Non sono stati condotti studi formali sulla cancerogenicità di canakinumab.
In uno studio sullo sviluppo embrione-fetale negli animali, canakinumab non ha mostrato effetti tossici sull'organismo materno, effetti embriotossici o teratogeni dopo somministrazione durante l'organogenesi.
Nessun effetto avverso con anticorpi murini anti-IL-1 beta murini è stato osservato in una serie di studi riproduttivi e su topi giovanili. Gli anticorpi anti-IL-1 beta murini non hanno mostrato effetti avversi sul feto o sulla crescita del neonato dopo somministrazione alla madre nei periodi tardivi della gravidanza, durante il parto e l'allattamento. Le alte dosi utilizzate in questi studi erano massimamente efficaci in termini di soppressione dell'attività di IL-1 beta.
Studi immunotossicologici su topi con anticorpi murini anti-IL-1 beta murini hanno mostrato che la neutralizzazione di IL-1 beta non ha alcun effetto sugli indicatori immunologici e non provoca alterazioni della funzione immunitaria nei topi.
Caratteristiche cliniche.
Indicazioni.
Sindromi febbrili periodiche
Ilaris è indicato per il trattamento dei seguenti sindromi febbrili periodici autoinfiammatori negli adulti, adolescenti e bambini di età pari o superiore a 2 anni.
Sindromi periodici associati alla criopirina
Trattamento dei sindromi periodici associati alla criopirina negli adulti, adolescenti e bambini di età pari o superiore a 2 anni con peso corporeo di almeno 7,5 kg, inclusi:
- Sindrome di Muckle-Wells;
- Malattia infiammatoria multisistemica neonatale / Sindrome neurologica cronica pediatrica cutaneo-articolare (CINCA);
- Forme gravi di sindrome autoinfiammatoria familiare da freddo / orticaria familiare da freddo con sintomi non tipici dell’orticaria da freddo.
Sindrome periodica associata al recettore del fattore di necrosi tumorale (TRAPS).
Sindrome da iperimmunoglobulinemia D (HIDS) / deficit di mevalonato chinasi (MKD).
Febbre familiare mediterranea (FMF).
Deve essere utilizzato in associazione con la colchicina, se necessario.
Ilaris è inoltre indicato per il trattamento delle seguenti malattie:
Morbo di Still
Ilaris è indicato per il trattamento dell’attività del morbo di Still, compreso il morbo di Still dell’adulto (AOSD) e l’artrite idiopatica giovanile sistemica (SJIA) in pazienti di età pari o superiore a 2 anni che hanno mostrato una risposta inadeguata alla terapia precedente con farmaci antiinfiammatori non steroidei (FANS) e corticosteroidi sistemici. Ilaris può essere utilizzato come monoterapia o in combinazione con metotrexato.
Artrite gotta
Trattamento sintomatico di pazienti adulti con artrite gotta ricorrente frequente (almeno 3 episodi negli ultimi 12 mesi) quando i farmaci antiinfiammatori non steroidei (FANS) e la colchicina sono controindicati, non tollerati o non sufficientemente efficaci e quando la ripetuta somministrazione di cicli di corticosteroidi non è accettabile.
Controindicazioni.
Ipersensibilità alla sostanza attiva o a uno qualsiasi degli eccipienti. Infezioni attive e gravi.
Interazioni con altri medicinali e altre forme di interazione.
Le interazioni tra canakinumab e altri medicinali non sono state valutate in studi formali.
È stato osservato un aumento della frequenza di infezioni gravi con l’uso combinato di un altro inibitore dell’IL-1 e inibitori del fattore di necrosi tumorale (TNF). L’uso concomitante di canakinumab con inibitori del TNF non è raccomandato poiché aumenta il rischio di infezioni gravi.
L’attività degli enzimi epatici CYP450 può essere inibita da citochine coinvolte nell’infiammazione cronica, come l’interleuchina-1 beta (IL-1 beta). Pertanto, l’attività del CYP450 può essere modificata durante un trattamento potente inibitorio delle citochine, ad esempio con canakinumab. Questo aspetto ha rilevanza clinica per i substrati del CYP450 con un indice terapeutico stretto, per i quali la dose viene aggiustata individualmente. All’inizio del trattamento con canakinumab e con questo tipo di medicinali, è necessario effettuare un monitoraggio terapeutico dell’effetto o della concentrazione della sostanza attiva e, se necessario, aggiustare la dose.
Non sono disponibili dati sull’effetto dei vaccini vivi attenuati o sulla trasmissione secondaria di infezioni da vaccini vivi attenuati nei pazienti trattati con canakinumab. Pertanto, i vaccini vivi attenuati non devono essere somministrati contemporaneamente a Ilaris, salvo nei casi in cui i benefici superino chiaramente i rischi. Se la vaccinazione con vaccini vivi attenuati è prevista dopo l’inizio del trattamento con canakinumab, si raccomanda un intervallo di almeno 3 mesi dall’ultima iniezione di canakinumab prima della successiva iniezione.
I risultati di uno studio condotto su volontari sani adulti hanno dimostrato che una singola dose di 300 mg di Ilaris non influenza l’induzione e il mantenimento della risposta anticorpale dopo vaccinazione antinfluenzale o con vaccino contro il meningococco basato su proteina glicosilata.
I risultati di uno studio aperto della durata di 56 settimane su pazienti con sindromi periodici associati alla criopirina di età inferiore a 4 anni hanno mostrato che tutti i pazienti che hanno ricevuto vaccini inattivati, conformi ai programmi vaccinali pediatrici standard, hanno sviluppato livelli protettivi di anticorpi.
Caratteristiche d'uso.
Tracciabilità
Per migliorare la tracciabilità dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto del prodotto somministrato.
Infezioni
L'uso di canakinumab è stato associato a un aumento dei casi di infezioni gravi. Pertanto, i pazienti devono essere attentamente monitorati per i sintomi di infezione durante e dopo il trattamento con canakinumab. I medici devono prestare cautela nell'uso di canakinumab in pazienti con infezioni attive, infezioni ricorrenti in anamnesi o condizioni predisponenti alle infezioni.
Trattamento dei sindromi periodici associati alla criopirina (CAP), sindrome TRAP, sindrome HID/MKD, FMF e malattia di Still (SJIA e AOSD)
Canakinumab non deve essere somministrato durante un'infezione attiva che richieda intervento medico.
Trattamento dell'artrite gottaica
Canakinumab non deve essere somministrato durante un'infezione attiva.
L'uso concomitante di Ilaris con inibitori del fattore di necrosi tumorale (TNF) non è raccomandato poiché aumenta il rischio di infezioni gravi (vedere la sezione «Interazioni con altri medicinali ed altre forme di interazione»).
Sono stati riportati singoli casi di infezioni insolite o opportuniste (inclusi aspergillosi, infezioni micobatteriche atipiche, herpes zoster) durante il trattamento con canakinumab. Tuttavia, non può essere escluso un legame causale tra canakinumab e questi eventi.
Screening della tubercolosi
Circa il 12% dei pazienti con sindromi periodici associati alla criopirina ha mostrato un risultato positivo al test cutaneo della tubercolina (PPD) negli studi clinici, pur ricevendo canakinumab senza segni clinici di infezione tubercolare latente o attiva.
Non è noto se l'uso di inibitori dell'interleuchina-1 (IL-1), come canakinumab, aumenti il rischio di riattivazione della tubercolosi. Prima dell'inizio della terapia, tutti i pazienti devono essere sottoposti a screening per tubercolosi attiva e latente. Il medico deve effettuare un'accurata anamnesi. Tutti i pazienti (possono applicarsi raccomandazioni locali) devono sottoporsi a test di screening appropriati (ad esempio, test cutaneo della tubercolina, test di rilascio dell'interferone gamma o radiografia del torace). I pazienti devono essere attentamente monitorati per la comparsa di sintomi di tubercolosi durante e dopo il trattamento con canakinumab. Il paziente deve essere informato che, in caso di comparsa di sintomi suggerenti tubercolosi (ad esempio tosse persistente, perdita di peso, febbre subfebrile) durante la terapia con canakinumab, deve rivolgersi immediatamente al medico. Se il test di Mantoux è positivo, specialmente in pazienti ad alto rischio, si devono considerare metodi alternativi di screening per l'infezione tubercolare.
Neutropenia e leucopenia
Neutropenia (conteggio assoluto dei neutrofili [ANC] < 1,5 × 109/l) e leucopenia sono state osservate con l'uso di medicinali che inibiscono l'IL-1, inclusi canakinumab. Il trattamento con canakinumab non deve essere iniziato in pazienti con neutropenia o leucopenia. Si raccomanda di valutare il livello dei globuli bianchi, compreso il numero di neutrofili, prima dell'inizio del trattamento e a 1 e 2 mesi dopo l'inizio. Durante il trattamento cronico o terapie ripetute, si raccomanda di valutare periodicamente il livello dei globuli bianchi durante il trattamento. Se un paziente entra in uno stato neutropenico o leucopenico, il livello dei globuli bianchi deve essere attentamente monitorato e si deve considerare la necessità di interrompere il trattamento.
Neoplasie maligne
Sono stati riportati casi di neoplasie maligne in pazienti trattati con canakinumab. Il rischio di sviluppare tumori maligni con l'uso di inibitori dell'IL-1 non è noto.
Reazioni di ipersensibilità
Sono stati riportati casi di reazioni di ipersensibilità con l'uso di canakinumab. La maggior parte di questi casi era di gravità lieve. Durante lo sviluppo clinico di Ilaris in oltre 2600 pazienti, non sono state osservate reazioni anafilattoidi o anafilattiche. Tuttavia, il rischio di reazioni gravi di ipersensibilità, non insolito con iniezioni proteiche, non può essere escluso.
Funzione epatica
Funzione epatica
Nel corso degli studi clinici sono stati riportati episodi brevi e asintomatici di aumento dei livelli sierici delle transaminasi o della bilirubina.
Vaccinazione
Non sono disponibili dati sul rischio di trasmissione secondaria di infezioni con vaccini vivi attenuati in pazienti che assumono canakinumab. Pertanto, i vaccini vivi non devono essere somministrati contemporaneamente a canakinumab, salvo nei casi in cui i benefici superino chiaramente i rischi.
Prima dell'inizio del trattamento con canakinumab, si raccomanda che adulti e bambini ricevano tutti i vaccini necessari, inclusi il vaccino antipneumococcico e il vaccino antinfluenzale inattivato (vedere la sezione «Interazioni con altri medicinali ed altre forme di interazione»).
Mutazione nel gene NLRP3 in pazienti con sindromi periodici associati alla criopirina
L'esperienza clinica in pazienti con sindromi periodici associati alla criopirina senza mutazione confermata nel gene NLRP3 è limitata.
Sindrome di attivazione macrofagica in pazienti con malattia di Still
La sindrome di attivazione macrofagica è una condizione nota e potenzialmente letale che può svilupparsi in pazienti con malattie reumatologiche, in particolare in quelli con malattia di Still. In caso di sviluppo o sospetto di sindrome di attivazione macrofagica, la valutazione e il trattamento devono essere avviati il più rapidamente possibile. I medici devono prestare particolare attenzione ai sintomi di infezione o al peggioramento della malattia di Still, noti come meccanismi scatenanti della sindrome di attivazione macrofagica. I dati degli studi clinici indicano che canakinumab probabilmente non aumenta il rischio di sviluppare la sindrome di attivazione macrofagica nei pazienti con malattia di Still, ma non consentono conclusioni definitive.
Reazioni da farmaco con eosinofilia e sintomi sistemici (DRESS)
Raramente, in pazienti trattati con Ilaris, principalmente con artrite idiopatica giovanile sistemica (SJIA), sono state riportate reazioni da farmaco con eosinofilia e sintomi sistemici (DRESS). I pazienti con DRESS potrebbero richiedere ospedalizzazione poiché questa condizione può portare a esito fatale. Se sono presenti segni e sintomi di DRESS e non è possibile stabilire un'eziologia alternativa, Ilaris non deve essere riutilizzato; si deve considerare un trattamento alternativo.
Uso durante la gravidanza o l'allattamento.
Donne in età fertile
Le donne devono utilizzare metodi contraccettivi efficaci durante il trattamento con canakinumab e per 3 mesi dopo l'ultima dose.
Gravidanza
I dati sull'uso di Ilaris in donne in gravidanza sono limitati. Studi sugli animali non indicano effetti sfavorevoli diretti o indiretti sulla funzione riproduttiva. Il rischio per il feto/la madre è sconosciuto. Pertanto, le donne in gravidanza o che desiderano rimanere incinte devono usare il medicinale solo dopo un'attenta valutazione del beneficio e del rischio potenziale.
Studi sugli animali mostrano che canakinumab attraversa la placenta ed è rilevabile nel feto. Non ci sono dati nell'uomo, ma poiché canakinumab è un'immunoglobulina di classe G (IgG1), si prevede un passaggio transplacentare. Il significato clinico di ciò è sconosciuto. Tuttavia, non è raccomandata la somministrazione di vaccini vivi ai neonati esposti a canakinumab in utero entro 16 settimane dall'ultima dose di canakinumab somministrata alla madre prima del parto. Le donne che hanno ricevuto canakinumab durante la gravidanza devono essere istruite a informare il medico del neonato prima di qualsiasi vaccinazione.
Allattamento
Non è noto se canakinumab sia escreto nel latte materno umano. L'uso di Ilaris in donne che allattano al seno deve essere considerato solo se il beneficio atteso per la madre supera qualsiasi rischio per il neonato.
Fertilità
Non sono stati condotti studi sull'eventuale impatto di Ilaris sulla fertilità umana.
Canakinumab non ha influenzato i parametri di fertilità nei maschi animali (C. jacchus). Anticorpi murini anti-IL-1 beta non hanno avuto effetti negativi sulla fertilità nei topi maschi o femmine.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell'uso di macchinari.
Ilaris ha un'influenza trascurabile sulla capacità di guidare autoveicoli o di usare macchinari. Il trattamento con Ilaris può causare capogiri/vertigini o astenia (vedere la sezione «Effetti indesiderati»). I pazienti che manifestano tali sintomi durante il trattamento con Ilaris devono attendere che questi scompaiano prima di guidare o usare macchinari.
Modalità e dosi di somministrazione.
Sindromi periodiche associate alla criopirina (CAP), sindrome associata al recettore del fattore di necrosi tumorale (TRAPS), sindrome da iperimmunoglobulinemia D (HIDS)/deficit di mevalonato chinasi (MKD), febbre familiare mediterranea (FMF) e malattia di Still
Il trattamento deve essere iniziato su prescrizione e sotto la supervisione di un medico esperto nella diagnosi e nel trattamento delle condizioni corrispondenti.
Dopo un’adeguata formazione sulla tecnica di iniezione, i pazienti o i loro caregiver possono auto-somministrare Ilaris, qualora il medico ritenga tale procedura appropriata e clinicamente indicata.
Dosaggio iniziale raccomandato di Ilaris per adulti, adolescenti e bambini a partire dai 2 anni di età affetti da sindromi periodiche associate alla criopirina (CAP):
Adulti e bambini di età pari o superiore a 4 anni:
- 150 mg per pazienti con peso corporeo > 40 kg;
- 2 mg/kg per pazienti con peso corporeo ≥ 15 kg e ≤ 40 kg;
- 4 mg/kg per pazienti con peso corporeo ≥ 7,5 kg e < 15 kg.
Bambini di età compresa tra 2 e 4 anni:
- 4 mg/kg per pazienti con peso corporeo ≥ 7,5 kg.
Queste dosi devono essere somministrate ogni otto settimane come dose singola mediante iniezione sottocutanea.
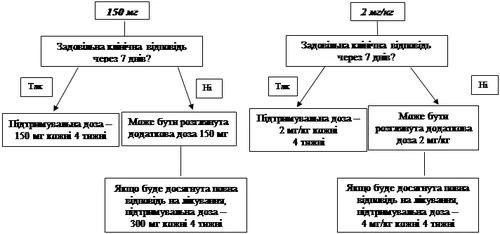
Se non si ottiene un adeguato effetto clinico (scomparsa dell’eruzione cutanea e di altri sintomi sistemici) entro 7 giorni dall’inizio del trattamento con la dose iniziale di 150 mg o 2 mg/kg, può essere somministrata una seconda dose di Ilaris pari a 150 mg o 2 mg/kg. Se si ottiene un effetto clinico, si deve mantenere un regime posologico intensificato di 300 mg o 4 mg/kg ogni 8 settimane. Se non si ottiene un adeguato effetto clinico entro 7 giorni da questo aumento di dose, può essere somministrata una terza dose di Ilaris pari a 300 mg o 4 mg/kg. Se si raggiunge un effetto clinico completo, si deve mantenere un regime posologico intensificato di 600 mg o 8 mg/kg ogni 8 settimane, sulla base di una valutazione clinica individuale.
Se non si ottiene un adeguato effetto clinico entro 7 giorni dall’inizio del trattamento con la dose iniziale di 4 mg/kg, può essere somministrata una seconda dose di Ilaris pari a 4 mg/kg. Se si ottiene un effetto clinico completo, si deve mantenere un regime posologico intensificato di 8 mg/kg ogni 8 settimane, sulla base di una valutazione clinica individuale.
L’esperienza clinica relativa all’uso di dosi con intervalli inferiori a 4 settimane e dosi superiori a 600 mg o 8 mg/kg è limitata.


 |
Malattia di Still (SJIA e AOSD)
La dose raccomandata di Ilaris nei pazienti con malattia di Still (artrite giovanile sistemica idiopatica, SJIA, e sindrome di Still dell'adulto, AOSD) con un peso corporeo ≥ 7,5 kg è di 4 mg/kg (massimo 300 mg) ogni quattro settimane mediante iniezione sottocutanea. La decisione di proseguire il trattamento con Ilaris in pazienti senza miglioramento clinico spetta al medico.
Artrite gottosa
Il trattamento deve essere effettuato sotto la supervisione di medici esperti nella diagnosi e nel trattamento dell’artrite gottosa e nell’uso di farmaci biologici. Il medicinale Ilaris deve essere somministrato da un operatore sanitario.
Deve essere iniziato il trattamento dell’iperuricemia con un’adeguata terapia che riduca o ottimizzi la concentrazione di urati (ULT). Canakinumab deve essere utilizzato come terapia su richiesta per il trattamento delle crisi di artrite gottosa.
È necessario controllare l’iperuricemia con un’appropriata terapia di riduzione dei livelli di urato. Ilaris deve essere utilizzato come terapia su richiesta per il trattamento delle artriti gottose.
La dose raccomandata di Ilaris nei pazienti adulti con artrite gottosa è di 150 mg per via sottocutanea come dose singola durante una crisi. Per ottenere l’effetto massimo, Ilaris deve essere somministrato il più presto possibile dopo l’inizio della crisi di artrite gottosa.
Nei pazienti che non hanno risposto al trattamento iniziale, non è consigliato un ulteriore utilizzo di Ilaris. Nei pazienti che hanno risposto al trattamento e nei quali è necessario un trattamento ripetuto, l’intervallo tra le somministrazioni deve essere di almeno 12 settimane.
Popolazioni particolari
Anziani
Non è necessario alcun aggiustamento della dose.
Non sono state osservate differenze significative nel profilo di sicurezza nei pazienti di età superiore a 65 anni.
Insufficienza epatica
Non sono disponibili dati sull’uso di Ilaris nei pazienti con compromissione epatica.
Insufficienza renale
Non è necessario alcun aggiustamento della dose nei pazienti con insufficienza renale. Tuttavia, l’esperienza clinica nell’uso del medicinale in questi pazienti è limitata.
Modalità di somministrazione
Per somministrazione sottocutanea.
Siti di iniezione: parte superiore della coscia, addome, spalla o natica. Si raccomanda di scegliere un sito diverso ogni volta che il farmaco viene somministrato, per ridurre il rischio di dolore. Si devono evitare aree con cute danneggiata, ematomi o eruzioni cutanee. L’iniezione in tessuto cicatriziale deve essere evitata poiché ciò potrebbe ridurre l’efficacia di canakinumab.
Ogni flaconcino è destinato all’uso singolo per la somministrazione individuale di una dose. Dopo un’adeguata formazione sulla corretta tecnica di iniezione, pazienti o caregiver possono somministrare canakinumab, qualora il medico lo ritenga appropriato e sotto continua supervisione medica.
Istruzioni per l’uso
Ilaris, soluzione iniettabile 150 mg/ml, è fornito in un flaconcino monodose per uso individuale.
Prima dell’iniezione, riscaldare il flaconcino a temperatura ambiente. La soluzione deve essere priva di particelle visibili, trasparente o opalescente. La soluzione deve essere incolore, ma può presentare una leggera sfumatura giallo-brunastra. Utilizzando un ago da 18 G o 21 G × 2 pollici (50 mm) (o di tipo equivalente disponibile sul mercato), riempire con attenzione una siringa da 1 ml con la quantità necessaria di soluzione, in base alla dose da somministrare.
Dopo aver riempito la siringa con la quantità necessaria di soluzione, coprire con il tappo, staccare l’ago di riempimento dalla siringa, collegare un ago da 27 G × 0,5 pollici (13 mm) (o di tipo equivalente disponibile sul mercato) e somministrare immediatamente la soluzione per via sottocutanea.
Smaltimento
Il medicinale non utilizzato o i rifiuti devono essere smaltiti in conformità con i requisiti locali.
Pazienti o caregiver devono essere istruiti sullo smaltimento di flaconcini, siringhe e aghi in conformità con i requisiti locali.
Popolazione pediatrica.
Sindromi periodiche associate alla criopirina (CAP), sindrome TRAPS, sindrome HID/MKD e FMF
Non sono state stabilite la sicurezza e l’efficacia di Ilaris nei pazienti di età inferiore a 2 anni con sindromi periodiche associate alla criopirina, sindrome TRAPS, sindrome HID/MKD e FMF. I dati attualmente disponibili sono descritti nelle sezioni «Farmacocinetica», «Farmacodinamica» e «Effetti indesiderati», ma non possono essere formulate raccomandazioni relative al dosaggio.
Artrite idiopatica giovanile sistemica (SJIA)
Non sono state stabilite la sicurezza e l’efficacia di Ilaris nei pazienti di età inferiore a 2 anni con artrite idiopatica giovanile sistemica.
Artrite gottosa
Non esiste esperienza nell’uso di Ilaris nei bambini per il trattamento dell’artrite gottosa.
Sovradosaggio.
Le informazioni relative al sovradosaggio sono limitate. Negli studi iniziali, pazienti e volontari sani hanno ricevuto dosi fino a 10 mg/kg per via endovenosa o sottocutanea senza alcun segno di tossicità acuta del farmaco.
In caso di sovradosaggio, si raccomanda il monitoraggio del paziente e, se necessario, l’inizio immediato di un’appropriata terapia sintomatica.
Effetti indesiderati
Sono stati arruolati circa 2300 pazienti, tra cui circa 250 bambini (di età compresa tra 2 e 17 anni), in studi clinici in aperto e in cieco, compresi pazienti con sindromi periodiche associate alla criopina, artrite idiopatica giovanile sistemica, artrite gottaica o altre malattie mediate da IL-1 beta, nonché volontari sani. Gli effetti indesiderati più comuni furono infezioni (ad esempio infezioni delle vie respiratorie superiori). La maggior parte degli eventi era di gravità da lieve a moderata. Il trattamento a lungo termine non ha influenzato il tipo o la frequenza degli effetti indesiderati.
Nei pazienti trattati con Ilaris sono stati osservati casi di reazioni di ipersensibilità.
Sono state riportate infezioni opportunistiche durante il trattamento con Ilaris.
Sindromi periodiche associate alla criopina
Nello studio clinico sono stati arruolati 211 pazienti adulti e pediatrici (con diagnosi di sindrome autoinfiammatoria familiare da freddo/orticaria familiare da freddo, sindrome di Muckle-Wells e malattia infiammatoria multisistemica neonatale/sindrome cronica pediatrica neurologica cutaneo-articolare). La sicurezza di Ilaris è stata confrontata con placebo nello studio principale di fase III, costituito da un periodo in aperto della durata di 8 settimane (parte 1), un periodo randomizzato, in doppio cieco e controllato con placebo della durata di 24 settimane (periodo di sospensione, parte 2) e un periodo in aperto di 16 settimane con trattamento con Ilaris (parte 3). Tutti i pazienti hanno ricevuto 150 mg di Ilaris per via sottocutanea oppure 2 mg/kg di peso corporeo in caso di peso corporeo ≥ 15 kg e ≤ 40 kg.
Artrite idiopatica giovanile sistemica
Nello studio clinico con Ilaris sono stati arruolati 201 pazienti di età compresa tra 2 e 20 anni con diagnosi di artrite idiopatica giovanile sistemica. La sicurezza di Ilaris è stata confrontata con placebo in due studi pivotali di fase III.
Artrite gottaica
Più di 700 pazienti con artrite gottaica sono stati arruolati in studi clinici randomizzati, in doppio cieco e controllati attivamente, della durata fino a 24 settimane, ai quali sono state somministrate dosi comprese tra 10 mg e 300 mg. Più di 250 pazienti sono stati trattati con la dose raccomandata di 150 mg nelle fasi II e III degli studi.
Gli effetti indesiderati sono elencati secondo le classi di sistemi e organi MedDRA e per frequenza. All'interno di ogni classe di sistemi e organi, gli effetti indesiderati sono classificati per categoria di frequenza, con i più comuni riportati per primi. Le categorie di frequenza sono definite come segue: molto comune (≥ 1/10); comune (≥ 1/100 fino a < 1/10); non comune (≥ 1/1.000 fino a < 1/100); raro (≥ 1/10.000 fino a < 1/1.000); molto raro (< 1/10.000); non noto (la frequenza non può essere stimata sulla base dei dati disponibili). All'interno di ciascun gruppo per frequenza, gli effetti indesiderati sono presentati in ordine decrescente di gravità.
Tabella 5. Effetti indesiderati
| Classi di sistemi e organi |
Sindrome CAP, sindrome TRAP, sindrome HID/MKD, FMF, artrite idiopatica giovanile sistemica, artrite gottaica |
| Infezioni e infestazioni |
|
| Molto frequente |
Infezioni delle vie respiratorie (inclusi polmonite, bronchiti, influenza, infezioni virali, sinusiti, riniti, faringiti, tonsilliti, nasofaringiti, infezioni delle alte vie respiratorie) Infezioni dell'orecchio Cellulite Gastroenterite Infezioni del tratto urinario |
| Frequente |
Candidosi vulvovaginale |
| Alterazioni del sistema nervoso |
|
| Frequente |
Capogiri/vertigini |
| Alterazioni del sistema gastrointestinale |
|
| Molto frequente |
Dolore addominale (parte superiore)1 |
| Non comune |
Malattia da reflusso gastroesofageo2 |
| Alterazioni della cute e del tessuto sottocutaneo |
|
| Molto frequente |
Reazioni nel sito di iniezione |
| Alterazioni del sistema muscoloscheletrico e del tessuto connettivo |
|
| Molto frequente |
Artralgia1 |
| Frequente |
Dolore muscoloscheletrico1 Dolore alla schiena2 |
| Alterazioni generali |
|
| Frequente |
Stanchezza/astenia2 |
| Esami diagnostici |
|
| Molto frequente |
Diminuzione della clearance renale della creatinina1,3 Proteinuria1,4 Leucopenia1,5 |
| Frequente |
Neutropenia |
| Non comune |
Diminuzione del numero di piastrine |
| 1Nell’artrite idiopatica giovanile sistemica. 2Nell’artrite gottaica. 3Secondo la clearance della creatinina stimata, la maggior parte dei casi è stata transitoria. 4La maggior parte dei casi è stata rappresentata da tracce transitorie o da una reazione a livello di 1+ di proteine nelle urine con strisce reattive. 5Vedere ulteriori informazioni riportate di seguito. |
|
In una sottopopolazione di giovani adulti con artrite idiopatica giovanile sistemica di età compresa tra 16 e 20 anni (n = 31), il profilo di sicurezza di canakinumab era coerente con quello osservato nei pazienti con artrite idiopatica giovanile sistemica di età inferiore ai 16 anni. Sulla base di rapporti pubblicati, ci si attende che il profilo di sicurezza nei pazienti con malattia di Still dell'adulto sia simile a quello dei pazienti con artrite idiopatica giovanile sistemica.
Dati a lungo termine e anomalie di laboratorio nei pazienti con sindromi periodiche associate alla criopirina
Nei trial clinici di Ilaris in pazienti con sindromi periodiche associate alla criopirina, i valori medi di emoglobina sono aumentati, mentre i livelli di globuli bianchi, neutrofili e piastrine sono diminuiti.
Raramente si sono osservati aumenti delle transaminasi.
Aumenti asintomatici e moderati del livello sierico di bilirubina, senza aumento concomitante delle transaminasi, sono stati riportati in pazienti con sindromi periodiche associate alla criopirina trattati con canakinumab.
Negli studi aperti a lungo termine con aumento della dose, le infezioni (gastroenterite, infezioni delle vie respiratorie, infezioni delle vie respiratorie superiori), il vomito e le vertigini si sono verificati più frequentemente nel gruppo trattato con 600 mg o 8 mg/kg rispetto ad altri gruppi di dosaggio.
Anomalie di laboratorio nei pazienti con sindrome TRAP, sindrome HID/MKD e FMF
Neutrofili
Sebbene una riduzione di grado ≥ 2 dei neutrofili si sia verificata nel 6,5% dei pazienti (frequente) e una riduzione di grado 1 nel 9,5% dei pazienti, tale riduzione è generalmente transitoria e non è stata identificata alcuna infezione associata alla neutropenia come reazione avversa.
Piastrine
Sebbene una riduzione del conteggio piastrinico (grado ≥ 2) si sia verificata nello 0,6% dei pazienti, non è stata identificata alcuna emorragia come reazione avversa. Una riduzione lieve e transitoria di grado 1 delle piastrine si è verificata nel 15,9% dei pazienti senza alcuna reazione avversa emorragica associata.
Anomalie di laboratorio nei pazienti con artrite idiopatica giovanile sistemica
Ematologia
Nel programma complessivo di trattamento dell’artrite idiopatica giovanile sistemica, riduzioni transitorie dei globuli bianchi ≤ 0,8 × LRI sono state osservate in 33 pazienti (16,5%). Riduzioni transitorie del conteggio assoluto dei neutrofili (ANC) a livelli inferiori a 1 × 109/l sono state osservate in 12 pazienti (6,0%). Riduzioni transitorie del conteggio piastrinico (< LRI) sono state osservate in 19 pazienti (9,5%).
ALT/AST
Nel programma complessivo di trattamento dell’artrite idiopatica giovanile sistemica, livelli elevati di ALT e/o AST (oltre 3 volte il limite superiore della norma - LSN) sono stati osservati in 19 pazienti (9,5%).
Anomalie di laboratorio nei pazienti con artrite gotta
Ematologia
Riduzioni dei globuli bianchi ≤ 0,8 × LRI (limite inferiore della norma) sono state registrate nel 6,7% dei pazienti trattati con canakinumab, rispetto all’1,4% dei pazienti trattati con acetone di triamcinolone. Riduzioni del conteggio assoluto dei neutrofili (ANC) a livelli inferiori a 1 × 109/l sono state osservate nel 2% dei pazienti negli studi comparativi. Sono stati inoltre osservati singoli casi con ANC < 0,5 × 109/l.
Una riduzione moderata (< LRI e > 75 × 109/l) e transitoria del conteggio piastrinico si è verificata con maggiore frequenza (12,7%) dopo somministrazione di canakinumab negli studi clinici controllati attivi, rispetto al farmaco di confronto (7,7%) nei pazienti con artrite gotta.
Acido urico
Un aumento dei livelli di acido urico (0,7 mg/dl a 12 settimane e 0,5 mg/dl a 24 settimane) è stato osservato dopo trattamento con canakinumab negli studi comparativi in pazienti con artrite gotta. In un altro studio in pazienti in trattamento con ULT, non è stato osservato alcun aumento dell’acido urico. Nessun aumento dei livelli di acido urico è stato osservato negli studi clinici nel gruppo di pazienti senza artrite gotta.
ALT/AST
Negli studi comparativi, negli studi con canakinumab e triamcinolone acetonide si sono osservati aumenti medi e mediani di alanina aminotransferasi (ALT) rispettivamente di 3,0 U/l e 2,0 U/l e di aspartato aminotransferasi (AST) rispettivamente di 2,7 U/l e 2,0 U/l rispetto ai livelli basali alla fine dello studio. Tuttavia, la frequenza di variazioni clinicamente significative (≥ 3 × LSN) è stata maggiore nei pazienti trattati con triamcinolone acetonide (2,5% per AST e ALT) rispetto al gruppo trattato con canakinumab (1,6% per ALT e 0,8% per AST).
Trigliceridi
Negli studi clinici controllati attivi con pazienti con artrite gotta, l’aumento medio dei livelli di trigliceridi è stato di 33,5 mg/dl nel gruppo trattato con canakinumab, rispetto a una lieve riduzione di −3,1 mg/dl nel gruppo trattato con triamcinolone acetonide. La frequenza di aumenti dei trigliceridi > 5 × LSN è stata del 2,4% per canakinumab e dell’0,7% per triamcinolone acetonide. Il significato clinico di questa osservazione è sconosciuto.
Dati a lungo termine dello studio di osservazione
In uno studio di lunga durata (esposizione media a canakinumab di 3,8 anni), 85 pazienti pediatrici di età ≥ 2 e ≤ 17 anni e 158 pazienti adulti di età ≥ 18 anni con artrite idiopatica giovanile sistemica sono stati trattati con canakinumab nella pratica clinica abituale. Il profilo di sicurezza di canakinumab osservato dopo un trattamento prolungato in queste condizioni era coerente con quello osservato negli studi interventistici nei pazienti con artrite idiopatica giovanile sistemica.
Popolazione pediatrica
80 pazienti pediatrici di età compresa tra 2 e 17 anni con sindromi periodiche associate alla criopirina sono stati inclusi nello studio. Nel complesso, non sono state osservate differenze clinicamente significative riguardo alla sicurezza e al profilo di tollerabilità di Ilaris nei pazienti pediatrici rispetto alla popolazione generale di pazienti con sindromi periodiche associate alla criopirina (comprendente adulti e pediatrici, N = 211), inclusa la frequenza e gravità complessiva degli episodi infettivi. Le infezioni delle vie respiratorie superiori sono state le infezioni più comuni.
Inoltre, 6 pazienti pediatrici di età inferiore ai 2 anni sono stati valutati in un piccolo studio clinico aperto. Il profilo di sicurezza di Ilaris è simile a quello osservato nei pazienti di età ≥ 2 anni.
Durante uno studio di 16 settimane, 102 pazienti con sindrome TRAP, sindrome HID/MKD e FMF (di età compresa tra 2 e 17 anni) hanno ricevuto canakinumab. Nel complesso, non sono state osservate differenze clinicamente significative nel profilo di sicurezza e tollerabilità di canakinumab nei pazienti pediatrici rispetto alla popolazione generale.
Periodo di validità. 3 anni.
Condizioni di conservazione.
Conservare a 2–8 °C nella confezione originale per proteggere dalla luce.
Non congelare. Conservare fuori dalla portata dei bambini.
Incompatibilità.
A causa della mancanza di studi di compatibilità, questo medicinale non deve essere mescolato con altri medicinali.
Confezionamento.
1 ml in flaconcino; 1 flaconcino in confezione.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore.
- Novartis Pharma GmbH (rilascio del lotto).
- Lek Pharmaceuticals d.d. (confezionamento secondario, rilascio del lotto).
- Novartis Pharmaceuticals, S.A. (rilascio del lotto).
Indirizzo del produttore e sede operativa.
- Roonstraße 25, Gostenhof, Norimberga, Baviera, 90429, Germania.
- Verovškova ulica 57, Lubiana, 1526, Slovenia.
- Gran Vía de las Cortes Catalanas 764, Barcellona, 08013, Spagna.