Gemlibra®
Ucrania
Contenido
INSTRUCCIONES para uso médico del medicamento HEMLIBRAÒ (HEMLIBRAÒ)
Composición:
Principio activo: emicizumab;
1 frasco contiene 12 mg/0,4 ml o 30 mg/1 ml, o 60 mg/0,4 ml, o 105 mg/0,7 ml, o 150 mg/1 ml de emicizumab;
Excipientes: L-histidina, ácido L-aspártico, L-arginina, poloxámero 188, agua para inyección.
Forma farmacéutica. Solución inyectable.
Principales propiedades fisicoquímicas: líquido estéril, transparente, incoloro o ligeramente amarillento, sin conservantes.
Grupo farmacoterapéutico.
Medicamentos que actúan sobre la sangre y el sistema hematopoyético. Agentes antihemorrágicos. Vitamina K y otros agentes hemostáticos. Otros agentes hemostáticos de uso sistémico. Emicizumab.
Código ATC B02B X06.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
El medicamento Hemlibra® une el factor IX activado y el factor X, restableciendo así la función deficiente del factor VIII activado, que es necesario para lograr una hemostasia eficaz.
Farmacocinética.
La emicizumab ha demostrado farmacocinética proporcional a la dosis en un rango de dosis de 0,3 mg/kg (0,1 parte de la dosis inicial recomendada aprobada) hasta 6 mg/kg tras la administración subcutánea. Tras múltiples inyecciones subcutáneas de emicizumab con una dosis de carga de 3 mg/kg una vez por semana durante las primeras 4 semanas, la concentración plasmática mínima media (± DE) de emicizumab fue de 52,6 ± 13,6 µg/ml, alcanzada en la semana 5. La información sobre la concentración plasmática media en estado estacionario (± DE) tras la administración de las dosis de mantenimiento recomendadas se presenta en la tabla 1.
Tabla 1
Concentraciones medias (± DE) en estado estacionario tras la administración de la dosis de carga de emicizumab según el régimen de tratamiento de mantenimiento
| Parámetros |
Dosis de mantenimiento |
||
| 1,5 mg/kg de peso corporal una vez por semana |
3 mg/kg de peso corporal una vez cada 2 semanas |
6 mg/kg de peso corporal una vez cada 4 semanas |
|
| Cmax, ss (μg/ml) |
55,1 ± 15,9 |
58,3 ± 16,4 |
67 ± 17,7 |
| AUCss,τ (μg/ml*día) |
376 ± 109 |
752 ± 218 |
1503 ± 437 |
| Cmín, ss (μg/ml) |
51,2 ± 15,2 |
46,9 ± 14,8 |
38,5 ± 14,2 |
| Relación Cmax/Cmín (μg/ml) |
1,08 ± 0,03 |
1,26 ± 0,12 |
1,85 ± 0,47 |
AUCss,τ – área bajo la curva de «concentración en plasma frente al tiempo» en estado de equilibrio durante el intervalo de dosificación (τ = 1, 2 ó 4 semanas); Cmax, ss – concentración máxima del medicamento en plasma en estado de equilibrio; Cmin, ss – concentración mínima del medicamento en plasma en estado de equilibrio.
Absorción
Tras la administración subcutánea, los valores medios (± DE) del período de semivida de absorción fueron de 1,6 ± 1 día. La biodisponibilidad absoluta tras la administración subcutánea a la dosis de 1 mg/kg osciló entre el 80,4 % y el 93,1 %. Se observaron perfiles farmacocinéticos similares tras la administración subcutánea en la región abdominal, del hombro y del muslo (véase la sección «Posología y forma de administración»).
Distribución
El volumen de distribución aparente medio (% coeficiente de variación [%CV]) fue de 10,4 l (26 %).
Eliminación
La depuración aparente media (%CV) fue de 0,27 l/día (28,4 %), y el período de semivida aparente medio (± DE) fue de 26,9 ± 9,1 días.
Poblaciones especiales
La farmacocinética de emicizumab no se ve afectada por la edad del paciente (de 1 a 77 años), raza (caucásica – 62,7 %, asiática – 22,9 % y afrodescendiente – 8 %), estado de inhibidores (inhibidores presentes en el 50 %), disfunción hepática leve (definida como nivel de bilirrubina total de 1–1,5 veces por encima del límite superior normal [LSN], cualquier nivel de aspartato aminotransferasa [AST]), disfunción hepática moderada (definida como nivel de bilirrubina total de 1,5–3 veces por encima del LSN y cualquier nivel de AST), así como disfunción renal leve (definida como aclaramiento de creatinina de 60–89 ml/min) y disfunción renal moderada (definida como aclaramiento de creatinina de 30–59 ml/min). No se ha estudiado el uso de emicizumab en pacientes con disfunción hepática o renal grave.
En niños menores de 6 meses de edad, las concentraciones predichas de emicizumab fueron un 19–33 % más bajas que en pacientes de mayor edad, especialmente con una dosis de mantenimiento de 3 mg/kg cada 2 semanas o de 6 mg/kg cada 4 semanas.
Niños
Se han establecido la eficacia y la seguridad del uso de Hemlibra® en niños. El uso de Hemlibra® en niños con hemofilia A está respaldado por los resultados de dos estudios aleatorizados (HAVEN 1 y HAVEN 3) y dos estudios no comparativos (HAVEN 2 y HAVEN 4). Todos los estudios clínicos incluyeron niños de un mismo grupo de edad: 47 adolescentes de 12 a 18 años. Solo en el estudio HAVEN 2 se incluyeron niños de varios grupos de edad: 55 niños de 2 a 12 años y 5 niños de 1 mes a 2 años. No se observaron diferencias en la eficacia del medicamento entre los distintos grupos de edad.
Las concentraciones mínimas de emicizumab en plasma en estado de equilibrio fueron comparables en adultos y niños mayores de 6 meses cuando se administraron dosis equivalentes ajustadas por peso corporal. En niños menores de 6 meses se esperan concentraciones más bajas de emicizumab.
Peso corporal. La depuración aparente y el volumen de distribución de emicizumab aumentan con el incremento del peso corporal (de 9 a 156 kg). La dosificación en miligramos por kilogramo (mg/kg) proporciona exposiciones similares a emicizumab en todo el rango de valores de peso corporal.
Inmunogenicidad
La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y especificidad del ensayo. Las diferencias en los métodos de análisis dificultan la comparación significativa de las tasas de formación de anticuerpos contra el medicamento en los estudios descritos a continuación con las tasas en otros estudios, incluyendo estudios con Hemlibra® u otros medicamentos de emicizumab.
Basado en datos de estudios clínicos con Hemlibra®, en el 5,1 % de los pacientes (34/668) se detectaron anticuerpos contra emicizumab. Los participantes recibieron emicizumab durante un rango intercuartílico medio (RIC) de 103,1 (82,4–148,1) semanas. Las muestras para la prueba de anticuerpos se obtuvieron al inicio del tratamiento y periódicamente durante todo el estudio.
Efecto de los anticuerpos contra el medicamento sobre la farmacocinética
Las muestras positivas para anticuerpos se analizaron posteriormente para detectar la presencia de anticuerpos neutralizantes contra emicizumab mediante un ensayo cromogénico modificado de FVIII. En total, 668 pacientes fueron evaluados para detectar anticuerpos contra emicizumab. En el 5,1 % de los pacientes (34/668) se detectaron anticuerpos contra emicizumab y en el 2,7 % de los pacientes (18/668) se desarrollaron anticuerpos contra emicizumab que fueron neutralizantes in vitro. De estos 2,7 % de pacientes (18/668), los anticuerpos neutralizantes contra emicizumab no tuvieron un impacto clínicamente significativo sobre la farmacocinética de Hemlibra® en el 2,1 % de los pacientes (14/668), mientras que una disminución en la concentración de emicizumab en plasma se observó en el 0,6 % de los pacientes (4/668).
Efecto de los anticuerpos contra el medicamento sobre la farmacodinamia
En un paciente (1/668; 0,1 %) se desarrollaron anticuerpos neutralizantes contra emicizumab, lo que provocó una disminución en la concentración de emicizumab en plasma y posterior pérdida de eficacia del medicamento (manifestada como hemorragia de ruptura) a las 5 semanas de tratamiento. El tratamiento de este paciente con Hemlibra® se interrumpió prematuramente (véase la sección «Precauciones de uso»).
Características clínicas.
Indicaciones.
Prevención rutinaria con el fin de evitar o reducir la frecuencia de episodios de hemorragia en adultos y niños, desde el nacimiento, con hemofilia A (déficit congénito del factor VIII de la coagulación sanguínea) con o sin formación de inhibidores contra el factor VIII.
Contraindicaciones.
Hipersensibilidad al emicizumab o a cualquiera de los excipientes del medicamento.
Interacción con otros medicamentos y otras formas de interacción.
Estudios de interacción con otros medicamentos
No se han realizado estudios de interacción del medicamento Hemlibra® con otros medicamentos.
Hipercoagulabilidad con uso concomitante de PPCA
La experiencia clínica sugiere que existe una interacción entre el medicamento Hemlibra® y el concentrado de complejo protrombínico activado (PPCA) (ver sección «Instrucciones de uso»).
Efecto del medicamento sobre los resultados de pruebas de laboratorio
El medicamento Hemlibra® restaura la actividad del cofactor tenasa del factor VIII activado (aFVIII). Las pruebas de laboratorio que evalúan la vía intrínseca de activación de la cascada de coagulación (por ejemplo, el tiempo de tromboplastina parcial activado [TTPa]) miden el tiempo total de coagulación, incluyendo el tiempo necesario para que la trombina active el FVIII en aFVIII, y producen una acortamiento excesivo del tiempo de coagulación cuando se utiliza Hemlibra®, ya que no se requiere activación por trombina. Este acortamiento excesivo del tiempo de la vía intrínseca de coagulación seguirá afectando los resultados de todos los análisis unifactoriales basados en la determinación del TTPa, en particular el análisis uniestadio de la actividad del FVIII; sin embargo, los análisis unifactoriales que utilizan métodos cromogénicos o inmunológicos no se ven alterados por el medicamento Hemlibra® y pueden utilizarse para el monitoreo de los parámetros de coagulación durante el tratamiento, con observaciones importantes respecto al análisis cromogénico de la actividad del FVIII, como se describe a continuación.
Los kits para el análisis cromogénico de la actividad del FVIII pueden fabricarse con proteínas de coagulación humanas o bovinas. Los kits que contienen factores humanos son sensibles al medicamento Hemlibra®, pero pueden sobreestimar el potencial hemostático clínico de Hemlibra®. Por el contrario, los kits basados en factores de coagulación bovinos son insensibles al medicamento Hemlibra® (la actividad no se mide) y pueden utilizarse para monitorear la actividad del FVIII endógeno o administrado por infusión, así como para la detección de inhibidores contra el FVIII.
El medicamento Hemlibra® no pierde actividad en presencia de inhibidores del FVIII, lo que dará lugar a un resultado falsamente negativo en la prueba de Bethesda para la inhibición funcional del FVIII mediante el método de coagulación. Como alternativa, puede utilizarse la prueba cromogénica de Bethesda con FVIII bovino, que es insensible al medicamento Hemlibra®.
Debido al prolongado período de semidesintegración de Hemlibra®, el efecto sobre los parámetros del perfil de coagulación puede persistir hasta 6 meses después de la administración de la última dosis (ver sección «Propiedades farmacológicas»).
Características de uso.
| ADVERTENCIA: MICRANGIOPATÍA TROMBÓTICA Y TROMBOEMBOLIA Se han notificado casos de microangiopatía trombótica y tromboembolia en pacientes que recibieron dosis profilácticas del medicamento HemlibraÒ, en estudios clínicos cuando se administró concentrado del complejo protrombínico activado (CPA) durante 24 horas o más, con una dosis acumulativa media > 100 UI/kg/24 h. Es necesario realizar un control riguroso para detectar la aparición de microangiopatía trombótica y eventos tromboembólicos durante la administración de CPA. Debe interrumpirse la administración de CPA y suspenderse la siguiente dosis del medicamento HemlibraÒ si aparecen síntomas. |
Microangiopatía trombótica asociada al uso de Hemlibra® y concentrado de complejo de protrombina activado (CCPA)
Se han notificado casos de microangiopatía trombótica (MAT) en pacientes que recibieron dosis profilácticas de Hemlibra® cuando se administró concentrado de complejo de protrombina activado (CCPA) durante 24 horas o más con una dosis acumulativa media > 100 UI/kg/24 h. En estudios clínicos, se notificó MAT en el 0,8 % de los pacientes (3 de 391) y en el 8,1 % de los pacientes (3 de 37) que recibieron al menos una dosis de CCPA. En el estudio ADAMTS13, los pacientes presentaron trombocitopenia, anemia hemolítica microangiopática y daño renal agudo sin signos evidentes de deficiencia.
Se observaron signos de mejoría dentro de una semana tras la interrupción del CCPA. Un paciente continuó recibiendo Hemlibra® tras la resolución de la MAT.
En los pacientes que reciben tratamiento profiláctico con Hemlibra®, se debe evaluar cuidadosamente el balance beneficio-riesgo si se necesita administrar CCPA. Debido al largo periodo de semivida de Hemlibra®, la posibilidad de interacción con CCPA puede persistir hasta 6 meses tras la última dosis. Durante la administración de CCPA, debe realizarse un monitoreo activo para detectar el desarrollo de MAT. Si aparecen signos clínicos y/o de laboratorio que sugieran MAT, se debe interrumpir inmediatamente el tratamiento con CCPA y suspender la profilaxis con Hemlibra® y tratar según las indicaciones clínicas. Se debe evaluar cuidadosamente el balance entre beneficios y riesgos de continuar la profilaxis con Hemlibra® tras la resolución de la MAT en cada caso individual.
Tromboembolismo asociado al uso de Hemlibra® y CCPA
Se han notificado casos de trombosis en pacientes que recibieron dosis profilácticas de Hemlibra® cuando se administró CCPA durante 24 horas o más con una dosis acumulativa media > 100 UI/kg/24 h. En estudios clínicos, se notificaron casos de trombosis en el 0,5 % de los pacientes (2 de 391) y en el 5,4 % de los pacientes (2 de 37) que recibieron al menos una dosis de CCPA.
Ningún caso de trombosis requirió tratamiento con anticoagulantes. Se observaron signos de mejoría o recuperación dentro de una semana tras la interrupción del CCPA. Un paciente continuó recibiendo Hemlibra® tras la desaparición de los signos de trombosis.
Para los pacientes que reciben tratamiento profiláctico con Hemlibra®, se debe evaluar cuidadosamente el balance beneficio-riesgo si se necesita administrar CCPA. Debido al largo periodo de semivida de Hemlibra®, la posibilidad de interacción con CCPA puede persistir hasta 6 meses tras la última dosis. Durante la administración de CCPA, debe realizarse un monitoreo activo para detectar el desarrollo de tromboembolismo. Si aparecen síntomas clínicos, hallazgos de imágenes o signos de laboratorio que sugieran tromboembolismo, se debe interrumpir inmediatamente el tratamiento con CCPA y suspender la profilaxis con Hemlibra® y tratar según las indicaciones clínicas. Se debe evaluar cuidadosamente el balance entre beneficios y riesgos de continuar la profilaxis con Hemlibra® tras la resolución del evento trombótico en cada caso individual.
Inmunogenicidad
El tratamiento con Hemlibra® puede inducir la formación de anticuerpos contra el medicamento. Se han notificado anticuerpos contra emicizumab en el 5,1 % de los pacientes (34 de 668) que recibieron Hemlibra® en estudios clínicos. En la mayoría de los pacientes con anticuerpos contra emicizumab no se observaron cambios en la concentración plasmática de Hemlibra® ni un aumento en el número de hemorragias; sin embargo, en casos raros (prevalencia < 1 %), la presencia de anticuerpos neutralizantes con disminución de la concentración plasmática puede asociarse con pérdida de eficacia (ver sección «Farmacocinética»).
Se debe observar a los pacientes en busca de signos clínicos de pérdida de eficacia del tratamiento (es decir, aumento en la frecuencia de hemorragias de escape), y ante su aparición, se debe evaluar inmediatamente la causa y considerar un cambio en el tratamiento si se sospecha la presencia de anticuerpos neutralizantes contra emicizumab.
Efecto sobre los parámetros de la coagulación
Hemlibra® afecta los resultados de las pruebas de laboratorio del camino intrínseco de activación de la cascada de coagulación, incluyendo el tiempo de trombina activado (TTA), el tiempo de tromboplastina parcial activado (TTPa) y todas las pruebas basadas en la determinación del TTPa, incluyendo la determinación en una etapa de la actividad del factor VIII (FVIII) (tabla 2). Por lo tanto, no se deben utilizar pruebas de laboratorio del camino intrínseco de activación de la cascada de coagulación mediante métodos de coagulación para monitorear la actividad de Hemlibra®, determinar la dosis para terapia de reemplazo, ni para anticoagulación o determinación de títulos de inhibidores de FVIII en pacientes que reciben Hemlibra® (ver sección «Interacción con otros medicamentos e interacciones de otro tipo»). En la tabla 2 se indican las pruebas de coagulación que se ven afectadas y no afectadas por Hemlibra®.
Tabla 2
Resultados de las pruebas de coagulación afectadas y no afectadas por Hemlibra®
| Resultados afectados por el medicamento HemlibraÒ |
Resultados no afectados por el medicamento HemlibraÒ |
| Tiempo de tromboplastina parcial activado (TTPa) Análisis de Bethesda (método coagulativo) para la determinación de títulos de inhibidores del FVIII Ensayo unifactorial en una etapa basado en el TTPa Resistencia a la proteína C activada (APC-R) basada en el TTPa Tiempo de coagulación activado (TCA) |
Análisis de Bethesda (con sustrato cromogénico bovino) para la determinación de títulos de inhibidores del FVIII Tiempo de trombina (TT) Ensayo unifactorial en una etapa basado en el tiempo de protrombina (TP) Ensayo cromogénico unifactorial para otros parámetros distintos del FVIII* Ensayos inmunológicos (por ejemplo, ELISA, métodos turbidimétricos) Pruebas genéticas para la determinación de factores de coagulación (por ejemplo, Factor V Leiden, protrombina 20210) |
* Para información importante sobre la determinación de la actividad del FVIII mediante el método cromogénico, véase la sección «Interacción con otros medicamentos y otras formas de interacción».
Uso durante el embarazo o la lactancia.
Embarazo
No hay datos disponibles sobre el riesgo de malformaciones congénitas graves o aborto espontáneo asociados con el uso de Hemlibra® en mujeres embarazadas. No se han realizado estudios de toxicidad reproductiva en animales con emicizumab. No se sabe si Hemlibra® puede causar daño fetal o afectar la fertilidad cuando se administra a mujeres embarazadas. Hemlibra® solo debe administrarse durante el embarazo si el beneficio potencial para la mujer supera el riesgo potencial para el feto.
En todos los embarazos existe un riesgo basal de malformaciones congénitas, pérdida fetal u otros resultados adversos. El riesgo basal estimado de malformaciones congénitas graves o aborto espontáneo en las poblaciones diana es desconocido. En la población general de Estados Unidos, el riesgo estimado de malformaciones congénitas graves o aborto espontáneo durante embarazos clínicamente confirmados es del 2-4 % y del 15-20 %, respectivamente.
Lactancia
No existe información sobre la excreción de emicizumab en la leche materna humana, ni sobre su posible impacto en el lactante ni sobre la secreción en la leche. Se sabe que la IgG humana está presente en la leche materna humana. Los beneficios de la lactancia para el desarrollo y la salud deben sopesarse frente a la necesidad clínica de la madre de recibir Hemlibra®, la posibilidad de efectos adversos en el lactante y la condición general de la madre.
Anticoncepción
Las mujeres en edad fértil deben utilizar métodos anticonceptivos durante el tratamiento con Hemlibra®.
Efecto sobre la capacidad para conducir y utilizar máquinas.
No hay datos que indiquen que el tratamiento con Hemlibra® aumente la probabilidad de reacciones adversas que puedan afectar la capacidad para conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
Únicamente para administración subcutánea.
La dosis recomendada de carga es de 3 mg/kg de peso corporal, administrada por vía subcutánea una vez por semana durante las primeras 4 semanas. Posteriormente, el medicamento se administra en dosis de mantenimiento:
- 1,5 mg/kg de peso corporal una vez por semana, o
- 3 mg/kg de peso corporal una vez cada dos semanas, o
- 6 mg/kg de peso corporal una vez cada cuatro semanas.
La elección de la dosis de mantenimiento debe basarse en la preferencia del profesional sanitario, considerando los regímenes que puedan facilitar una mejor adherencia del paciente al tratamiento.
El uso profiláctico de medicamentos de derivación debe interrumpirse un día antes del inicio del tratamiento con Hemlibra® con fines profilácticos.
El uso profiláctico de medicamentos con factor VIII (FVIII) puede continuar durante la primera semana de tratamiento con Hemlibra® con fines profilácticos.
Dosificación olvidada
Si se omite una dosis de Hemlibra®, debe administrarse tan pronto como sea posible, y luego continuar con el horario habitual de administración. No administre dos dosis en el mismo día para compensar la dosis olvidada.
Recomendaciones especiales de dosificación
Niños
No se recomienda ajuste de dosis en niños. No existen datos de estudios en niños menores de 1 año.
Pacientes de edad avanzada
Los estudios clínicos con Hemlibra® incluyeron un número insuficiente de pacientes de 65 años o más para determinar si responden al tratamiento de manera diferente a los pacientes más jóvenes. Otros informes sobre experiencia clínica no han revelado diferencias en la respuesta al tratamiento entre pacientes de edad avanzada y pacientes más jóvenes.
Preparación y administración
Hemlibra® está indicado para su uso únicamente bajo supervisión médica. Tras una formación adecuada sobre la técnica de inyecciones subcutáneas, el paciente puede autoadministrarse la inyección o bien puede ser administrada por un cuidador, si el médico lo considera apropiado. La autoadministración no se recomienda en niños menores de 7 años.
A continuación se proporcionan instrucciones más detalladas sobre la preparación y el uso de Hemlibra® (véase la sección «Instrucciones de uso»).
Antes de la administración, el medicamento Hemlibra® debe inspeccionarse visualmente para detectar la presencia de partículas sólidas o cambios de color. Hemlibra® para administración subcutánea es una solución de color incoloro a ligeramente amarillento. No debe utilizarse si se observan partículas visibles o cambios en el color.
Para extraer Hemlibra® del frasco y administrarlo subcutáneamente, se requieren una jeringa, una aguja de transferencia con filtro y una aguja de inyección.
Véanse más abajo las «Instrucciones de uso» para obtener orientaciones sobre el manejo de Hemlibra® cuando se usan varios frascos conjuntamente.
No se debe mezclar en una misma inyección Hemlibra® procedente de frascos que contengan concentraciones diferentes del medicamento (es decir, 30 mg/ml y 150 mg/ml).
Véanse más abajo los criterios para seleccionar la presentación médica recomendada. Las dosis de Hemlibra® inferiores a 1 ml deben administrarse con una jeringa de 1 ml. Puede utilizarse una jeringa de 1 ml que cumpla los siguientes criterios: jeringa transparente de polipropileno o policarbonato con conector Luer-Lock (si no está disponible, puede usarse una jeringa con conector Luer Slip), graduación (marcaciones) de 0,01 ml, estéril, destinada únicamente para inyecciones, de un solo uso, sin látex y apirógena.
Las inyecciones de Hemlibra® con un volumen de entre 1 ml y 2 ml deben realizarse con jeringas de 2 ml o 3 ml que cumplan los siguientes criterios: jeringa transparente de polipropileno o policarbonato con conector Luer-Lock (si no está disponible, puede usarse una jeringa con conector Luer Slip), graduación (marcaciones) de 0,1 ml, estéril, destinada únicamente para inyecciones, de un solo uso, sin látex y apirógena.
Debe utilizarse una aguja de transferencia con filtro que cumpla los siguientes criterios: aguja de acero inoxidable con conexión Luer-Lock (si no está disponible, puede usarse una aguja con conexión Luer Slip), estéril, tamaño 18G, longitud de 1 a 1½ pulgadas, con punta biselada unilateral o romo (o semirromo), de un solo uso, sin látex, con filtro de 5 micrómetros y apirógena.
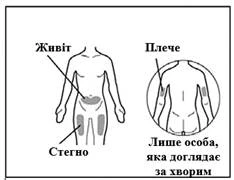
Puede utilizarse una aguja de inyección que cumpla los siguientes criterios: aguja de acero inoxidable con conexión Luer-Lock (si no está disponible, puede usarse una aguja con conexión Luer Slip), estéril, tamaño 26G (rango aceptable: 25–27G), longitud preferiblemente de 3/8 de pulgada o con longitud máxima de ½ pulgada, de un solo uso, sin látex, apirógena y con dispositivo de seguridad. Cada nueva inyección de Hemlibra® debe administrarse en una zona anatómica diferente (cara externa del brazo, muslo o cualquier cuadrante del abdomen), distinta de la utilizada para la inyección anterior. No se debe inyectar nunca en lunares, cicatrices ni en áreas sensibles, con hematomas, enrojecidas, duras o dañadas. La inyección de Hemlibra® en la cara externa del brazo debe realizarse exclusivamente por un cuidador o personal sanitario.
Debe desecharse cualquier cantidad de Hemlibra® no utilizada que quede en un frasco de un solo uso.
Almacenamiento de Hemlibra®:
- Almacene Hemlibra® a una temperatura de entre 2 y 8 °C en su envase original para protegerlo de la luz. No congele. No agite.
- Antes de la inyección, los frascos cerrados de Hemlibra® pueden retirarse del refrigerador y volver a colocarlos después. La temperatura y el tiempo total acumulado de almacenamiento de Hemlibra® fuera del refrigerador no deben superar los 30 °C ni los 7 días (a una temperatura de hasta 30 °C).
- Tras extraer el contenido del frasco, cualquier cantidad de Hemlibra® no utilizada debe desecharse si no se utiliza inmediatamente.
Instrucciones de uso
Comprobación del medicamento Hemlibra® y de los componentes del kit suministrado
-
Reúna todos los componentes indicados a continuación para preparar y realizar la inyección.
-
Compruebe la fecha de caducidad final en la caja de cartón, en la etiqueta del frasco y en el kit suministrado, tal como se indica a continuación. No utilice el medicamento ni los componentes indicados a continuación si esta fecha ya ha expirado.
-
No utilice el medicamento si:
- el medicamento está turbio, opaco o tiene un color intenso;
- el medicamento contiene partículas;
- falta el tapón sobre el cierre.
-
Compruebe los componentes del kit suministrado en busca de daños. No los utilice si están dañados o han caído al suelo.
-
Coloque los componentes del kit suministrado sobre una superficie de trabajo limpia, bien iluminada y plana.
| Los componentes del envase son: |
||
|
|
|
|
|
|
| Los siguientes elementos no forman parte del envase: |
||
Nota: si para la administración de la dosis prescrita es necesario utilizar el contenido de más de un frasco, debe utilizarse una toallita antiséptica nueva para cada frasco.
|
|
|
|
||
Nota: si para la administración de la dosis prescrita
|
||
|
||
|
|
|
| Preparación del frasco y del lugar de inyección
|
|
|
|
||





Preparación del jeringuilla para la inyección
- No se debe almacenar el medicamento Hemlibra® en la jeringa.
- El medicamento Hemlibra® en la jeringa debe administrarse inmediatamente por vía subcutánea.
Información importante sobre el procedimiento tras la inyección:
- No friccionar el lugar de la inyección tras su administración.
- Si aparece una gota de sangre en el lugar de la inyección, puede aplicar una compresa estéril de algodón o gasa y presionar durante al menos 10 segundos hasta que la hemorragia se detenga.
- Si se forma un hematoma (una pequeña zona de sangrado bajo la piel), también puede aplicar una bolsa de hielo sobre el lugar de la inyección, presionando suavemente. Si la hemorragia no se detiene, póngase en contacto con su médico.
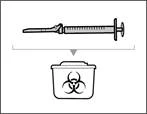
Eliminación de los viales, agujas y jeringas usados del medicamento Hemlibra®
Importante: mantenga siempre el contenedor para la eliminación de objetos punzantes en un lugar fuera del alcance de los niños.
- Inmediatamente después de su uso, coloque las agujas y jeringas usadas en un contenedor para la eliminación de objetos punzantes. No deseche agujas ni jeringas abiertas junto con la basura doméstica.
- Si no dispone de un contenedor para la eliminación de objetos punzantes, puede utilizar un recipiente de uso doméstico que:
- esté fabricado con plástico resistente;
- pueda cerrarse herméticamente con una tapa resistente a perforaciones, para evitar que los objetos punzantes salgan del contenedor;
- sea estable y pueda mantenerse de pie durante su uso;
- no tenga fugas;
- esté marcado adecuadamente para indicar que contiene desechos peligrosos.
- Cuando su contenedor para la eliminación de objetos punzantes esté casi lleno, siga las normas locales sobre el procedimiento correcto para desecharlo.
- No deseche ningún contenedor usado para la eliminación de objetos punzantes junto con la basura doméstica, salvo que las normativas locales lo permitan. No reutilice su contenedor para la eliminación de objetos punzantes.
Instrucción paso a paso para la administración de la inyección del medicamento Hemlibra®
| Paso 1. Retirar la tapa del frasco y limpiar la parte superior del tapón |
||
|
|
|
|
|
|
|
|
|
|
|
|
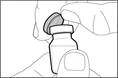
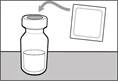
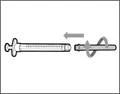
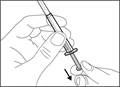
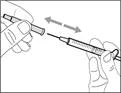
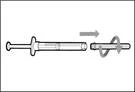
- Sujete la jeringa por el cilindro de manera que la aguja de transferencia quede orientada hacia arriba.
- Tire cuidadosamente del tapón de la aguja de transferencia alejándolo de usted (véase la figura 16). No deseche el tapón. Coloque el tapón de la aguja de transferencia sobre una superficie plana y limpia. Deberá volver a colocarlo sobre la aguja de transferencia después de extraer la solución del medicamento.
- No toque la punta de la aguja ni la apoye sobre ninguna superficie una vez retirado el tapón. || Paso 4. Introducir aire en el frasco | |
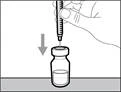
| * Mantenga el frasco sobre una superficie de trabajo plana e introduzca la aguja de transferencia con la jeringa verticalmente hacia abajo, en el centro del tapón del frasco (véase la figura 17). |
|
|
|
||||||
| Paso 5. Transferir el medicamento Hemlibra® al jeringuero
|
|||||||
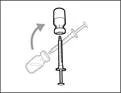
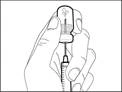
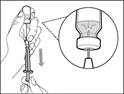
Importante: si la dosis prescrita supera la cantidad de medicamento Hemlibra® en un frasco, retire todo el medicamento Hemlibra® y continúe con la sección «Combinación de frascos».
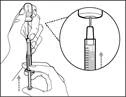
| Paso 6. Eliminar las burbujas de aire |
|
|
|
Nota: Antes de pasar al siguiente paso, debe asegurarse de que haya suficiente medicamento HEMLIBRA® en la jeringa para completar la dosis total. Si no consigue extraer todo el medicamento HEMLIBRA®, coloque el frasco en posición vertical para acceder al resto del contenido. |
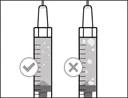
| No utilice la aguja de transferencia para administrar la inyección del medicamento |
||||
|
|
|
|||
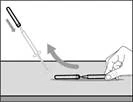
| Paso 8. Limpiar el área de la piel para la inyección |
||||
|
|
|
|||
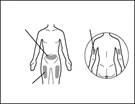
| Paso 10. Conectar la aguja de inyección a la jeringa |
||||
|
|
|
|||
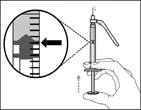
| Paso 13. Ajustar el émbolo hasta la marca correspondiente a la dosis prescrita |
||||
|
|
|
|||
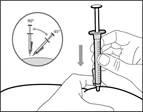
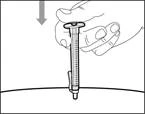
| Paso 14. Inyección subcutánea (bajo la piel) |
||||
|
|
|
|||
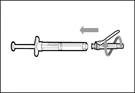
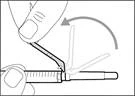
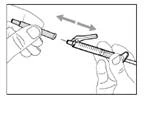
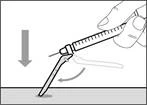
- Desplace la membrana protectora 90º hacia adelante en dirección opuesta al cilindro de la jeringa. * Sosteniendo la jeringa con una mano, presione firmemente hacia abajo la membrana protectora con un movimiento rápido y seguro hasta escuchar un clic (véase fig. 32). ||
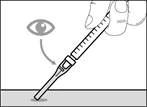
| * Si no ha escuchado el clic, compruebe si la aguja está completamente cubierta por la membrana protectora (véase fig. 33). * Mantenga siempre los dedos detrás de la membrana protectora y lejos de la aguja. * No desmonte la aguja de inyección de la jeringa. |
Paso 17. Deseche la jeringa y la aguja usadas (véase fig. 34)
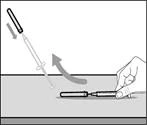
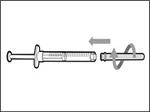
- Retire la jeringa y la aguja de transferencia del primer frasco. * Con una mano, introduzca la aguja de transferencia dentro del tapón y diríjala hacia arriba para que el tapón cubra la aguja (véase fig. 35). * Tan pronto como el tapón cubra la aguja de transferencia, con una mano diríjala hacia la jeringa para cerrarla firmemente y evitar lesiones accidentales por la aguja. | || Paso B. Retirar la aguja de transferencia | | | |
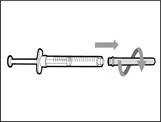
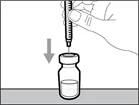
| | * Mantenga el nuevo frasco sobre una superficie plana de trabajo e introduzca la nueva aguja de transferencia con la jeringa en el centro del tapón del frasco en dirección descendente (véase fig. 39). | | |
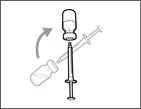
| | | * Mantenga la aguja de transferencia dentro del frasco e inviértalo boca abajo (véase fig. 40). | | |
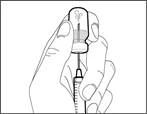
| | | * Dirigiendo la aguja hacia arriba, introduzca el aire de la jeringa en el frasco por encima de la solución del medicamento. * Continúe presionando con el dedo el émbolo de la jeringa (véase fig. 41). * No introduzca aire en la solución del medicamento, ya que podría provocar la formación de burbujas de aire o espuma en el producto. | | | Paso F. Aspirar el medicamento Hemlibra® en la jeringa | | | | | |
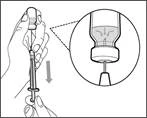
| | * Desplace la punta de la aguja hacia abajo de forma que quede dentro de la solución. * Tire lentamente del émbolo hacia atrás, evitando la formación de burbujas de aire o espuma. Llene la jeringa con una cantidad mayor del medicamento de la necesaria para la dosis prescrita (véase fig. 42). * Tenga cuidado: no extraiga el émbolo de la jeringa. Nota: Antes de continuar con los siguientes pasos, asegúrese de que la jeringa contiene suficiente cantidad del medicamento Hemlibra® para la dosis completa. Si no puede aspirar todo el medicamento Hemlibra®, invierta el frasco a posición vertical para extraer el remanente. | | | | No utilice la aguja de transferencia para la inyección del medicamento Hemlibra®, ya que podría causar lesiones como dolor o hemorragia. | | | | | | | Repita los pasos A–F para cada frasco adicional hasta obtener una cantidad del medicamento Hemlibra® mayor que la necesaria para la dosis prescrita. Tras completarlos, deje la aguja de transferencia en el frasco y vuelva al paso 6. Continúe con los pasos restantes (pasos 7–17). | | | | | | | | | |
Niños.
El medicamento se puede administrar a niños según las instrucciones indicadas en la sección «Instrucciones de uso y dosis».
Sobredosificación.
Existe una experiencia limitada con respecto a la sobredosificación con Hemlibra®. Una sobredosificación accidental podría provocar hipercoagulación.
En caso de sobredosificación accidental, debe buscarse inmediatamente atención médica y mantenerse bajo vigilancia médica.
Reacciones adversas
Las siguientes reacciones adversas graves se describen en otras secciones del prospecto:
- Microangiopatía trombótica asociada al uso de Hemlibra® y concentrado de complejo de activación de protrombina (CAPK) (ver sección «Precauciones de uso»).
- Tromboembolismo asociado al uso de Hemlibra® y CAPK (ver sección «Precauciones de uso»).
Datos de los estudios clínicos
Dado que los estudios clínicos se realizan en condiciones diferentes, la frecuencia de reacciones adversas observadas durante los estudios clínicos con un medicamento no puede compararse directamente con la frecuencia de reacciones adversas observadas durante los estudios clínicos con otro medicamento, y puede no reflejar la frecuencia observada en la práctica clínica.
En general, las reacciones adversas observadas en niños que recibieron tratamiento con Hemlibra® fueron similares en tipo a las observadas en pacientes adultos con hemofilia A.
Las reacciones adversas que se indican a continuación se han identificado basándose en datos combinados de dos estudios aleatorizados con adultos y adolescentes (HAVEN 1 y HAVEN 3), un estudio no comparativo con adultos y adolescentes (HAVEN 4), un estudio no comparativo con niños (HAVEN 2) y un estudio de selección de dosis, en los que un total de 391 hombres con hemofilia A recibieron al menos una dosis de Hemlibra® con fines de profilaxis rutinaria. Doscientos ochenta y un pacientes (72 %) eran adultos de 18 años o más, 50 (13 %) eran adolescentes de 12 a 18 años, 55 (14 %) eran niños de 2 a 12 años y 5 pacientes (1 %) eran niños de 1 mes a 2 años de edad. La mediana de duración del tratamiento en todos los estudios fue de 34,1 semanas (0,1–224,4 semanas).
Las reacciones adversas más frecuentes notificadas, observadas en ≥ 10 % de los pacientes que recibieron tratamiento con Hemlibra®, fueron reacciones en el lugar de inyección, cefalea y artralgia.
Cuatro pacientes (1 %) de los estudios clínicos que recibieron profilaxis con Hemlibra® interrumpieron el tratamiento debido a reacciones adversas, que incluyeron microangiopatía trombótica, necrosis cutánea, tromboflebitis de venas superficiales, cefalea y reacciones en el lugar de inyección.
Tabla 3
Reacciones adversas notificadas en ≥ 5 % de los pacientes, según los datos combinados de los estudios clínicos con Hemlibra®
| Sistema del organismo |
Reacción adversa |
Número de pacientes n (%) (N = 391) |
| Alteraciones generales y en el sitio de inyección |
Reacciones en el sitio de inyección * |
85 (22 %) |
| Pirexia |
23 (6 %) |
|
| Trastornos del sistema nervioso |
Cefalea |
57 (15 %) |
| Trastornos del sistema gastrointestinal |
Diárea |
22 (6 %) |
| Trastornos del sistema músculo-esquelético y del tejido conectivo |
Artalgia |
59 (15 %) |
* Incluyen equimosis en el lugar de inyección, molestias en el lugar de inyección, eritema en el lugar de inyección, hematoma en el lugar de inyección, induración en el lugar de inyección, dolor en el lugar de inyección, prurito en el lugar de inyección, erupción en el lugar de inyección, reacciones en el lugar de inyección, hinchazón en el lugar de inyección, urticaria en el lugar de inyección y sensación de calor en el lugar de inyección.
Características del tratamiento con PCAC en todos los estudios clínicos
Durante el tratamiento con PCAC se observaron 130 casos en 36 pacientes, de los cuales en 37 casos (10 %) el PCAC se administró a una dosis acumulativa media > 100 U/kg/24 h durante 24 horas o más; dos de los 13 casos estuvieron asociados con la aparición de eventos trombóticos y tres de los 13 casos estuvieron asociados con la aparición de MT (microangiopatía trombótica) (tabla 5). En los demás casos no se observaron eventos trombóticos ni MT durante el tratamiento con PCAC.
Tabla 4
Características del tratamiento* con PCAC en todos los estudios clínicos
| Duración del tratamiento con CAPC |
Dosis acumulativa media de CAPC a las 24 horas (U/kg/24 h) |
||
| < 50 |
50–100 |
> 100 |
|
| < 24 horas |
11 |
76 |
18 |
| 24–48 horas |
0 |
6 |
3a |
| > 48 horas |
1 |
5 |
10a,b,b,b |
* Un caso de tratamiento con FEIBA se define como todas las dosis de FEIBA recibidas por el paciente por cualquier motivo y hasta un intervalo de tratamiento de 36 horas.
a Evento trombótico.
b Microangiopatía trombótica.
Descripción de reacciones adversas individuales
Reacciones en el sitio de inyección
En general, se notificaron reacciones en el sitio de inyección (RSI) en 85 pacientes (22 %). Todas las RSI observadas durante los estudios clínicos con el medicamento Hemlibra® fueron de intensidad leve a moderada, y el 93 % desaparecieron sin tratamiento. Las RSI más frecuentemente notificadas fueron eritema en el sitio de inyección (11 %), prurito en el sitio de inyección (4 %) y dolor en el sitio de inyección (4 %).
Otras reacciones menos frecuentes (< 1 %)
Rabdomiólisis
Se notificó rabdomiólisis en dos pacientes adultos con aumento asintomático de los niveles séricos de creatincinasa, sin síntomas asociados de afectación renal o del sistema músculo-esquelético. En ambos casos, la rabdomiólisis ocurrió tras un incremento de la actividad física.
Experiencia tras la comercialización
Durante la experiencia tras la comercialización del medicamento Hemlibra®, se han identificado las siguientes reacciones adversas. Dado que los informes sobre estas reacciones se han recibido de forma voluntaria de una población de tamaño desconocido, no siempre es posible estimar con fiabilidad su frecuencia ni establecer una relación causal con la exposición al medicamento.
Alteraciones de la piel y del tejido celular subcutáneo: erupción cutánea, urticaria, angioedema.
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite continuar con el seguimiento de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar de todos los casos sospechosos de reacciones adversas y de falta de eficacia del medicamento a través del sistema automatizado de información sobre farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua».
Período de validez
24 meses.
Condiciones de almacenamiento.
Conservar a una temperatura de entre 2 y 8 °C en el envase original para protegerlo de la luz. No congelar. No agitar. Mantener fuera del alcance de los niños.
Incompatibilidad.
No se ha observado incompatibilidad del medicamento Hemlibra® con las jeringas y agujas recomendadas.
Envase.
12 mg/0,4 ml o 30 mg/1 ml o 60 mg/0,4 ml o 105 mg/0,7 ml o 150 mg/1 ml por vial.
1 vial por caja de cartón.
Categoría de dispensación.
Bajo receta.
Fabricante.
F. Hoffmann-La Roche Ltd.
Domicilio del fabricante y dirección del lugar de actividad.
Wurmisweg, 4303 Kaiseraugst, Suiza