Imunat
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO IMUNAT (IMMUNATE)
Composición:
principios activos: factor VIII de coagulación sanguínea humana, factor de von Willebrand (vWF:RCo);
1 frasco con polvo contiene:
| Sustancia activa |
Contenido de sustancia por frasco |
||
| 250/190 U.I. |
500/375 U.I. |
1000/750 U.I. |
|
| Factor de coagulación sanguínea humano VIII (corregido por contenido de albúmina)* |
250 U.I. (50 U.I./ml) |
500 U.I. (100 U.I./ml) |
1 000 U.I. (100 U.I./ml) |
| Actividad específica** |
(70 ± 30) U.I./mg de proteína |
||
| Factor de von Willebrand (vWF:RCo)*** |
190 U.I. (38 U.I./ml) |
375 U.I. (75 U.I./ml) |
750 U.I. (75 U.I./ml) |
disolvente: agua para inyección – 5 ml (para 250/190 UI y 500/375 UI) y 10 ml (para 1000/750 UI);
excipientes: albúmina humana, glicina, clorhidrato de lisina, cloruro de sodio, citrato trisódico dihidrato, cloruro de calcio dihidrato.
____________________________________________________________________
* La actividad del factor VIII se determinó frente al estándar internacional de la OMS para concentrados de factor VIII.
** Sin estabilizante (albúmina).
La actividad específica máxima en una relación factor VIII:antígeno del factor de von Willebrand de 1:1 es de 100 UI por mg de proteína.
*** La actividad del factor de von Willebrand se determinó frente al estándar internacional de la OMS para concentrados de factor VIII y factor de von Willebrand en plasma.
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Propiedades físico-químicas principales: polvo o sustancia frágil de color blanco o amarillo pálido.
Grupo farmacoterapéutico. Agentes hemostáticos. Factores de coagulación sanguínea. Factor de von Willebrand en combinación con el factor de coagulación VIII.
Código ATC B02B D06.
Propiedades inmunológicas y biológicas.
Farmacodinamia.
El complejo factor VIII/factor de von Willebrand está compuesto por dos moléculas (factor de coagulación sanguínea VIII y factor de von Willebrand) que tienen funciones fisiológicas diferentes.
El factor VIII activado actúa como cofactor del factor IX activado, acelerando la transformación del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Posteriormente, la trombina convierte el fibrinógeno en fibrina, formándose así el coágulo de fibrina. La hemofilia A es un trastorno hereditario ligado al sexo en la coagulación sanguínea debido a niveles reducidos de actividad del factor VIII, lo que provoca hemorragias abundantes en articulaciones, músculos y órganos internos, que aparecen espontáneamente o como consecuencia de traumatismos accidentales o quirúrgicos. Los niveles plasmáticos de factor VIII se incrementan mediante terapia sustitutiva, que corrige temporalmente el déficit del factor y la tendencia a sangrar.
Además del papel protector del factor VIII como proteína, el factor de von Willebrand (FVW) media la adhesión de las plaquetas a los sitios de daño vascular y desempeña un papel en la agregación plaquetaria, siendo esencial para la terapia sustitutiva en pacientes con enfermedad de von Willebrand.
Farmacocinética.
Los parámetros farmacocinéticos obtenidos a partir de un estudio farmacocinético en pacientes mayores de 12 años se muestran en las tablas 1 y 2.
La tabla 1 describe las propiedades farmacocinéticas respecto al factor VIII de coagulación.
| Parámetro |
|||||
| Número de pacientes |
Valor medio |
Desviación estándar |
Mediana |
IC del 90 % |
|
| AUC0–48 h ([MO × h]/ml) |
18 |
11,4 |
2,8 |
11,6 |
10,9–12,7 |
| AUC0–∞ h ([MO × h]/ml) |
18 |
12,2 |
3,1 |
12,4 |
11,1–13,2 |
| Cmax (MO/ml) |
18 |
1,0 |
0,3 |
0,9 |
0,8–1,0 |
| Tmax (h) |
18 |
0,3 |
0,1 |
0,3 |
0,3–0,3 |
| Período de semivida terminal (h) |
18 |
12,7 |
3,2 |
12,2 |
10,8–15,3 |
| Depuración (ml/h) |
18 |
283 |
146 |
232 |
199–254 |
| Tiempo medio de retención del fármaco (h) |
18 |
15,3 |
3,6 |
15,3 |
12,1–17,2 |
| Vss (ml) |
18 |
4166 |
2021 |
3613 |
2815–4034 |
| Incremento de recuperación del nivel de actividad ([MO/ml]/[MO/kg]) |
18 |
0,020 |
0,006 |
0,019 |
0,016–0,020 |
Tabla 1
En la tabla 2 se describen las propiedades farmacocinéticas según el antígeno FB.
Tabla 2
| Parámetro |
|||||
| Número de pacientes |
Mediana |
IC del 90 % |
|||
| AUC0–∝([MO × h]/ml) |
15 |
24,6 |
12,8–48,3 |
||
| Cmax (MO/ml) |
17 |
1,40 |
1,15–1,51 |
||
| Tmax (h) |
17 |
0,28 |
0,25–1,00 |
||
| Período de semivida terminal (h) |
16 |
13,6 |
10,5–47,2 |
||
| Depuración (ml/h) |
15 |
136 |
68–178 |
||
| Tiempo medio de retención del fármaco (h) |
15 |
23,1 |
12,4–57,1 |
||
| Vss (ml) |
15 |
3156 |
2391–4672 |
||
| Incremento de recuperación del nivel de actividad ([MO/ml]/[MO/kg]) |
17 |
0,028 |
0,024–0,030 |
||
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit congénito del factor VIII, hemofilia A con inhibidor del factor VIII, déficit adquirido del factor VIII debido a la aparición espontánea de inhibidores del factor VIII).
Enfermedad de von Willebrand con déficit del factor VIII.
Nota. La eficacia y seguridad del medicamento Inmunat en el síndrome de von Willebrand-Jürgens han sido investigadas clínicamente solo en un número reducido de pacientes. Esto debe tenerse especialmente en cuenta en el tipo 3 de esta enfermedad.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento.
Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios sobre interacciones con el medicamento Inmunat. No se han notificado interacciones específicas del factor de coagulación sanguínea humana VIII con otros medicamentos.
Características de uso.
Como con todos los medicamentos administrados por vía intravenosa, pueden presentarse reacciones de hipersensibilidad de tipo alérgico. Los pacientes deben informarse sobre los signos tempranos de reacciones de hipersensibilidad, en particular hipotensión, taquicardia, dolor torácico, disnea, edema (especialmente edema facial y de párpados), urticaria, erupciones cutáneas, hiperemia y prurito, así como anafilaxia hasta anafilaxia con shock.
Ante la aparición de síntomas de hipersensibilidad, se debe recomendar a los pacientes que interrumpan inmediatamente la administración del medicamento y consulten a su médico. En caso de presentarse shock, se debe aplicar el tratamiento médico estándar para el shock.
También se han notificado otras reacciones relacionadas con la infusión de Immunate, tales como escalofríos, fiebre o náuseas.
Dado que la cantidad de sodio en la dosis diaria máxima puede superar los 200 mg, esto debe tenerse en cuenta en personas que siguen una dieta baja en sodio.
Frascos de 250 y 500 UI
Immunate contiene aproximadamente 9,8 mg de sodio por frasco. Esto equivale al 0,5 % de la ingesta diaria máxima recomendada de sodio de 2 g por la OMS para adultos.
Frascos de 1000 UI
Immunate contiene aproximadamente 19,6 mg de sodio por frasco. Esto equivale al 1 % de la ingesta diaria máxima recomendada de sodio de 2 g por la OMS para adultos.
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida durante el tratamiento de pacientes con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad anticoagulante del factor VIII, cuya concentración se determina cuantitativamente en unidades de Bethesda (UB) por 1 ml de plasma sanguíneo mediante un análisis modificado. El riesgo de desarrollo de inhibidores se correlaciona con la gravedad de la enfermedad y con la exposición al factor VIII. Este riesgo es más alto durante los primeros 50 días de tratamiento, pero persiste durante toda la vida, aunque sea raro.
Los pacientes que reciben tratamiento con medicamentos que contienen factor VIII de coagulación sanguínea deben estar bajo un seguimiento cuidadoso para detectar la aparición de inhibidores mediante observaciones clínicas adecuadas y análisis de laboratorio (véase también la sección «Reacciones adversas»).
El riesgo de desarrollo de inhibidores depende de diversos factores relacionados con las características específicas de cada paciente. Entre los principales factores de riesgo se incluyen el tipo de mutación del gen del factor VIII, antecedentes familiares y etnia del paciente.
La mayoría de los informes sobre inhibidores provienen de pacientes previamente no tratados.
Immunate se fabrica a partir de plasma humano. Las medidas estándar para prevenir la transmisión de infecciones mediante medicamentos elaborados a partir de sangre o plasma humano incluyen la selección de donantes, el cribado de muestras individuales y de pools de plasma para marcadores específicos de infección, así como etapas eficaces de producción para la inactivación/eliminación de virus. A pesar de estas medidas, no puede excluirse por completo la posibilidad de transmisión de agentes infecciosos con medicamentos derivados de sangre o plasma humano. Esto también incluye virus desconocidos o nuevos y otros agentes infecciosos.
Se considera que las medidas adoptadas son eficaces contra virus envueltos, como el virus de la inmunodeficiencia humana (VIH), los virus de la hepatitis B y C, así como contra el virus de la hepatitis A no envuelto. Las medidas empleadas podrían tener un valor limitado frente a virus no envueltos, como el parvovirus B19. La infección por parvovirus B19 puede ser peligrosa para mujeres embarazadas (infección fetal) y para personas con inmunodeficiencia o eritropoyesis aumentada (por ejemplo, anemia hemolítica).
Durante el tratamiento prolongado de la enfermedad de von Willebrand con un medicamento que contiene factor VIII, el nivel de VIII:C puede elevarse excesivamente. Existe riesgo de eventos trombóticos durante el tratamiento de pacientes con síndrome de von Willebrand, especialmente en aquellos con factores de riesgo clínicos o de laboratorio conocidos. Por lo tanto, los pacientes deben estar bajo vigilancia para detectar signos tempranos de trombosis. En pacientes con antecedentes de tromboembolismo venoso, un alto nivel endógeno de factor VIII se ha asociado con un mayor riesgo de eventos trombóticos futuros.
En pacientes que reciben un medicamento que contiene factor de von Willebrand y factor VIII, deben controlarse los niveles plasmáticos del factor VIII:C para evitar un nivel persistente y excesivo de factor VIII:C en plasma, lo que podría aumentar el riesgo de eventos trombóticos.
Debe realizarse profilaxis de tromboembolismo venoso de acuerdo con las recomendaciones vigentes.
En pacientes con enfermedad de von Willebrand, especialmente en aquellos con tipo 3, pueden desarrollarse anticuerpos neutralizantes (inhibidores) contra el factor de von Willebrand.
Estos anticuerpos pueden aparecer en estrecha relación con reacciones anafilácticas. Por esta razón, los pacientes con reacción anafiláctica deben evaluarse para detectar la presencia de este inhibidor. Si no se alcanzan los niveles esperados de actividad vWF:RCo en plasma o si la hemorragia no se controla con la dosis adecuada, debe realizarse un análisis para determinar la presencia de un inhibidor del factor de von Willebrand. En pacientes con alto nivel de inhibidor, la terapia con factor de von Willebrand puede ser ineficaz, por lo que deben considerarse otras opciones terapéuticas. El tratamiento debe realizarse bajo supervisión médica por un profesional con experiencia en trastornos de la hemostasia.
A los pacientes que reciben regular o repetidamente medicamentos con factor VIII derivado de plasma humano, se les debe considerar la vacunación adecuada (hepatitis A y B).
Se recomienda que, cada vez que se use Immunate, se registre en el diario del paciente el nombre y número de lote del medicamento, con el fin de establecer una relación entre el paciente y el lote del producto.
Immunate contiene isoaglutininas de grupos sanguíneos (anti-A y anti-B). En pacientes con grupo sanguíneo A, B o AB puede producirse hemólisis tras administraciones repetidas en intervalos cortos o tras la administración de dosis muy elevadas. Dosis muy elevadas en un corto período de tiempo podrían usarse en el marco del tratamiento de inducción de tolerancia inmunológica para el tratamiento de hemofilia con inhibidores del factor VIII.
Antes de prescribir Immunate, debe confirmarse que el trastorno de la coagulación se debe efectivamente a un déficit del factor VIII (hemofilia A) o a un déficit del factor de von Willebrand (enfermedad de von Willebrand).
Uso durante el embarazo y la lactancia.
No se han realizado estudios sobre el efecto del factor VIII en la función reproductiva de animales. Debido a la baja frecuencia de hemofilia A en mujeres, no existe experiencia clínica sobre el uso de factor VIII durante el embarazo y la lactancia. Por lo tanto, el factor VIII debe usarse durante el embarazo y la lactancia solo si hay indicaciones claramente justificadas.
Para información sobre la infección por parvovirus B19, véase la sección «Características de uso».
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
No existe información sobre el efecto de Immunate sobre la velocidad de reacción al conducir vehículos o manejar maquinaria.
Vía de administración y dosis
El tratamiento debe realizarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia A.
Dosis estándar
La dosis y la duración de la terapia sustitutiva dependen del grado de déficit del factor VIII, de la localización y volumen del sangrado, así como del estado clínico del paciente.
La cantidad administrada de unidades del factor VIII se expresa en unidades internacionales (UI), definidas según el estándar internacional actual de la OMS para medicamentos basados en el factor VIII. La actividad del factor VIII en el plasma sanguíneo se expresa bien en porcentaje (en relación con el valor normal del plasma humano), bien en unidades internacionales (en relación con el estándar internacional para el factor VIII en plasma sanguíneo).
Una unidad internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
Dosis en la hemofilia A
El cálculo de la dosis requerida del factor VIII se realiza según la relación empíricamente demostrada: 1 unidad internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en el plasma sanguíneo aproximadamente en un 1,5–2 % respecto a la actividad normal. La dosis requerida se determina mediante la siguiente fórmula:
Cantidad necesaria de unidades (UI) = peso corporal (kg) × incremento deseado del factor VIII (%) × 0,5
La dosis y la frecuencia de administración deben ajustarse siempre para garantizar la eficacia clínica en cada paciente individual.
Sangrado y cirugía
En caso de episodios adicionales de sangrado, la actividad del factor VIII no debe descender por debajo del nivel objetivo de actividad en el plasma (en % respecto a la normalidad o en UI/dl) durante el período adecuado. La tabla 3 que se indica a continuación puede utilizarse para determinar la dosis en episodios de sangrado y durante intervenciones quirúrgicas.
Tabla 3
| Grado de hemorragia/tipo de procedimiento quirúrgico |
Nivel necesario del factor VIII (% de la normalidad) (UI/dl) |
Frecuencia de administración de la dosis (horas)/duración del tratamiento (días) |
| Hemorragia |
||
| Hemartrosis precoz, hemorragia muscular o hemorragia en la cavidad bucal |
20–40 |
Repetir cada 12–24 horas durante al menos 1 día hasta la cesación de la hemorragia, evidenciada por la ausencia de dolor y la cicatrización de la herida. |
| Hemartrosis más pronunciada, hemorragia muscular o hematoma |
30–60 |
Repetir la infusión cada 12–24 horas durante 3–4 días o más, hasta la desaparición del dolor y la corrección de alteraciones funcionales graves. |
| Hemorragias amenazantes para la vida |
60–100 |
Repetir la infusión cada 8–24 horas hasta la desaparición del riesgo. |
| Intervención quirúrgica |
||
| Pequeña intervención quirúrgica, incluyendo extracción dental |
30–60 |
Cada 24 horas durante al menos 1 día hasta la cicatrización completa de la herida. |
| Gran intervención quirúrgica |
80–100 (antes y después de la intervención quirúrgica) |
Repetir la infusión cada 8–24 horas hasta una adecuada cicatrización de la herida; posteriormente, continuar el tratamiento durante al menos 7 días más para mantener el nivel de actividad del factor VIII entre el 30–60 % (UI/dl). |
La dosis y frecuencia de administración deben adaptarse a la respuesta clínica en cada caso individual. En determinadas circunstancias (por ejemplo, presencia de un título bajo de inhibidor del factor VIII), especialmente al comienzo del tratamiento, pueden ser necesarias dosis superiores a las calculadas mediante la fórmula.
Si no se logra controlar la hemorragia con la dosis calculada, se debe determinar el nivel de factor VIII en el plasma sanguíneo y administrar una dosis suficiente del medicamento Immunate para alcanzar una respuesta clínica adecuada.
Durante el tratamiento, se recomienda evaluar la actividad del factor VIII con el fin de determinar la necesidad de ajustar la dosis y la frecuencia de las infusiones repetidas. En el caso de intervenciones quirúrgicas mayores, es extremadamente importante garantizar un control preciso durante la terapia sustitutiva mediante pruebas cuantitativas de la actividad del factor VIII en el plasma sanguíneo. La respuesta al factor VIII puede variar en cada paciente individual, con diferentes valores de vida media y grado de recuperación in vivo.
Profilaxis a largo plazo
Para la profilaxis prolongada de hemorragias en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, con intervalos de administración de 2 a 3 días. En algunos casos, especialmente en pacientes jóvenes, puede ser necesario reducir los intervalos entre administraciones o utilizar dosis más altas del medicamento.
Pacientes con inhibidores del factor VIII
Los pacientes deben controlarse regularmente respecto al desarrollo de inhibidores del factor VIII. Si no se alcanzan los niveles esperados de actividad del factor VIII en el plasma o si la hemorragia no se controla con una dosis adecuada, se debe realizar una prueba para detectar la presencia de un inhibidor del factor VIII. En pacientes con niveles altos de inhibidor, la terapia con factor VIII puede ser ineficaz, por lo que deben considerarse otras opciones terapéuticas. El manejo de estos pacientes debe realizarse bajo supervisión médica por profesionales con experiencia en el tratamiento de la hemofilia, especialmente en casos de aparición de inhibidores del factor VIII (véase también la sección «Instrucciones de uso»).
Eventos cardiovasculares
En pacientes con factores de riesgo cardiovasculares preexistentes, la terapia sustitutiva con factor VIII puede aumentar el riesgo cardiovascular.
Enfermedad de von Willebrand con déficit de factor VIII
Immunate está indicado para el tratamiento y la profilaxis del déficit de factor VIII en pacientes con enfermedad de von Willebrand con déficit de factor VIII, y en casos en los que el tratamiento únicamente con desmopresina es ineficaz o está contraindicado. La terapia sustitutiva con Immunate para el control de hemorragias, así como la profilaxis perioperatoria de hemorragias, se realiza según las mismas recomendaciones que en la hemofilia A.
La experiencia con el uso del medicamento en niños es limitada.
Los pacientes deben vigilarse respecto al desarrollo de inhibidores del factor von Willebrand si no se alcanzan los niveles esperados de actividad del factor von Willebrand en el plasma sanguíneo o si la hemorragia no se controla con la dosis adecuada. En pacientes con niveles altos de inhibidor, la terapia con factor von Willebrand puede ser ineficaz y deben considerarse otras opciones terapéuticas (véase también la sección «Instrucciones de uso»).
En caso de uso de un medicamento que contenga factor von Willebrand y factor VIII, el médico tratante debe tener en cuenta que un tratamiento continuo puede provocar un aumento excesivo del nivel de factor VIII:C. Debe considerarse la posibilidad de reducir la dosis y/o prolongar los intervalos entre administraciones tras un tratamiento de entre 24 y 48 horas, con el fin de evitar niveles demasiado altos de factor VIII:C.
Niños
El medicamento debe administrarse con precaución a niños menores de 6 años que hayan tenido una exposición limitada a los medicamentos del factor VIII, ya que los datos clínicos en este grupo de pacientes son limitados. Sin embargo, a partir de informes de casos clínicos, puede concluirse que su eficacia y seguridad están respaldadas.
Vía de administración
Las instrucciones sobre la reconstitución del medicamento antes de la administración se describen en la sección «Vía de administración y dosis».
Immunate debe administrarse lentamente por vía intravenosa. La velocidad máxima de infusión no debe exceder los 2 ml por minuto.
Preparación de la solución
Immunate debe reconstituirse inmediatamente antes de la administración. La solución debe utilizarse inmediatamente, ya que el medicamento no contiene conservantes.
Antes de la administración, los medicamentos reconstituidos deben examinarse visualmente para detectar la presencia de partículas sólidas y cambios de color. No deben utilizarse soluciones turbias o que contengan sedimentos.
Se recomienda lavar los dispositivos implantados de acceso venoso con solución salina isotónica antes y después de la infusión del medicamento Immunate.
Reconstitución del polvo: utilizar técnica aséptica, como se describe a continuación
- Calentar el frasco cerrado con el diluyente (agua estéril para inyección) hasta temperatura ambiente (no superior a 37 °C).
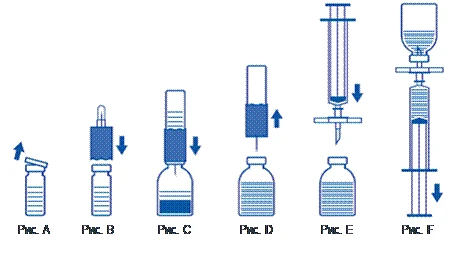
- Retirar las tapas protectoras del frasco con el polvo y del frasco con el diluyente (fig. A) y limpiar los tapones de goma de ambos frascos.
- Colocar el borde ondulado del dispositivo de transferencia en el frasco con el diluyente y presionar (fig. B).
- Retirar la cubierta protectora del otro extremo del conjunto de transferencia, evitando tocar el extremo abierto.
- Invertir el conjunto de transferencia con el frasco de diluyente unido sobre el frasco con el polvo e introducir el extremo libre de la aguja en el tapón de goma del frasco con el polvo (fig. C). El diluyente será aspirado al frasco con el polvo por efecto del vacío.
- Aproximadamente un minuto después, separar los dos frascos retirando el conjunto de transferencia con el frasco de diluyente del frasco con el polvo (fig. D). Dado que el medicamento se disuelve fácilmente, solo debe agitarse suavemente, si acaso, el frasco con el concentrado. NO AGITAR EL FRASCO CON EL POLVO. NO INVERTIR EL FRASCO CON EL POLVO HASTA QUE ESTÉ LISTO PARA EXTRAER SU CONTENIDO.
- Tras la reconstitución, la solución preparada debe examinarse visualmente antes de la administración para detectar inclusiones y cambios de color. Sin embargo, incluso con un estricto cumplimiento del procedimiento de reconstitución, ocasionalmente pueden observarse algunas partículas finas. Estas partículas serán eliminadas mediante el filtro incluido en el conjunto, sin que se reduzca la eficacia del medicamento.
Utilice técnica aséptica, como se describe a continuación
- Para evitar la administración de partículas de goma procedentes del tapón (riesgo de microembolismo), debe utilizarse el conjunto de filtro incluido. Para extraer el medicamento reconstituido, coloque el conjunto de filtro en la jeringa desechable suministrada e introdúzcala a través del tapón de goma (fig. E).
- Desconecte momentáneamente la jeringa del conjunto de filtro. El aire entrará en el frasco con el polvo y cualquier espuma desaparecerá. A continuación, aspire la solución a través del conjunto de filtro (fig. F).
- Desconecte la jeringa del conjunto con filtro y administre lentamente la solución por vía intravenosa (velocidad máxima de administración: 2 ml por minuto) utilizando el conjunto de infusión con ala suministrado (o la aguja desechable suministrada).
Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
Niños
El medicamento debe administrarse con precaución a niños menores de 6 años que hayan tenido una exposición limitada a los medicamentos del factor VIII, ya que los datos clínicos en este grupo de pacientes son limitados. Sin embargo, a partir de informes de casos clínicos, puede concluirse que su eficacia y seguridad están respaldadas.
Sobredosis
No se han descrito síntomas relacionados con sobredosis de factor de coagulación VIII humano.
Principalmente, existe el riesgo de presentar fenómenos tromboembólicos. En pacientes con grupos sanguíneos A, B o AB existe riesgo de hemólisis (véase también la sección «Instrucciones de uso»).
Reacciones adversas.
Las reacciones adversas indicadas a continuación se detectaron durante estudios clínicos y se notificaron en el período poscomercialización tras la obtención de la autorización de comercialización del medicamento Inmunat. La frecuencia se indica según la siguiente escala: muy frecuentes (≥ 10 %); frecuentes (≥ 1 % — < 10 %); poco frecuentes (≥ 0,1 % — < 1 %); raras (≥ 0,01 % — < 0,1 %); muy raras (< 0,01 %).
Estudios clínicos
La frecuencia de todas las reacciones adversas indicadas a continuación, notificadas en estudios clínicos, fue poco frecuente (≥ 0,1 % — < 1 %).
Enfermedades del sistema inmunitario
Poco frecuentes: reacciones alérgicas.
Notificaciones espontáneas tras la obtención de la autorización de comercialización
La frecuencia de todas las reacciones adversas indicadas a continuación fue muy rara (< 0,01 %).
Enfermedades de la sangre y del sistema linfático
Coagulopatía, inhibición del factor VIII.
Trastornos psiquiátricos
Ansiedad.
Enfermedades del corazón
Palpitaciones, taquicardia.
Trastornos gastrointestinales
Vómitos, náuseas.
Enfermedades generales y condiciones en el lugar de administración
Dolor en el pecho, molestias en el pecho, edema (especialmente edema periférico y edema facial), irritación en el lugar de la inyección (especialmente sensación de ardor), escalofríos, dolor, pirexia.
Enfermedades del sistema inmunitario
Hipersensibilidad.
Enfermedades del sistema nervioso
Cefalea, mareo, parestesias.
Enfermedades de las vías respiratorias, órganos torácicos y mediastino
Tos, disnea.
Enfermedades vasculares
Hipotensión arterial, hiperemia, palidez.
Enfermedades del ojo
Conjuntivitis, edema de párpados.
Enfermedades de la piel y tejidos subcutáneos
Eritema, exantema, neurodermatitis, prurito, erupción cutánea, erupción eritematosa, erupción generalizada, urticaria, hiperhidrosis.
Enfermedades del músculo esquelético, tejido conjuntivo y huesos
Mialgia.
Aún no se han recibido notificaciones sobre las siguientes reacciones adversas, pero podrían ocurrir con el uso del medicamento Inmunat.
Enfermedades de la sangre y del sistema linfático
Hemólisis en pacientes con grupo sanguíneo A, B o AB.
Enfermedades generales y condiciones en el lugar de administración
Disminución de la respuesta terapéutica.
Descripción de efectos adversos específicos
Se han observado raramente reacciones de hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, sensación de ardor y hormigueo en el lugar de la infusión, escalofríos, eritema, urticaria generalizada, cefalea, erupción cutánea, hipotensión arterial, somnolencia, náuseas, ansiedad, taquicardia, opresión en el pecho, hormigueo, vómitos, sibilancias), y en algunos casos pueden progresar a una anafilaxia grave (incluyendo shock). Se debe recomendar a los pacientes que consulten al médico si aparecen estos síntomas.
En casos raros se ha observado pirexia.
En pacientes con hemofilia A pueden desarrollarse anticuerpos neutralizantes (inhibidores) frente al factor VIII. Si se desarrollan estos inhibidores, el estado se manifiesta como una respuesta clínica insuficiente. En tales casos, se recomienda consultar a un centro especializado en el tratamiento de la hemofilia.
La hemólisis puede ocurrir tras la administración de dosis elevadas (por ejemplo, cuando se requiere alcanzar un nivel de factor VIII en plasma superior al 100 %) en pacientes con grupo sanguíneo A, B o AB.
Para información sobre la seguridad vírica, véase la sección «Precauciones especiales de uso».
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la comercialización del medicamento es de gran importancia. Permite realizar un seguimiento continuo de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar cualquier caso sospechoso de reacción adversa o falta de eficacia del medicamento a través del sistema automatizado de información de farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Periodo de validez
Medicamento: 2 años.
Disolvente (agua para inyección): 5 años.
Condiciones de conservación
Conservar entre 2 y 8 °C. ¡No congelar! Conservar en el embalaje original para protegerlo de la luz. Mantener fuera del alcance de los niños. Dentro del periodo de validez indicado, el medicamento puede conservarse a temperatura ambiente (no superior a 25 °C) durante un máximo de 6 meses.
Tras la reconstitución, el medicamento puede conservarse a temperatura ambiente (no superior a 25 °C) durante un máximo de 6 horas.
Incompatibilidades
No se debe mezclar Inmunat con otros medicamentos, ya que esto podría afectar negativamente su seguridad y eficacia. Se deben utilizar únicamente los kits de infusión proporcionados, ya que la adsorción del factor de coagulación VIII humano en la superficie interna de algunos dispositivos de infusión podría provocar una falta de eficacia del tratamiento.
Envase
1 vial con polvo (250/190 UI, 500/375 UI o 1000/750 UI) junto con 1 vial con disolvente (agua para inyección, 5 ml o 10 ml) y un kit para disolución y administración (1 dispositivo de transferencia/filtración, 1 jeringa desechable (5 ml o 10 ml), 1 aguja desechable, 1 sistema de infusión) en una caja.
Categoría de dispensación
Medicamento sujeto a prescripción médica.
Fabricante
Takeda Manufacturing Austria AG, Austria.
Dirección del fabricante y lugar de actividad
Industriestrasse 67, 1221 Viena, Austria.