Edoxadar
UcrainaIndice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE Edoxadar (Edoxadar)
Composizione:
Principio attivo: edoxaban;
1 compressa rivestita con film contiene edoxaban 30,000 mg, equivalente a 40,410 mg di edoxaban tosilato monoidrato oppure 60,000 mg, equivalente a 80,820 mg di edoxaban tosilato monoidrato.
Eccipienti: mannitolo (E 421), idrossipropilcellulosa, crospovidone, amido pregelatinizzato (di mais), biossido di silicio colloidale anidro, stearato di magnesio; rivestimento:
compresse da 30 mg: carmellosa sodica; maltodestrina; glucosio monoidrato; lecitina (di soia); carbonato di calcio; ossido di ferro rosso (E 172);
compresse da 60 mg: carmellosa sodica; maltodestrina; glucosio monoidrato; lecitina (di soia); carbonato di calcio; ossido di ferro giallo (E 172).
Forma farmaceutica. Compresse rivestite con film.
Principali proprietà fisico-chimiche:
compresse da 30 mg: compresse rosa, di forma rotonda, rivestite con film, con impresso «30» su un lato e liscio sull'altro lato;
compresse da 60 mg: compresse gialle, di forma rotonda, rivestite con film, con impresso «60» su un lato e liscio sull'altro lato.
Gruppo farmacoterapeutico. Agenti antitrombotici. Inibitori diretti del fattore Xa. Edoxaban. Codice ATC B01A F03.
Proprietà farmacologiche
Farmacodinamica
Meccanismo d'azione
Edoxadar è un inibitore diretto, selettivo e reversibile del fattore Xa, una serina proteasi che rappresenta l'ultimo passaggio della cascata della coagulazione. Edoxadar inibisce sia il fattore Xa libero che l'attività della protrombinasi. L'inibizione del fattore Xa nella cascata della coagulazione determina una riduzione della produzione di trombina, prolunga il tempo di coagulazione del sangue e riduce il rischio di formazione del trombo.
Effetti farmacodinamici
Edoxadar induce rapidamente un effetto farmacodinamico entro 1-2 ore, corrispondente al valore di concentrazione massima nel plasma (Cmax) di edoxadar. Gli effetti farmacodinamici, misurati mediante analisi dell'attività anti-fattore Xa, sono prevedibili e correlano con la dose e la concentrazione di edoxadar. Come conseguenza dell'inibizione del fattore Xa, edoxadar prolunga anche il tempo di protrombina (PT) e il tempo di tromboplastina parziale attivato (aPTT). Le variazioni osservate nei parametri di coagulazione con l'uso di dosi terapeutiche sono minime e variabili, pertanto non sono raccomandate per il monitoraggio dell'effetto anticoagulante di edoxadar.
Effetto sui marcatori di coagulazione nel passaggio da rivaroxaban, dabigatran o apixaban a edoxadar
In studi clinici farmacologici su soggetti sani, è stato somministrato rivaroxaban 20 mg una volta al giorno, dabigatran 150 mg due volte al giorno o apixaban 5 mg due volte al giorno, seguito da una singola dose di edoxadar 60 mg al quarto giorno. È stato valutato l'effetto sul PT e altri biomarcatori della coagulazione (ad esempio, attività anti-fattore Xa, aPTT). Dopo il passaggio a edoxadar al quarto giorno, il valore di PT era equivalente a quello osservato al terzo giorno di trattamento con rivaroxaban e apixaban. I valori di aPTT erano più elevati dopo somministrazione di edoxadar nei soggetti precedentemente trattati con dabigatran, rispetto ai soggetti trattati solo con edoxadar. Si ritiene che questo risultato sia dovuto all'effetto residuo del trattamento con dabigatran, tuttavia non ha comportato un prolungamento del tempo di sanguinamento.
Sulla base di questi dati, nel passaggio da questi anticoagulanti a edoxadar, la prima dose di edoxadar può essere somministrata al posto della successiva dose programmata dell'anticoagulante precedente.
Efficacia e sicurezza clinica
Prevenzione dell'ictus e dell'embolia sistemica
Il programma clinico di studi con edoxadar nella fibrillazione atriale è stato progettato per dimostrare l'efficacia e la sicurezza di due gruppi di dosi di edoxadar rispetto al warfarin nella prevenzione dell'ictus e dell'embolia sistemica in pazienti con fibrillazione atriale non valvolare (FANV) e con rischio moderato o elevato di ictus e di eventi embolici sistemici. Il punto finale primario di efficacia era la combinazione di ictus e eventi embolici sistemici. I punti finali secondari di efficacia includevano: combinazione di ictus, eventi embolici sistemici e morte cardiovascolare; evento cardiovascolare avverso serio (ECAS), definito come combinazione di infarto miocardico non fatale, ictus non fatale, embolia sistemica non fatale e esito fatale per cause cardiovascolari o emorragia; combinazione di ictus, embolia sistemica e esito fatale per qualsiasi causa.
L'obiettivo dell'analisi primaria di efficacia era dimostrare che edoxadar non fosse inferiore al warfarin riguardo all'ictus primario o agli eventi embolici sistemici verificatisi durante il trattamento o entro tre giorni dalla somministrazione dell'ultima dose nella popolazione modificata dei pazienti che avevano iniziato il trattamento. Edoxadar alla dose di 60 mg ha dimostrato un'efficacia non inferiore al warfarin riguardo al parametro primario di efficacia, ovvero ictus o evento embolico sistemico.
Il punto finale primario di sicurezza era rappresentato dalle emorragie maggiori.
È stato osservato un significativo riduzione del rischio di emorragie maggiori, emorragie intracraniche e altri tipi di emorragie nel gruppo trattato con edoxadar 60 mg rispetto al gruppo trattato con warfarin.
Nel gruppo trattato con edoxadar 60 mg si è osservata anche una significativa riduzione del numero di emorragie fatali rispetto al gruppo trattato con warfarin, principalmente dovuta alla riduzione degli eventi di emorragia intracranica fatale.
Trattamento della trombosi venosa profonda (TVP) e dell'embolia polmonare (EP), nonché prevenzione delle recidive di TVP ed EP [TVP (tromboembolia venosa)]
Il programma clinico di studi con edoxadar nella TVP è stato progettato per dimostrare l'efficacia e la sicurezza dell'uso di edoxadar nel trattamento della TVP e dell'EP e nella prevenzione delle recidive di TVP ed EP.
Lo studio ha dimostrato che edoxadar non è inferiore al warfarin riguardo al parametro primario di efficacia, ovvero le recidive di TVP.
I risultati di efficacia nelle sottopopolazioni predefinite (con l'uso di dosi ridotte se necessario), inclusi i fattori età, peso corporeo, sesso e funzionalità renale, sono stati coerenti con i risultati principali di efficacia nell'intera popolazione dello studio.
Il punto finale primario di sicurezza era rappresentato dall'emorragia clinicamente significativa (maggiore o non maggiore ma clinicamente significativa).
Nel gruppo edoxadar si è osservata una significativa riduzione del rischio di emorragie clinicamente significative, emorragie maggiori o emorragie non maggiori ma clinicamente significative rispetto al gruppo warfarin.
Prevenzione dell'ictus e dell'embolia sistemica in pazienti con FANV e clearance della creatinina (CCr) > 100 ml/min
Alla luce dell'insieme dei dati disponibili su pazienti con FANV e alta clearance della creatinina (CCr > 100 ml/min) trattati con edoxadar 60 mg, il tasso annuo previsto di ictus ischemico/embolia sistemica è ≤ 1%. L'aumento della dose di edoxadar (oltre 60 mg) in pazienti con FANV e alta CCr (> 100 ml/min) non fornisce una maggiore protezione contro l'ictus e può essere associato a un aumento degli effetti collaterali. Pertanto, in questi pazienti si raccomanda un regime di trattamento con edoxadar 60 mg una volta al giorno dopo un'attenta valutazione del rischio individuale di tromboembolia e di emorragia.
Pazienti sottoposti a cardioversione
Durante gli studi è stata osservata una bassa frequenza di emorragie maggiori e non maggiori ma clinicamente significative e di eventi tromboembolici nei pazienti sottoposti a cardioversione.
Popolazione pediatrica
L'Agenzia europea per i medicinali ha rinviato l'obbligo di presentare i risultati degli studi con edoxadar in una o più sottopopolazioni pediatriche per la prevenzione della tromboembolia arteriosa, il trattamento della tromboembolia e la prevenzione della tromboembolia (vedi sezione «Modalità di somministrazione e posologia»).
Farmacocinetica
Assorbimento. Dopo l'assorbimento, la concentrazione massima di edoxadar nel plasma viene raggiunta entro 1-2 ore. La biodisponibilità assoluta è di circa il 62%. L'assunzione di cibo aumenta in modo non uniforme la massima esposizione, ma ha un impatto minimo sull'effetto complessivo. Edoxadar è stato somministrato negli studi sia a digiuno che con cibo. Edoxadar ha una scarsa solubilità a valori di pH ≥ 6,0. La co-somministrazione di inibitori della pompa protonica non ha un effetto clinicamente significativo sull'effetto di edoxadar.
Distribuzione. Edoxadar presenta una distribuzione bifasica. Il volume di distribuzione è mediamente di 107 l (deviazione standard 19,9 l).
Il legame alle proteine plasmatiche in vitro è di circa il 55%. Non si osservano accumuli clinicamente significativi di edoxadar (coefficiente di accumulo 1,14) con somministrazione una volta al giorno. Le concentrazioni di equilibrio vengono raggiunte entro 3 giorni.
Biometabolismo. Nel plasma, edoxadar viene principalmente rilevato nella forma invariata. Edoxadar è metabolizzato principalmente per idrolisi (mediata dalla carbossilesterasi 1), coniugazione o ossidazione mediata da CYP3A4/5 (< 10%). Edoxadar è un substrato del trasportatore di efflusso P-glicoproteina (P-gp), ma non è un substrato dei trasportatori di uptake come il trasportatore peptidico organico anionico OATP1B1, i trasportatori di anioni organici OAT1 o OAT3 o il trasportatore di cationi organici OCT2. Il suo metabolita attivo è un substrato di OATP1B1.
Eliminazione. In soggetti sani, la clearance totale calcolata è di 22 (± 3) l/ora; il 50% del farmaco viene eliminato dai reni (11 l/ora). La clearance renale rappresenta circa il 35% della dose somministrata. Il resto della clearance è dovuto al metabolismo e all'eliminazione biliare/intestinale. L'emivita di eliminazione dopo somministrazione orale è di 10-14 ore.
Linearità. Con dosi comprese tra 15 mg e 60 mg, edoxadar mostra una farmacocinetica approssimativamente proporzionale alla dose.
Popolazioni speciali
Pazienti anziani
L'analisi farmacocinetica di popolazione ha dimostrato che, tenendo conto della funzionalità renale e del peso corporeo, l'età del paziente non ha un effetto aggiuntivo clinicamente significativo sulla farmacocinetica di edoxadar.
Alterazioni della funzionalità renale
L'area sotto la curva concentrazione-tempo (AUC) nei pazienti con disfunzione renale lieve (CCr > 50-80 ml/min), moderata (CCr 30-50 ml/min) e grave (CCr < 30 ml/min, senza dialisi) aumentava rispettivamente del 32%, 74% e 72% rispetto ai pazienti con funzionalità renale normale. Nei pazienti con disfunzione renale, il profilo dei metaboliti cambia e si forma una maggiore quantità di metaboliti attivi.
È stata osservata una correlazione lineare tra la concentrazione plasmatica di edoxadar e l'attività anti-fattore Xa, indipendentemente dalla funzionalità renale.
Nei pazienti con insufficienza renale terminale sottoposti a dialisi peritoneale, l'esposizione totale era del 93% superiore rispetto a soggetti sani.
La modellizzazione farmacocinetica di popolazione indica che l'esposizione aumenta di circa due volte nei pazienti con disfunzione renale grave rispetto ai pazienti con funzionalità renale normale.
Attività anti-fattore Xa in funzione della CCr
Il trattamento con edoxadar non richiede monitoraggio regolare; l'effetto anticoagulante può essere valutato mediante un saggio quantitativo calibrato per l'attività anti-fattore Xa, utile in situazioni eccezionali in cui informazioni sull'esposizione a edoxadar possono aiutare nelle decisioni cliniche, ad esempio in caso di sovradosaggio o interventi chirurgici d'urgenza.
Dopo una sessione di emodialisi di 4 ore, si osserva una riduzione dell'esposizione totale a edoxadar inferiore al 9%.
Alterazioni della funzionalità epatica
Nei pazienti con disfunzione epatica lieve o moderata, i parametri farmacocinetici e farmacodinamici sono stati simili a quelli osservati nel gruppo di controllo di soggetti sani. L'uso di edoxadar in pazienti con disfunzione epatica grave non è stato studiato.
Sesso
L'analisi farmacocinetica di popolazione ha dimostrato che il sesso del paziente non ha un effetto aggiuntivo clinicamente significativo sulla farmacocinetica di edoxadar.
Origine etnica
L'analisi farmacocinetica di popolazione ha dimostrato che i livelli di esposizione massima e totale nei pazienti asiatici e nei pazienti non appartenenti a paesi asiatici sono comparabili.
Peso corporeo
L'analisi farmacocinetica di popolazione ha mostrato che nei pazienti con basso peso corporeo (55 kg), i valori di Cmax e AUC aumentavano rispettivamente del 40% e del 13% rispetto ai pazienti con peso corporeo medio (84 kg). In uno studio (con indicazioni per FANV e TVP), i pazienti con peso corporeo ≤ 60 kg hanno ricevuto una dose di edoxadar ridotta del 50%, mostrando un'efficacia simile e un minor numero di emorragie rispetto al trattamento con warfarin.
Rapporto farmacocinetica/farmacodinamica
PT, INR (rapporto normalizzato internazionale), aPTT e attività anti-fattore Xa correlano linearmente con la concentrazione di edoxadar.
Caratteristiche cliniche
Indicazioni
- Per la prevenzione dell’ictus e dell’embolia sistemica in adulti con fibrillazione atriale non valvolare (FANV) e uno o più fattori di rischio, come scompenso cardiaco congestizio, ipertensione arteriosa, diabete mellito, ictus o attacco ischemico transitorio (AIT) in anamnesi, età ≥ 75 anni.
- Per il trattamento della trombosi venosa profonda (TVP) e dell’embolia polmonare (EP), nonché per la prevenzione delle recidive di TVP ed EP negli adulti (vedere la sezione «Informazioni importanti sull’uso» per i pazienti con EP e instabilità emodinamica).
Controindicazioni
- Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
- Emorragia clinicamente significativa in atto.
- Malattie epatiche associate a coagulopatia e rischio emorragico clinicamente rilevante.
- Lesioni o condizioni caratterizzate da un elevato rischio di emorragia, tra cui ulcera peptica attiva o recente, presenza di neoplasie con alto rischio emorragico, trauma recente al cervello o al midollo spinale, intervento chirurgico recente al cervello, midollo spinale o occhio, emorragia intracranica recente, varici esofagee diagnosticata o sospetta, malformazioni artero-venose, aneurismi vascolari o gravi anomalie vascolari intraspinali o intracraniche.
- Ipertensione arteriosa grave non controllata.
- Associazione con altri anticoagulanti, ad esempio eparina non frazionata (ENF), eparine a basso peso molecolare (EBPM) (enoxaparina, dalteparina, ecc.), derivati dell’eparina (fondaparinux, ecc.), anticoagulanti orali (warfarin, dabigatran, rivaroxaban, apixaban, ecc.), ad eccezione di specifici casi di cambio di terapia anticoagulante (vedere la sezione «Modalità e posologia di somministrazione») o somministrazione di ENF a dosi necessarie per mantenere la pervietà di un catetere venoso centrale o arterioso.
- Gravidanza o allattamento al seno (vedere la sezione «Uso in gravidanza o durante l’allattamento»).
Interazioni con altri medicinali e altre forme di interazione
Edoxadar è principalmente assorbito nella parte superiore del tratto gastrointestinale. Pertanto, farmaci o malattie che aumentano l’evacuazione gastrica e la peristalsi intestinale possono ridurre la dissoluzione e l’assorbimento di edoxaban.
Inibitori del P-gp (glicoproteina P)
Edoxadar è un substrato del trasportatore di efflusso P-gp. In studi farmacocinetici, la somministrazione concomitante di edoxaban con inibitori del P-gp come ciclosporina, dronedarone, eritromicina, ketoconazolo, chinidina o verapamil ha portato ad un aumento della concentrazione plasmatica di edoxaban.
La somministrazione concomitante di edoxaban con ciclosporina, dronedarone, eritromicina o ketoconazolo richiede una riduzione della dose a 30 mg una volta al giorno.
La somministrazione concomitante di edoxaban con chinidina, verapamil o amiodarone non richiede riduzione della dose.
L’associazione di edoxaban con altri inibitori del P-gp, in particolare inibitori delle proteasi dell’HIV, non è stata studiata.
Edoxadar in dose di 30 mg una volta al giorno può essere somministrato concomitantemente con i seguenti inibitori del P-gp:
- Ciclosporina: la somministrazione concomitante di una dose singola di ciclosporina 500 mg con una dose singola di edoxaban 60 mg ha aumentato l’AUC e la Cmax di edoxaban.
- Dronedarone 400 mg due volte al giorno per 7 giorni con una dose singola concomitante di edoxaban 60 mg al quinto giorno ha aumentato l’AUC e la Cmax di edoxaban.
- Eritromicina 500 mg quattro volte al giorno per 8 giorni con una dose singola concomitante di edoxaban 60 mg al settimo giorno ha aumentato l’AUC e la Cmax di edoxaban.
- Ketoconazolo 400 mg una volta al giorno per 7 giorni con una dose singola concomitante di edoxaban 60 mg al quarto giorno ha aumentato l’AUC e la Cmax di edoxaban.
Edoxadar in dose di 60 mg una volta al giorno può essere somministrato concomitantemente con i seguenti inibitori del P-gp:
- Chinidina: con l’uso di chinidina 300 mg una volta al giorno al primo e al quarto giorno e tre volte al giorno al secondo e terzo giorno, insieme a una dose concomitante singola di edoxaban 60 mg al terzo giorno, si è osservato un aumento dell’AUC di edoxaban nell’arco della giornata.
- Verapamil: con l’uso di verapamil 240 mg una volta al giorno per undici giorni, insieme a una dose singola di edoxaban 60 mg al decimo giorno, si è osservato un aumento dell’AUC e della Cmax di edoxaban di circa il 53%.
- Amiodarone: la somministrazione concomitante di amiodarone 400 mg una volta al giorno con edoxaban 60 mg una volta al giorno ha determinato un aumento dell’AUC e della Cmax. Tale aumento non è stato considerato clinicamente significativo. È noto che in uno studio su pazienti con FANV, efficacia e sicurezza si sono dimostrate analoghe nei pazienti che assumevano contemporaneamente amiodarone e in quelli che non lo assumevano.
- Claritromicina: la claritromicina (500 mg due volte al giorno) per 10 giorni con una singola dose di edoxaban 60 mg al nono giorno ha aumentato l’AUC e la Cmax di edoxaban rispettivamente di circa il 53% e il 27%.
Induttori del P-gp
La somministrazione concomitante di edoxaban con l’induttore del P-gp rifampicina ha determinato una riduzione dell’AUC media di edoxaban e un accorciamento della semivita, con possibile riduzione degli effetti farmacodinamici. La somministrazione concomitante di edoxaban con altri induttori del P-gp (ad esempio fenitoina, carbamazepina, fenobarbital o Hypericum perforatum) potrebbe causare una riduzione delle concentrazioni plasmatiche di edoxaban. Si raccomanda cautela nell’uso concomitante di edoxaban con induttori del P-gp.
Substrati del P-gp
Digossina: con l’uso di edoxaban 60 mg una volta al giorno per 1–14 giorni insieme a digossina 0,25 mg due volte al giorno (all’8° e 9° giorno) e 0,25 mg una volta al giorno (all’10° e 11° giorno), si è osservato un aumento della Cmax di edoxaban del 17%, senza alcun effetto significativo sull’AUC o sul clearance renale a stato stazionario. Nello studio sull’impatto di edoxaban sulla farmacocinetica della digossina, si è osservato un aumento della Cmax della digossina di circa il 28% e dell’AUC del 7%. Tale aumento non è stato considerato clinicamente significativo. Non è necessario alcun aggiustamento della dose quando edoxaban viene somministrato concomitantemente con digossina.
Anticoagulanti, agenti antiaggreganti, farmaci antiinfiammatori non steroidei
(FANS) e inibitori selettivi della ricaptazione della serotonina / inibitori della ricaptazione della noradrenalina (SSRI/SNRI)
Anticoagulanti: la somministrazione concomitante di edoxaban con altri anticoagulanti è controindicata a causa dell’aumentato rischio di emorragia (vedere la sezione «Controindicazioni»).
ASA (acido acetilsalicilico): la somministrazione concomitante di ASA (100 mg o 325 mg) ed edoxaban aumenta il tempo di sanguinamento. La somministrazione concomitante di alte dosi di ASA (325 mg) ha determinato un aumento dei valori di Cmax e AUC di edoxaban a stato stazionario. Non è raccomandata la somministrazione concomitante a lungo termine di alte dosi di ASA ed edoxaban. La somministrazione concomitante di dosi di ASA superiori a 100 mg deve avvenire esclusivamente sotto controllo medico.
Negli studi clinici era consentita la somministrazione concomitante di ASA (a basse dosi, ≤ 100 mg/giorno), altri agenti antiaggreganti e tienopiridinici, con un aumento del rischio di emorragie maggiori di circa due volte rispetto alla monoterapia, con tassi simili di aumento sia nel gruppo edoxaban che in quello warfarin. La somministrazione concomitante di basse dosi di ASA non ha influenzato l’effetto massimo o totale di edoxaban dopo una dose singola.
Edoxadar può essere somministrato concomitantemente con basse dosi di ASA.
Inibitori dell’aggregazione piastrinica: in uno studio, la somministrazione concomitante di monoterapia con tienopiridinici (ad esempio clopidogrel) ha determinato un aumento della frequenza di emorragie clinicamente significative, sebbene con edoxaban il rischio fosse relativamente inferiore rispetto a warfarin.
L’esperienza con l’uso di edoxaban in combinazione con doppia terapia antiaggregante o con fibrinolitici è molto limitata.
FANS: la somministrazione concomitante di naprossene ed edoxaban prolunga il tempo di sanguinamento rispetto alla monoterapia con ciascun farmaco. Il naprossene non influisce sulla Cmax e sull’AUC di edoxaban. È noto che negli studi clinici l’uso concomitante di FANS ha determinato un aumento del numero di emorragie clinicamente significative. Non è raccomandato l’uso prolungato di FANS con edoxaban.
SSRI/SNRI: come con altri anticoagulanti, il rischio di emorragia aumenta nei pazienti che assumono concomitantemente SSRI o SNRI, a causa dell’effetto di questi ultimi sulla funzione piastrinica (vedere la sezione «Informazioni importanti sull’uso»).
Caratteristiche di impiego
Edoxabano 15 mg non deve essere utilizzato come monoterapia, poiché ciò potrebbe portare a una riduzione dell'efficacia. Questo dosaggio è indicato esclusivamente nel passaggio da edoxabano 30 mg [con uno o più fattori clinici che aumentano l'esposizione (vedere tabella 1)] ad AVK (antagonista della vitamina K), in associazione con la dose appropriata di AVK (vedere tabella 2, sezione «Modalità di somministrazione e posologia»).
Rischio di emorragia
L’edoxabano aumenta il rischio di emorragia e può causare emorragie gravi, potenzialmente letali. Come per tutti gli anticoagulanti, si raccomanda cautela nell’uso di edoxabano nei pazienti con condizioni associate a un aumentato rischio di emorragia. In caso di emorragia grave, il trattamento deve essere interrotto.
Negli studi clinici è noto che emorragie delle mucose (ad esempio epistassi, emorragie gastrointestinali, emorragie del tratto urinario e genitale) e anemia si verificano più frequentemente con terapie prolungate con edoxabano rispetto al trattamento con AVK. Per questo motivo, oltre a un adeguato monitoraggio clinico, in alcuni casi è opportuno effettuare controlli di laboratorio degli indici di emoglobina/ematocrito per rilevare emorragie occulte.
Alcune categorie di pazienti, come descritto di seguito, presentano un rischio aumentato di emorragia. Tali pazienti devono essere sottoposti a un attento monitoraggio dopo l’inizio del trattamento per rilevare sintomi di complicanze emorragiche e anemia. In caso di qualsiasi calo dei livelli di emoglobina o della pressione arteriosa di eziologia sconosciuta, è necessario identificare la fonte dell’emorragia.
L’effetto anticoagulante dell’edoxabano non può essere controllato in modo affidabile mediante esami di laboratorio standard. Non esiste un agente specifico per l’inversione dell’effetto anticoagulante dell’edoxabano.
L’emodialisi non influenza in modo significativo l’eliminazione dell’edoxabano.
Pazienti anziani
L’uso concomitante di edoxabano e ASA (acido acetilsalicilico) nei pazienti anziani richiede cautela a causa del rischio aumentato di emorragia.
Pazienti con compromissione renale
L’AUC plasmatica nei pazienti con compromissione renale lieve (clearance della creatinina > 50–80 ml/min), moderata (clearance della creatinina 30–50 ml/min) e grave (clearance della creatinina < 30 ml/min, senza dialisi) è aumentata rispetto ai pazienti con funzione renale normale (vedere sezione «Modalità di somministrazione e posologia»).
Non è raccomandato il trattamento con edoxabano nei pazienti con clearance della creatinina < 15 ml/min o nei pazienti sottoposti a dialisi.
Funzione renale in caso di FAN
Con l’uso di edoxabano si è osservata una tendenza alla riduzione dell’efficacia all’aumentare della clearance della creatinina, rispetto all’uso appropriato di warfarin. Pertanto, nei pazienti con FAN e alta clearance della creatinina, l’edoxabano deve essere utilizzato solo dopo un’attenta valutazione del rischio individuale di tromboembolia e di emorragia.
Valutazione della funzione renale: il controllo della clearance della creatinina deve essere effettuato all’inizio del trattamento in tutti i pazienti e successivamente in base alle indicazioni cliniche.
Pazienti con compromissione epatica
L’edoxabano non è raccomandato nei pazienti con gravi alterazioni della funzione epatica.
L’edoxabano deve essere usato con cautela nei pazienti con insufficienza epatica lieve o moderata.
I pazienti con livelli elevati degli enzimi epatici (alanina aminotransferasi / aspartato aminotransferasi (ALT/AST) superiori al limite superiore della norma di oltre 2 volte) o con livelli di bilirubina totale superiori al limite superiore della norma di 1,5 volte o più non sono stati inclusi negli studi clinici. Pertanto, l’edoxabano deve essere usato con cautela nel trattamento di questa popolazione di pazienti. Prima di iniziare il trattamento con edoxabano è necessario eseguire un esame della funzionalità epatica. Si raccomanda di effettuare controlli periodici della funzionalità epatica nei pazienti trattati con edoxabano per oltre un anno.
Sospensione del trattamento per interventi chirurgici e altre procedure
Se la terapia anticoagulante deve essere sospesa per ridurre il rischio di emorragia durante interventi chirurgici o altre procedure, l’assunzione di edoxabano deve essere interrotta il più presto possibile, almeno 24 ore prima dell’intervento.
Se l’intervento non può essere rinviato, si deve valutare la presenza di un rischio aumentato di emorragia e l’urgenza dell’intervento. Il trattamento con edoxabano deve essere ripreso dopo l’intervento chirurgico o altre procedure, non appena raggiunto un emostasi adeguato, tenendo presente che l’effetto anticoagulante di edoxabano si manifesta entro 1–2 ore dalla somministrazione.
Se durante o dopo un intervento chirurgico non è possibile assumere farmaci per via orale, si deve considerare la possibilità di somministrare un anticoagulante per via parenterale, seguito da un passaggio alla somministrazione orale di edoxabano una volta al giorno.
Interazioni con altri farmaci che influenzano la coagulazione
L’uso concomitante di farmaci che influenzano la coagulazione aumenta il rischio di emorragia. Tra questi farmaci rientrano ASA, inibitori dell’aggregazione piastrinica, altri agenti antiaggreganti, fibrinolitici, SSRI e FANS con uso prolungato (vedere sezione «Interazioni con altri farmaci e altre forme di interazione»).
Valvole cardiache artificiali e stenosi mitralica di grado moderato o grave
L’effetto di edoxabano non è stato studiato nei pazienti con valvole cardiache artificiali, nei pazienti nei primi 3 mesi dopo l’impianto di una valvola bioprostetica, con o senza fibrillazione atriale, né nei pazienti con stenosi mitralica di grado moderato o grave. Pertanto, l’uso di edoxabano per il trattamento di tali pazienti non è raccomandato.
Pazienti con EP e instabilità emodinamica o pazienti che richiedono trombolisi o embolectomia polmonare
L’edoxabano non è raccomandato come alternativa all’eparina non frazionata (ENF) nei pazienti con EP e instabilità emodinamica o che potrebbero richiedere trombolisi o embolectomia polmonare, poiché la sicurezza e l’efficacia di edoxabano in queste situazioni cliniche non sono state stabilite.
Pazienti con cancro attivo
L’efficacia e la sicurezza di edoxabano nel trattamento e/o nella prevenzione della TVP in pazienti con cancro attivo non sono state stabilite.
Pazienti con sindrome da anticorpi antifosfolipidi
Non è raccomandato l’uso di anticoagulanti orali diretti, inclusa edoxabano, nei pazienti con trombosi anamnestica e sindrome da anticorpi antifosfolipidi diagnosticata. In particolare, nei pazienti con risultati positivi confermati per tutti e tre gli anticorpi antifosfolipidi (anticoagulante lupico, anticorpi anti-cardiolipina, anticorpi anti-beta-2-glicoproteina I), la terapia con anticoagulanti orali diretti aumenta il rischio di eventi trombotici ricorrenti rispetto alla terapia con AVK.
Parametri di laboratorio
Sebbene il trattamento con edoxabano non richieda un monitoraggio regolare, l’effetto anticoagulante può essere valutato mediante un dosaggio quantitativo calibrato dell’attività anti-fattore Xa, i cui risultati possono essere utili per decisioni cliniche in determinate situazioni, ad esempio in caso di sovradosaggio o intervento chirurgico d’urgenza.
L’edoxabano prolunga i valori dei test standard di coagulazione come il tempo di protrombina (PT), il rapporto normalizzato internazionale (INR) e il tempo di tromboplastina parziale attivato (aPTT) a causa dell’inibizione del fattore Xa. Tuttavia, le variazioni di questi test in seguito all’assunzione del farmaco nell’intervallo terapeutico previsto sono minime, caratterizzate da un’elevata variabilità e non sono utili per il monitoraggio dell’effetto anticoagulante di edoxabano.
Informazioni importanti sugli eccipienti
Il medicinale contiene glucosio; pertanto, i pazienti con diagnosticata intolleranza ad alcuni zuccheri devono consultare il medico prima di assumere questo medicinale.
Uso durante la gravidanza o l’allattamento
Effetto sulla funzione riproduttiva. Le donne in età fertile devono evitare la gravidanza durante il trattamento con edoxabano.
Gravidanza. L’efficacia e la sicurezza dell’uso di edoxabano nelle donne in gravidanza non sono state studiate. Gli studi sugli animali hanno dimostrato tossicità riproduttiva. A causa della potenziale tossicità riproduttiva, del rischio significativo di emorragia e del passaggio di edoxabano attraverso la barriera placentare, il medicinale Edoxadara è controindicato durante la gravidanza (vedere sezione «Controindicazioni»).
Allattamento. L’efficacia e la sicurezza dell’uso di edoxabano nelle donne durante l’allattamento non sono state studiate. Negli studi sugli animali è stato dimostrato che edoxabano passa nel latte materno. Pertanto, il medicinale Edoxadara è controindicato durante l’allattamento (vedere sezione «Controindicazioni»). È necessario prendere una decisione riguardo all’interruzione dell’allattamento o all’interruzione/astensione dal trattamento.
Fertilità. Non sono stati condotti studi specifici per valutare l’effetto di edoxabano sulla fertilità umana. Negli studi sulla fertilità condotti su ratti maschi e femmine non è stato osservato alcun effetto.
Capacità di influenzare la velocità di reazione nella guida di veicoli o nell’uso di macchinari
Il medicinale Edoxadara non ha alcun effetto o ha un effetto trascurabile sulla capacità di guidare veicoli o di usare macchinari.
Modalità e posologia di somministrazione
Prevenzione dell'ictus e dell'embolia sistemica
La dose raccomandata è di 60 mg di edoxaban una volta al giorno.
La terapia con edoxaban nei pazienti con FA non valvolare viene effettuata a lungo termine.
Trattamento della TVP e dell'EP e prevenzione delle recidive di TVP ed EP (VTE)
La dose raccomandata è di 60 mg di edoxaban una volta al giorno, dopo un trattamento iniziale parenterale con anticoagulante per almeno 5 giorni. Non somministrare contemporaneamente edoxaban e un anticoagulante parenterale.
La durata del trattamento della TVP e dell'EP (VTE) e della prevenzione delle recidive di VTE deve essere stabilita individualmente, dopo un'attenta valutazione del rapporto beneficio/rischio emorragico. Un trattamento a breve termine (almeno per 3 mesi) è indicato in presenza di fattori di rischio transitori (ad esempio intervento chirurgico recente, trauma, immobilizzazione), mentre un trattamento più prolungato deve essere prescritto in caso di fattori di rischio persistenti o in caso di TVP o EP idiopatica.
La dose raccomandata nei pazienti con FA non valvolare e VTE è di 30 mg di edoxaban una volta al giorno nei pazienti con uno o più dei seguenti fattori clinici (vedere anche tabella 1):
- insufficienza renale di grado moderato o grave (clearance della creatinina 15–50 ml/min);
- basso peso corporeo: ≤ 60 kg;
- co-somministrazione con inibitori del glicoproteina P (P-gp): ciclosporina, dronedarone, eritromicina o ketoconazolo.
Tabella 1
Riepilogo delle indicazioni posologiche in FA non valvolare e VTE (TVP ed EP)
| Indicazioni generali per il dosaggio |
|
| Dose raccomandata |
60 mg di edoxaban una volta al giorno |
| Indicazioni per il dosaggio nei pazienti con uno o più dei seguenti fattori clinici: |
|
| insufficienza renale di grado moderato o grave (clearance della creatinina 15-50 ml/min) |
30 mg una volta al giorno |
| basso peso corporeo: ≤ 60 kg |
|
| inibitori della glicoproteina P: ciclosporina, dronedarone, eritromicina, ketoconazolo |
|
Salto della dose
In caso di salto della dose di edoxaban, tale dose deve essere assunta immediatamente e il giorno successivo si deve continuare il trattamento secondo il regime di una dose al giorno, come raccomandato. Non si deve assumere una dose doppia entro lo stesso giorno per compensare la dose dimenticata.
Passaggio all’edoxaban e passaggio dall’edoxaban
I pazienti con FA e TVP devono ricevere una terapia anticoagulante continua. In alcune situazioni può rendersi necessario sostituire la terapia anticoagulante (tabella 2).
Tabella 2
Sostituzione dei trattamenti anticoagulanti in caso di FA e TVP (TVP e EP)
Passaggio all’edoxaban
| Medicamento attuale |
Raccomandazioni |
| Antagonista della vitamina K (AVK) |
Interrompere l'uso dell'AVK e iniziare l'uso di Edoxadar quando si raggiunge un valore di INR ≤ 2,5 |
| Altri anticoagulanti orali, eccetto AVK
|
Interrompere l'assunzione di dabigatran, rivarossaban o apixaban e iniziare l'assunzione di Edoxadar nel momento in cui sarebbe stata assunta la dose successiva dell'anticoagulante orale |
| Anticoagulanti parenterali |
Non somministrare questi medicinali contemporaneamente. Anticoagulanti sottocutanei (ad esempio, Eparina a basso peso molecolare, fondaparinux): interrompere la somministrazione sottocutanea dell'anticoagulante e iniziare l'assunzione di Edoxadar nel momento in cui era programmata la successiva iniezione sottocutanea. Eparina non frazionata endovenosa: interrompere l'infusione e iniziare l'assunzione di Edoxadar dopo 4 ore. |
Conversione da edoxaban
| Medicinale da cui si passa |
Indicazioni |
| AVK |
Esiste la possibilità di un effetto anticoagulante inadeguato durante il passaggio da edoxaban ad AVK. Quando si passa a un anticoagulante alternativo, è necessario garantire un'adeguata anticoagulazione. Assunzione orale: ai pazienti che assumono una dose di 60 mg si deve somministrare una dose di edoxaban di 30 mg una volta al giorno insieme alla dose appropriata di AVK. Ai pazienti che assumono una dose di 30 mg (a causa della presenza di uno o più dei seguenti fattori clinici: insufficienza renale di grado moderato o grave, basso peso corporeo o assunzione concomitante di specifici inibitori della P-gp) deve essere somministrata una dose di edoxaban di 15 mg una volta al giorno insieme alla dose appropriata di AVK. I pazienti non devono assumere una dose di carico di AVK al fine di raggiungere rapidamente un valore stabile dell'INR compreso tra 2 e 3. Si raccomanda di prendere in considerazione la possibilità di utilizzare una dose di mantenimento di AVK e di considerare se il paziente ha precedentemente assunto AVK, oppure di utilizzare un algoritmo terapeutico affidabile con AVK basato sui valori dell'INR, in conformità con la pratica locale accettata. Dopo aver raggiunto un valore dell'INR ≥ 2,0, l'edoxaban deve essere sospeso. La maggior parte dei pazienti (85 %) raggiunge un valore dell'INR ≥ 2,0 entro 14 giorni di terapia concomitante con edoxaban e AVK. Dopo 14 giorni si raccomanda di sospendere l'edoxaban e continuare l'aggiustamento della dose di AVK per raggiungere un valore dell'INR compreso tra 2 e 3. Nei primi 14 giorni di terapia concomitante si raccomanda di misurare l'INR almeno 3 volte immediatamente prima dell'assunzione della dose giornaliera di edoxaban, per minimizzare l'effetto dell'edoxaban sui valori misurati dell'INR. L'assunzione concomitante di edoxaban e AVK può aumentare i valori dell'INR dopo l'assunzione della dose di edoxaban di circa il 46 %. Somministrazione parenterale: interrompere l'assunzione di edoxaban e somministrare un anticoagulante parenterale e AVK nel momento previsto per l'assunzione della successiva dose programmata di edoxaban. Dopo aver raggiunto un valore stabile dell'INR ≥ 2,0, l'anticoagulante parenterale deve essere sospeso e la terapia con AVK deve essere continuata. |
| Altri anticoagulanti orali, diversi dagli AVK |
Interrompere l'assunzione di edoxaban e iniziare la terapia con un altro anticoagulante orale non appartenente alla classe degli AVK nel momento previsto per l'assunzione della successiva dose programmata di edoxaban. |
| Anticoagulanti parenterali |
Non somministrare questi medicinali contemporaneamente. È necessario interrompere l'assunzione di edoxaban e iniziare la somministrazione di un anticoagulante parenterale durante il momento previsto per l'assunzione della successiva dose programmata di edoxaban. |
Gruppi particolari di pazienti
Pazienti anziani
Non è necessaria alcuna modifica della dose (vedere il paragrafo «Farmacocinetica»).
Compromissione renale
Prima di iniziare il trattamento con edoxaban, è necessario valutare la funzionalità renale in tutti i pazienti mediante la determinazione della clearance della creatinina (CCr), al fine di evitare l’uso di edoxaban nei pazienti con insufficienza renale allo stadio terminale (CCr < 15 ml/min), stabilire la dose appropriata di edoxaban per i pazienti con CCr 15–50 ml/min (30 mg una volta al giorno), per i pazienti con CCr > 50 ml/min (60 mg una volta al giorno), nonché valutare l’opportunità dell’uso di edoxaban nei pazienti con valori elevati di CCr.
La valutazione della funzionalità renale deve essere effettuata anche quando ci si aspetta un cambiamento della funzionalità renale durante il trattamento (ad esempio in caso di ipovolemia, disidratazione o trattamento concomitante con alcuni farmaci).
La funzionalità renale (CCr in ml/min) si calcola mediante la formula di Cockcroft-Gault riportata di seguito:
Per ottenere i valori di creatinina in µmol/l:
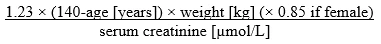
Per ottenere i valori di creatinina in mg/dl:

Questo metodo è raccomandato per la valutazione della CCr nei pazienti prima e durante il trattamento con edoxaban.
Per i pazienti con compromissione renale lieve (CCr > 50–80 ml/min), la dose raccomandata è di 60 mg di edoxaban una volta al giorno.
Per i pazienti con compromissione renale moderata o grave (CCr 15–50 ml/min), la dose raccomandata è di 30 mg di edoxaban una volta al giorno.
Non è raccomandato l’uso di questo medicinale nei pazienti con insufficienza renale allo stadio terminale (CCr < 15 ml/min) o nei pazienti sottoposti a dialisi.
Compromissione epatica
L’edoxaban è controindicato nei pazienti con malattie epatiche associate a coagulopatia e rischio clinicamente significativo di emorragia (vedere il paragrafo «Controindicazioni»).
L’edoxaban non è raccomandato per il trattamento di pazienti con compromissione epatica grave.
L’edoxaban deve essere usato con cautela nei pazienti con compromissione epatica lieve o moderata; per questi pazienti la dose raccomandata di edoxaban è di 60 mg una volta al giorno.
Pazienti con livelli elevati di enzimi epatici (alanina aminotransferasi (ALT) o aspartato aminotransferasi (AST) superiori a due volte il limite superiore della norma (LSN)) o bilirubina totale (superiore a 1,5 volte il LSN) non sono stati inclusi negli studi clinici. Pertanto, il medicinale Edoxadar deve essere usato con cautela nel trattamento di questo gruppo di pazienti. Prima di iniziare il trattamento con edoxaban, è necessario effettuare un esame della funzionalità epatica.
Peso corporeo
Per i pazienti con peso corporeo ≤ 60 kg, la dose raccomandata è di 30 mg di edoxaban una volta al giorno.
Sesso
Non è necessaria alcuna modifica della dose (vedere il paragrafo «Farmacocinetica»).
Uso concomitante di Edoxadar e inibitori della glicoproteina P (P-gp)
Per i pazienti che assumono contemporaneamente edoxaban e inibitori della P-gp come ciclosporina, dronedarone, eritromicina o ketoconazolo, la dose raccomandata di edoxaban è di 30 mg una volta al giorno. Quando si assume amiodarone, chinidina o verapamil, non è necessario ridurre la dose.
L’uso concomitante di edoxaban con altri inibitori della P-gp, inclusi gli inibitori della proteasi dell’HIV, non è stato studiato.
Pazienti sottoposti a cardioversione
È consentito iniziare o continuare il trattamento con il medicinale Edoxadar nei pazienti che potrebbero necessitare di cardioversione.
Nel caso di cardioversione guidata da ecocardiografia transesofagea in pazienti che in precedenza non avevano ricevuto anticoagulanti, il trattamento con Edoxadar deve essere iniziato almeno 2 ore prima della cardioversione per garantire un livello adeguato di anticoagulazione. La cardioversione deve essere eseguita non oltre 12 ore dopo l’assunzione della dose di edoxaban nel giorno della procedura.
Per tutti i pazienti sottoposti a cardioversione: prima della cardioversione è necessario confermare che abbiano assunto edoxaban secondo prescrizione. Nella decisione riguardo all’inizio e alla durata del trattamento, è necessario seguire le raccomandazioni sull’uso degli anticoagulanti nei pazienti sottoposti a cardioversione.
Modalità di somministrazione
Per uso orale.
Edoxadar può essere assunto con o senza cibo.
Popolazione pediatrica
La sicurezza e l’efficacia di questo medicinale nei bambini e negli adolescenti di età inferiore a 18 anni non sono state stabilite. I dati non sono disponibili.
Sovradosaggio
Un sovradosaggio di edoxaban può causare emorragia. L’esperienza relativa a casi di sovradosaggio è molto limitata.
Non esiste un antidoto specifico in grado di neutralizzare l’effetto farmacodinamico dell’edoxaban.
In caso di sovradosaggio di edoxaban, per ridurre l’assorbimento, si può considerare un precoce utilizzo di carbone attivo. Questa raccomandazione si basa sul trattamento standard del sovradosaggio di farmaci e sui dati disponibili su composti simili, poiché l’uso di carbone attivo per ridurre l’assorbimento di edoxaban non è stato specificamente studiato.
Trattamento dell’emorragia. In caso di complicanze emorragiche, si deve sospendere la somministrazione della dose successiva di edoxaban o interrompere il trattamento. L’emivita di eliminazione dell’edoxaban è di circa 10–14 ore. Il trattamento deve essere personalizzato in base all’intensità e alla localizzazione dell’emorragia. Se necessario, si deve effettuare un trattamento sintomatico adeguato, ad esempio compressione meccanica in caso di epistassi grave, emostasi chirurgica con procedure di controllo dell’emorragia, ripristino dell’equilibrio idroelettrolitico e supporto emodinamico, trasfusione di sangue (concentrato di eritrociti o plasma fresco congelato, a seconda della condizione insorta: anemia o coagulopatia) o piastrine.
Poiché emorragie potenzialmente letali non possono essere controllate con interventi come trasfusioni o emostasi, è stato dimostrato che l’infusione di un concentrato del complesso protrombinico a 4 fattori alla dose di 50 UI/kg neutralizza l’effetto di edoxaban entro 30 minuti dal termine dell’infusione.
Può essere preso in considerazione anche l’uso del fattore VIIa ricombinante (r-FVIIa). Tuttavia, l’esperienza clinica con questo farmaco in pazienti in trattamento con edoxaban è limitata.
In caso di emorragia significativa, potrebbe essere necessaria una consulenza ematologica.
Il protammina solfato e la vitamina K non dovrebbero influenzare l’attività anticoagulante di edoxaban.
Non esiste esperienza nell’uso di agenti antifibrinolitici (acido tranexamico, acido aminocaproico) in pazienti in trattamento con edoxaban. Non vi è alcuna giustificazione scientifica circa l’utilità, né esperienza nell’uso di emostatici sistemici (desmopressina, aprotinina) in pazienti in trattamento con edoxaban. Data l’elevata legatura alle proteine plasmatiche, si prevede che l’edoxaban non venga eliminato dall’organismo mediante dialisi.
Effetti indesiderati
Gli effetti indesiderati più comuni riportati con edoxaban sono stati epistassi, ematuria e anemia. Il sanguinamento può verificarsi in qualsiasi sede e può essere grave, anche fatale.
Gli effetti indesiderati riportati di seguito sono classificati per sistemi e organi (secondo la classificazione MedDRA) e per frequenza: molto comune (≥1/10); comune (≥1/100 – <1/10); non comune (≥1/1000 – <1/100); raro (≥1/10000 – <1/1000); molto raro (<1/10000); frequenza non nota (non può essere stimata sulla base dei dati disponibili).
Apparato emolinfopoietico: comune – anemia; non comune – trombocitopenia.
Apparato immunitario: raro – reazioni anafilattiche, angioedema; non comune – ipersensibilità.
Sistema nervoso: comune – capogiri, cefalea; non comune – emorragia intracerebrale; rado – emorragia subaracnoidea.
Organi della vista: non comune – emorragia oculare, emorragie congiuntivali/sclerali.
Apparato cardiaco: raro – emorragia nella cavità pericardica.
Apparato vascolare: non comune – altri sanguinamenti.
Apparato respiratorio, torace e mediastino: comune – epistassi; non comune – emottisi.
Apparato gastrointestinale: comune – dolore addominale, sanguinamenti del tratto gastrointestinale superiore e inferiore, sanguinamenti orali/faringei, nausea; rado – emorragia peritoneale.
Apparato epatobiliare: comune – aumento dei livelli ematici di bilirubina e gamma-glutamiltransferasi; non comune – aumento dei livelli ematici di fosfatasi alcalina e transaminasi.
Pelle e tessuto sottocutaneo: comune – emorragia cutanea nei tessuti molli; eruzione cutanea, prurito; non comune – orticaria.
Apparato muscoloscheletrico e tessuto connettivo: raro – emorragia muscolare (senza sindrome da compartimento), emorragia intra-articolare.
Apparato urinario: comune – ematuria macroscopica/emorragia uretrale; frequenza non nota – nefropatia da anticoagulante.
Apparato riproduttivo e ghiandole mammarie: comune – emorragia vaginale¹.
Condizioni generali e reazioni nel sito di somministrazione: comune – sanguinamento nel sito di somministrazione del farmaco.
Esami di laboratorio: comune – alterazioni dei parametri delle prove di funzionalità epatica.
Traumi, avvelenamenti e complicanze procedurali: non comune – sanguinamenti in sede di interventi chirurgici; rado – emorragia subdurale.
¹Le emorragie vaginali si sono verificate principalmente in donne sotto i 50 anni e sono state rare in donne sopra i 50 anni (dati degli studi).
Descrizione di specifici effetti indesiderati
Anemia emorragica
L’uso di edoxaban aumenta il rischio di sanguinamenti interni o esterni in qualsiasi tessuto o organo, che possono portare ad anemia post-emorragica. I segni, i sintomi e la gravità (incluso l’esito fatale) variano in base alla localizzazione e all’intensità del sanguinamento e/o dell’anemia (vedere la sezione «Sovradosaggio»). Dagli studi è emerso che i sanguinamenti delle mucose (ad esempio epistassi, sanguinamenti gastrointestinali, sanguinamenti degli organi del tratto urinario e genitale) e l’anemia si verificano più frequentemente con il trattamento prolungato con edoxaban rispetto al trattamento con AVK. Per questo motivo, oltre al monitoraggio clinico appropriato, in casi appropriati si raccomanda il controllo di laboratorio di emoglobina/ematocrito per rilevare sanguinamenti interni. In alcuni gruppi di pazienti il rischio di sanguinamento può essere maggiore, ad esempio in pazienti con ipertensione arteriosa grave non controllata e/o in pazienti che assumono contemporaneamente un medicinale che influenza la coagulazione del sangue. Può aumentare l’intensità e/o la durata delle emorragie mestruali. I segni di complicanze emorragiche possono includere debolezza, pallore, capogiri, cefalea, edema di origine sconosciuta, dispnea, shock di origine sconosciuta.
Sono stati riportati complicanze note come conseguenze di sanguinamenti gravi, come la sindrome da compartimento e l’insufficienza renale secondaria a ipoperfusione. Pertanto, nella valutazione del paziente a cui viene prescritto un anticoagulante, va considerato il rischio di sanguinamento.
Segnalazione di sospetti effetti indesiderati
La segnalazione di sospetti effetti indesiderati dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di effetti indesiderati e/o di mancata efficacia del medicinale attraverso il Sistema Informatizzato Automatizzato di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità. 2 anni.
Condizioni di conservazione
Conservare nella confezione originale a una temperatura non superiore a 25 °C.
Tenere fuori dalla portata dei bambini.
Confezionamento
10 compresse in un blister; 3 blister in una confezione.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore.
Faros MT Limited
Pharos Pharmaceutical Oriented Services Ltd.
Sede del produttore e indirizzo del luogo di esercizio dell’attività
XF62Ex, Casam Industries Hell Farr, Hell Farr, Birżebbuġa, BBG 3000, Malta.
Lesvou Street End, Tese Loggos Industrial Zone, Metamorfossi, 144 52, Grecia.
Titolare dell’autorizzazione all’immissione in commercio
PJSC «Pharmaceutical Company “Darnitsa».
Sede del titolare dell’autorizzazione all’immissione in commercio
Ucraina, 02093, Kiev, via Borispol’ska, 13.