Hemlibra
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT HEMLIBRAÒ (HEMLIBRAÒ)
Composition:
Active substance: emicizumab;
1 vial contains 12 mg/0.4 mL or 30 mg/1 mL or 60 mg/0.4 mL or 105 mg/0.7 mL or 150 mg/1 mL of emicizumab;
Excipients: L-histidine, L-aspartic acid, L-arginine, poloxamer 188, water for injections.
Pharmaceutical form. Solution for injection.
Main physicochemical properties: sterile, clear liquid, colorless to slightly yellowish, without preservatives.
Pharmacotherapeutic group.
Agents affecting the blood and hematopoietic system. Antihemorrhagics. Vitamin K and other hemostatics. Other systemic hemostatics. Emicizumab.
ATC code B02BX06.
Pharmacological properties.
Pharmacodynamics.
Mechanism of action
Hemlibra® binds activated factor IX and factor X, restoring the deficient function of activated factor VIII, which is necessary for effective hemostasis.
Pharmacokinetics.
Emicizumab demonstrated dose-proportional pharmacokinetics over a dose range of 0.3 mg/kg (0.1 of the approved recommended starting dose) to 6 mg/kg following subcutaneous administration. After multiple subcutaneous injections of emicizumab at a loading dose of 3 mg/kg once weekly for the first 4 weeks, the mean (± SD) trough plasma concentration of emicizumab was 52.6 ± 13.6 µg/mL, reached by week 5. Information on the steady-state mean (± SD) plasma concentration of emicizumab with recommended maintenance dosing regimens is presented in Table 1.
Table 1
Mean (± SD) steady-state concentrations following emicizumab loading dose according to maintenance treatment regimen
| Parameters |
Maintenance dose |
||
| 1.5 mg/kg body weight once weekly |
3 mg/kg body weight once every 2 weeks |
6 mg/kg body weight once every 4 weeks |
|
| Cmax,ss (μg/mL) |
55.1 ± 15.9 |
58.3 ± 16.4 |
67 ± 17.7 |
| AUCss,τ (μg/mL*day) |
376 ± 109 |
752 ± 218 |
1503 ± 437 |
| Cmin,ss (μg/mL) |
51.2 ± 15.2 |
46.9 ± 14.8 |
38.5 ± 14.2 |
| Cmax/Cmin ratio (μg/mL) |
1.08 ± 0.03 |
1.26 ± 0.12 |
1.85 ± 0.47 |
AUCss,τ – area under the plasma concentration-time curve at steady state over the dosing interval (τ = 1, 2, or 4 weeks); Cmax,ss – maximum plasma concentration of the drug at steady state; Cmin,ss – minimum plasma concentration of the drug at steady state.
Absorption
After subcutaneous administration, mean (± SD) absorption half-life values were 1.6 ± 1 day. Absolute bioavailability following subcutaneous administration at a dose of 1 mg/kg ranged from 80.4% to 93.1%. Similar pharmacokinetic profiles were observed after subcutaneous administration in the abdominal, shoulder, and thigh regions (see section “Dosage and administration”).
Distribution
The mean apparent volume of distribution (% coefficient of variation [%CV]) was 10.4 L (26%).
Elimination
The mean apparent clearance (%CV) was 0.27 L/day (28.4%), and the mean apparent elimination half-life (± SD) was 26.9 ± 9.1 days.
Special Populations
The pharmacokinetics of emicizumab are not influenced by patient age (1 to 77 years), race (Caucasian – 62.7%, Mongoloid – 22.9%, and Negroid – 8%), inhibitor status (inhibitors present in 50%), mild hepatic impairment (defined as total bilirubin levels 1–1.5 times above the upper limit of normal (ULN) with any level of aspartate aminotransferase (AST)), moderate hepatic impairment (defined as total bilirubin levels 1.5–3 times above ULN with any AST level), mild renal impairment (defined as creatinine clearance of 60–89 mL/min), or moderate renal impairment (defined as creatinine clearance of 30–59 mL/min). The use of emicizumab in patients with severe hepatic or renal impairment has not been studied.
In children under 6 months of age, predicted emicizumab concentrations were 19–33% lower than in older patients, particularly when receiving a maintenance dose of 3 mg/kg body weight once every 2 weeks or 6 mg/kg body weight once every 4 weeks.
Children
The efficacy and safety of Hemlibra® in children have been established. The use of Hemlibra® in children with hemophilia A is supported by results from two randomized trials (HAVEN 1 and HAVEN 3) and two non-comparative trials (HAVEN 2 and HAVEN 4). Across all clinical trials, only one age group of pediatric patients was included: 47 adolescents aged 12 to 18 years. Only in the HAVEN 2 trial were children from multiple age groups enrolled: 55 children aged 2 to 12 years and 5 children aged 1 month to 2 years. No differences in drug efficacy were observed among children of different age groups.
Minimum steady-state plasma concentrations of emicizumab were comparable between adults and children aged 6 months and older receiving body weight-adjusted equivalent doses. Lower emicizumab concentrations are expected in children under 6 months of age.
Body Weight. Apparent clearance and volume of distribution of emicizumab increase with increasing body weight (from 9 to 156 kg). Dosing in milligrams per kilogram (mg/kg) provides similar emicizumab exposures across the entire body weight range.
Immunogenicity
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the frequency of antibody formation to the drug between the studies described below and other studies, including studies of Hemlibra® or other emicizumab products.
Based on clinical trial data for Hemlibra®, anti-emicizumab antibodies were detected in 5.1% of patients (34/668). Participants received emicizumab for a median interquartile range (IQR) of 103.1 (82.4–148.1) weeks. Samples for anti-drug antibody testing were collected at the beginning of treatment and periodically throughout the study.
Impact of Anti-Drug Antibodies on Pharmacokinetics
Antibody-positive samples were further tested for the presence of neutralizing anti-emicizumab antibodies using a modified chromogenic FVIII assay. Overall, 668 patients were tested for anti-emicizumab antibodies. Anti-emicizumab antibodies were detected in 5.1% of patients (34/668), and in 2.7% of patients (18/668), the antibodies were neutralizing in vitro. Among these 2.7% of patients (18/668), neutralizing anti-emicizumab antibodies had no clinically significant impact on the pharmacokinetics of Hemlibra® in 2.1% of patients (14/668), while decreased emicizumab plasma concentrations were observed in 0.6% of patients (4/668).
Impact of Anti-Drug Antibodies on Pharmacodynamics
One patient (1/668; 0.1%) developed neutralizing anti-emicizumab antibodies, resulting in decreased emicizumab plasma concentration and subsequent loss of drug efficacy (manifested as breakthrough bleeding) 5 weeks after initiation of treatment. Treatment with Hemlibra® in this patient was discontinued prematurely (see section “Special precautions for use”).
Clinical Characteristics.
Indications.
Routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children, from birth onwards, with haemophilia A (congenital factor VIII deficiency) with or without factor VIII inhibitors.
Contraindications.
Hypersensitivity to emicizumab or to any of the excipients of the medicinal product.
Interaction with other medicinal products and other forms of interactions.
Drug interaction studies
No drug interaction studies have been conducted with Hemlibra® and other medicinal products.
Hypocoagulability when used concomitantly with aPCC
Clinical experience indicates that there is an interaction between Hemlibra® and activated prothrombin complex concentrate (aPCC) (see section "Special warnings and precautions for use").
Effect of the medicinal product on laboratory test results
Hemlibra® restores the cofactor activity of activated factor VIII (aFVIII). Laboratory tests assessing the intrinsic pathway of coagulation cascade activation (e.g., activated partial thromboplastin time (aPTT)) measure overall clotting time, including the time required for thrombin-mediated activation of FVIII to aFVIII, and result in an exaggerated shortening of clotting time with Hemlibra® treatment, since thrombin activation is not required. This exaggerated shortening of the intrinsic pathway clotting time will continue to affect results of all one-stage factor assays based on aPTT measurement, including one-stage factor VIII activity assays. However, single-factor assays using chromogenic or immunological methods are not influenced by Hemlibra® and may be used for monitoring coagulation parameters during treatment, with important considerations regarding the chromogenic factor VIII activity assay as described below.
Chromogenic factor VIII activity assays may be manufactured using either human or bovine coagulation proteins. Assays containing human coagulation factors are sensitive to Hemlibra®, but may overestimate the clinical haemostatic potential of Hemlibra®. In contrast, assays based on bovine coagulation factors are insensitive to Hemlibra® (activity is not measured) and can be used to monitor endogenous or infused FVIII activity, as well as for detection of inhibitors against FVIII.
Hemlibra® retains activity in the presence of FVIII inhibitors, which may lead to falsely negative results in the Bethesda assay for functional FVIII inhibition using a clotting method. In contrast, a chromogenic Bethesda assay using bovine FVIII, which is insensitive to Hemlibra®, may be used.
Due to the long half-life of Hemlibra®, its effect on coagulation parameters may persist for up to 6 months after the last dose (see section "Pharmacological properties").
Special precautions for use.
| WARNING: THROMBOTIC MICROANGIOPATHY AND THROMBOEMBOLISM Cases of thrombotic microangiopathy and thromboembolism have been reported in clinical trials in patients receiving prophylactic doses of HemlibraÒ when activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more at a mean cumulative dose > 100 IU/kg/24 hours. Monitoring for thrombotic microangiopathy and thromboembolic events is required when aPCC is used. Administration of aPCC should be discontinued and the next dose of HemlibraÒ should be delayed if symptoms occur. |
Thrombotic microangiopathy associated with HEMLIBRA® and activated prothrombin complex concentrate (aPCC)
Cases of thrombotic microangiopathy (TMA) have been reported in patients receiving prophylactic treatment with HEMLIBRA® when activated prothrombin complex concentrate (aPCC) was administered for 24 hours or more at a mean cumulative dose > 100 IU/kg/24 hours. In clinical trials, TMA was reported in 0.8% of patients (3 out of 391) and in 8.1% of patients (3 out of 37) who received at least one dose of aPCC. In the ADAMTS13 study, patients exhibited thrombocytopenia, microangiopathic hemolytic anemia, and acute kidney injury without signs of overt organ failure.
Signs of clinical improvement were observed within one week after discontinuation of aPCC. One patient continued HEMLIBRA® treatment after resolution of TMA.
For patients receiving prophylactic treatment with HEMLIBRA®, the benefits and risks should be carefully weighed if aPCC administration is required. Due to the long half-life of HEMLIBRA®, the potential for interaction with aPCC may persist for up to 6 months after the last dose. Monitoring for the development of TMA is recommended during aPCC administration. Treatment with aPCC should be discontinued immediately and prophylaxis with HEMLIBRA® should be interrupted if clinical and/or laboratory signs suggestive of TMA occur; appropriate treatment should be initiated according to clinical judgment. The potential benefits and risks of continuing HEMLIBRA® prophylaxis after resolution of TMA should be evaluated on a case-by-case basis.
Thromboembolism associated with HEMLIBRA® and aPCC
Cases of thrombosis have been reported in patients receiving prophylactic doses of HEMLIBRA® when aPCC was administered for 24 hours or more at a mean cumulative dose > 100 IU/kg/24 hours. In clinical trials, thrombosis was reported in 0.5% of patients (2 out of 391) and in 5.4% of patients (2 out of 37) who received at least one dose of aPCC.
No thrombotic events required anticoagulant therapy. Signs of improvement or recovery were observed within one week after discontinuation of aPCC. One patient continued HEMLIBRA® treatment after resolution of thrombosis.
For patients receiving prophylactic treatment with HEMLIBRA®, the benefits and risks should be carefully weighed if aPCC administration is required. Due to the long half-life of HEMLIBRA®, the potential for interaction with aPCC may persist for up to 6 months after the last dose. Monitoring for thromboembolic events is recommended during aPCC administration. Treatment with aPCC should be discontinued immediately and prophylaxis with HEMLIBRA® should be interrupted if clinical symptoms, imaging findings, or laboratory signs suggestive of thromboembolism occur; appropriate treatment should be initiated according to clinical judgment. The potential benefits and risks of continuing HEMLIBRA® prophylaxis after resolution of a thrombotic event should be evaluated on a case-by-case basis.
Immunogenicity
Treatment with HEMLIBRA® may induce the formation of antibodies against the medicinal product. Antibodies to emicizumab were reported in 5.1% of patients (34 out of 668) treated with HEMLIBRA® in clinical trials. Most patients with anti-emicizumab antibodies did not experience changes in HEMLIBRA® plasma concentrations or increased bleeding episodes; however, in rare cases (< 1% prevalence), the presence of neutralizing antibodies with reduced plasma concentrations may be associated with loss of efficacy (see section "Pharmacokinetics").
Patients should be monitored for clinical signs of loss of efficacy (e.g., increased breakthrough bleeding episodes). If such signs occur, the cause should be promptly evaluated and a change in therapy should be considered if neutralizing antibodies to emicizumab are suspected.
Effect on coagulation test parameters
HEMLIBRA® affects laboratory tests of the intrinsic pathway of coagulation cascade activation, including activated clotting time (ACT), activated partial thromboplastin time (aPTT), and all assays based on aPTT, including one-stage factor VIII (FVIII) activity assays (Table 2). Therefore, coagulation tests based on the intrinsic pathway using clotting-based methods should not be used for monitoring HEMLIBRA® activity, determining replacement therapy dosing, anticoagulation monitoring, or measuring FVIII inhibitor titers in patients receiving HEMLIBRA® (see section "Interaction with other medicinal products and other forms of interaction"). Coagulation tests affected and not affected by HEMLIBRA® are listed in Table 2.
Table 2
Coagulation test results affected and not affected by HEMLIBRA®
| Test results affected by HemlibraÒ |
Test results not affected by HemlibraÒ |
| Activated partial thromboplastin time (aPTT) Bethesda assay (clotting method) for determination of FVIII inhibitor titers One-stage, one-factor assay based on aPTT aPTT-based activated protein C resistance (APC-R) Activated clotting time (ACT) |
Bethesda assay (with bovine chromogenic substrate) for determination of FVIII inhibitor titers Thrombin time (TT) One-stage, prothrombin time (PT)-based, one-factor assay One-factor chromogenic assay for factors other than FVIII* Immunological assays (e.g., ELISA, turbidimetric methods) Genetic tests for coagulation factors (e.g., Factor V Leiden, prothrombin 20210) |
* For important information regarding the determination of FVIII activity using the chromogenic method, see section "Interaction with other medicinal products and other forms of interaction".
Use during pregnancy or breastfeeding.
Pregnancy
There are no available data on the risk of major congenital malformations or miscarriage associated with the use of Hemlibra® in pregnant women. Reproductive studies with emicizumab in animals have not been conducted. It is not known whether Hemlibra® can cause fetal harm or affect reproductive capacity when administered to pregnant women. Hemlibra® should be used during pregnancy only if the potential benefit to the woman outweighs the potential risk to the fetus.
In every pregnancy, there is a background risk of congenital malformations, fetal loss, or other adverse outcomes. The estimated background risk of major congenital malformations or miscarriage in the target populations is unknown. In the general US population, the estimated risk of major congenital malformations or miscarriage during clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Breastfeeding
There is no information available on the excretion of emicizumab in human breast milk, its potential effect on the breastfed child, or its effect on milk production. It is known that human IgG is present in human breast milk. The benefits of breastfeeding for child development and health should be weighed against the mother's clinical need for Hemlibra®, the potential for any adverse effects in the breastfed child, and the mother's overall health condition.
Contraception
Women of reproductive potential should use contraceptive methods during treatment with Hemlibra®.
Effect on ability to drive and use machines.
There are no data indicating that treatment with Hemlibra® leads to an increased incidence of adverse reactions that could impair the ability to drive or operate machinery.
Method of Administration and Dosage
For subcutaneous use only.
The recommended loading dose is 3 mg/kg body weight administered subcutaneously once weekly for the first 4 weeks. After this period, the drug should be administered at the maintenance dose:
- 1.5 mg/kg body weight once weekly, or
- 3 mg/kg body weight once every two weeks, or
- 6 mg/kg body weight once every four weeks.
The choice of maintenance dose should be based on the preference of the healthcare provider, considering regimens that may enhance patient adherence to the treatment schedule.
Prophylactic use of bypassing agents should be discontinued one day prior to initiating prophylactic treatment with Hemlibra®.
Prophylactic administration of factor VIII (FVIII) products may continue during the first week of initiating prophylactic treatment with Hemlibra®.
Missed Dose
If a dose of Hemlibra® is missed, it should be administered as soon as possible, followed by resuming the regular dosing schedule. Do not administer two doses on the same day to make up for a missed dose.
Special Dosage Recommendations
Children
Dose adjustment in children is not recommended. There are no data available from studies in children under 1 year of age.
Elderly Patients
Clinical trials of Hemlibra® included an insufficient number of patients aged 65 years and older to determine whether they respond differently compared to younger patients. Other reported clinical experience has not identified differences in responses between elderly and younger patients.
Preparation and Administration
Hemlibra® is intended for use only under the supervision of a physician. After appropriate training in subcutaneous injection techniques, a patient may self-administer the injection or have it administered by a caregiver, if the physician determines this to be appropriate. Self-administration is not recommended for children under 7 years of age.
Detailed instructions for the preparation and administration of Hemlibra® are provided below (see section "Instructions for Use").
Prior to administration, visually inspect Hemlibra® for the presence of particulate matter or discoloration. Hemlibra® for subcutaneous administration is a solution ranging from colorless to slightly yellow. Do not use the product if visible particles are present or if discoloration occurs.
To withdraw Hemlibra® from the vial and administer it subcutaneously, a syringe, a filter needle for transfer, and an injection needle are required.
Refer to the section "Instructions for Use" below for guidance on handling Hemlibra® when multiple vials are used in combination.
Do not mix Hemlibra® from vials with different concentrations (i.e., 30 mg/mL and 150 mg/mL) in the same injection.
See below for criteria for selecting the recommended medical device options. Doses of Hemlibra® up to 1 mL should be administered using a 1 mL syringe. A 1 mL syringe meeting the following criteria may be used: clear syringe made of polypropylene or polycarbonate with a Luer-Lock tip (if unavailable, a syringe with a Luer-Slip tip may be used), graduated in 0.01 mL increments, sterile, for injection use only, single-use, latex-free, and pyrogen-free.
Injections of Hemlibra® with volumes from 1 mL to 2 mL should be administered using 2 mL or 3 mL syringes meeting the following criteria: clear syringe made of polypropylene or polycarbonate with a Luer-Lock tip (if unavailable, a syringe with a Luer-Slip tip may be used), graduated in 0.1 mL increments, sterile, for injection use only, single-use, latex-free, and pyrogen-free.
A filter transfer needle meeting the following criteria must be used: stainless steel needle with Luer-Lock connection (if unavailable, a Luer-Slip connection may be used), sterile, 18G size, 1 to 1½ inches in length, with a single-beveled or blunt (or semi-blunt) tip, single-use, latex-free, equipped with a 5-micron filter, and pyrogen-free.
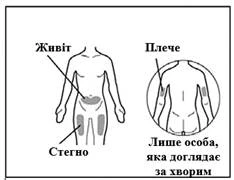
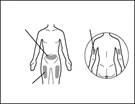
An injection needle meeting the following criteria may be used: stainless steel needle with Luer-Lock connection (if unavailable, a Luer-Slip connection may be used), sterile, 26G size (acceptable range: 25–27G), preferably 3/8 inch in length with a maximum of ½ inch, single-use, latex-free, pyrogen-free, and equipped with a safety shield. Each new injection of Hemlibra® should be administered at a different anatomical site (outer surface of the upper arm, thigh, or any abdominal quadrant), distinct from the site used for the previous injection. Injections must never be administered into moles, scars, or tender, bruised, red, hardened, or damaged skin areas. Administration of Hemlibra® into the outer surface of the upper arm should only be performed by a caregiver or healthcare professional.
Any unused portion of Hemlibra® remaining in a single-use vial must be discarded.
Storage of Hemlibra®:
- Store Hemlibra® at 2–8°C in the original packaging to protect from light. Do not freeze. Do not shake.
- Prior to injection, closed vials of Hemlibra® may be removed from refrigeration and returned to the refrigerator. The temperature and total cumulative time outside the refrigerator must not exceed 30°C and 7 days (at temperatures up to 30°C).
- After drawing the solution from the vial, any remaining Hemlibra® in the vial must be discarded if not used immediately.
Instructions for Use
Inspection of Hemlibra® and Components of the Supply Kit
-
Gather all components listed below to prepare and administer the injection.
-
Check the expiration date on the carton, vial label, and supply kit as indicated below. Do not use the product or the components listed below if this date has passed.
-
Do not use the product if:
- the medicinal product is cloudy, opaque, or deeply colored;
- the medicinal product contains particles;
- the cap over the stopper is missing.
-
Inspect the components of the supply kit for damage. Do not use if damaged or if dropped on the floor.
-
Place the components of the supply kit on a clean, well-lit, flat working surface.
| Contents of the pack include: |
||
|
|
|
|
|
|
| Items not included in the pack: |
||
Note: If more than one vial is required to administer the prescribed dose, use a new alcohol swab for each vial.
|
|
|
|
||
Note: If more than one vial is required to administer the prescribed dose
|
||
|
||
|
|
|
| Preparing the vial and injection site
|
|
|
|
||





Preparation of the injection syringe
- Hemlibra® must not be stored in the syringe.
- Hemlibra® in the syringe must be administered immediately by subcutaneous injection.
Important information on post-injection care:
- Do not rub the injection site after administration.
- If there is any bleeding at the injection site, apply a sterile cotton ball or gauze and press firmly for at least 10 seconds until bleeding stops.
- If a hematoma (a small area of bleeding under the skin) develops, an ice pack may also be applied gently to the injection site. If bleeding does not stop, please contact your doctor.
Disposal of used Hemlibra® vials, needles, and syringes
Important: Always keep the sharps disposal container in a location inaccessible to children.
- Immediately after use, place used needles and syringes into the sharps disposal container. Do not dispose of any open needles or syringes with household waste.
- If you do not have a sharps disposal container, you may use a household container that:
- is made of heavy-duty plastic;
- can be tightly closed with a puncture-resistant lid to prevent sharps from protruding;
- is stable and can stand upright during use;
- is leak-proof;
- is clearly labeled to indicate the presence of hazardous waste inside.
- When your sharps disposal container is nearly full, follow local guidelines for the proper method of disposing of the sharps container.
- Do not dispose of any used sharps disposal container with household waste, unless permitted by local regulations. Do not reuse your sharps disposal container.
Step-by-step instructions for administering Hemlibra® injection
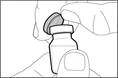
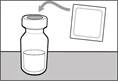
| Step 1. Remove the cap from the vial and clean the top of the stopper |
||
|
|
|
|
|
|
|
|
|
|
|
|
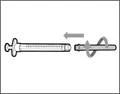
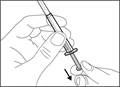
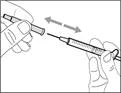
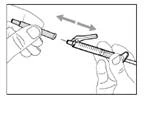
- Hold the syringe by the barrel with the transfer needle pointing upwards.
- Carefully pull the transfer needle cap away from you (see Fig. 16). Do not discard the cap. Place the transfer needle cap on a clean, flat surface. After drawing the medication solution, the cap should be replaced back onto the transfer needle.
- Do not touch the tip of the needle and do not place it on any surface after removing the cap. || Step 4. Insert air into the vial | | |
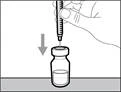
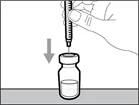
| * Hold the vial on a flat working surface and insert the transfer needle with syringe straight down through the center of the vial stopper (see Fig. 17). |
|
|
|
||||||
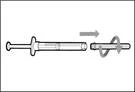
| Step 5. Transfer Hemlibra® medicine into the syringe
Important: If the prescribed dose exceeds the amount of Hemlibra® in the vial, draw up all the Hemlibra® and proceed to the section "Combining vials". |
|||||||
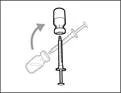
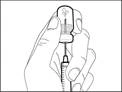
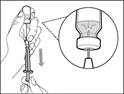
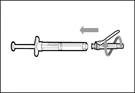
| Step 6. Remove air bubbles |
|
|
|
Note: Before proceeding to the next step, ensure that sufficient Hemlibra® is present in the syringe to administer the full dose. If you are unable to draw up all of the Hemlibra®, return the vial to an upright position to access the remaining solution. |
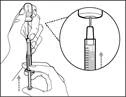
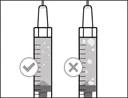
| Do not use the transfer needle to administer the HemlibraÒ medication, as this may cause injury such as pain and bleeding. |
||||
|
|
|
|||
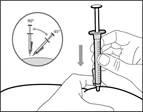
| Step 8. Clean the injection site on the skin |
||||
|
|
|
|||
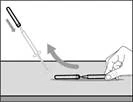
| Step 10. Attach the injection needle to the syringe |
||||
|
|
|
|||
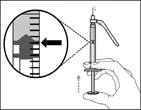
| Step 13. Adjust the plunger to the mark corresponding to the prescribed dose |
||||
|
|
|
|||
| Step 14. Subcutaneous (under the skin) injection |
||||
|
|
|
|||
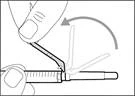
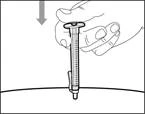
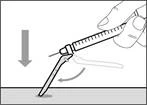
- Move the protective shield forward 90º in the direction away from the syringe barrel. * Holding the syringe in one hand, press the protective shield downward firmly and confidently onto the broad surface until a click is heard (see Fig. 32). ||
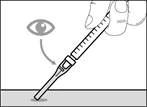
| * If you did not hear a click, check whether the needle is completely covered by the protective shield (see Fig. 33). * Always keep your fingers behind the protective shield and away from the needle. * Do not detach the injection needle from the syringe. |
Step 17. Dispose of (discard) the used syringe and needle (see Fig. 34)
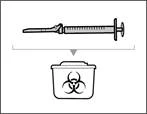
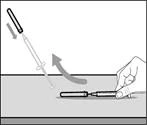
- Remove the syringe and transfer needle from the first vial. * With one hand, insert the transfer needle into the cap and direct it upward so that the cap covers the needle (see Fig. 35). * As soon as the cap covers the transfer needle, use one hand to guide it toward the syringe to close it securely and prevent accidental needlestick injury. | || Step B. Remove the transfer needle | | | |
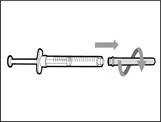
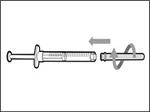
| | * Hold the new vial on a flat working surface and insert the new transfer needle with the syringe centrally into the vial stopper, directing downward (see Fig. 39). | | |
| | | * Keep the transfer needle in the vial and turn the vial upside down (see Fig. 40). | | |
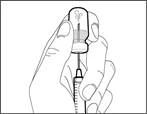
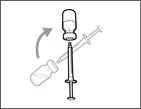
| | | * With the needle pointing upward, inject air from the syringe into the vial above the Hemlibra® solution. * Continue pressing with your finger on the syringe plunger (see Fig. 41). * Do not inject air into the Hemlibra® solution, as this may cause formation of air bubbles or foam in the product. | | | Step F. Draw Hemlibra® into the syringe | | | | | |
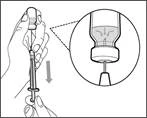
| | * Position the needle tip downward so that it is immersed in the solution. * Slowly pull back on the plunger, avoiding formation of air bubbles or foam. Fill the syringe with more than the required amount for the prescribed dose (see Fig. 42). * Caution: Do not pull the plunger out of the syringe. Note: Before proceeding to the next steps, ensure that sufficient Hemlibra® is drawn into the syringe to provide the full dose. If not all of the Hemlibra® can be drawn up, return the vial to an upright position to access the remaining solution. | | | | Do not use the transfer needle to administer the Hemlibra® injection, as this may cause injury such as pain and bleeding. | | | | | | | Repeat steps A–F for each additional vial until you have drawn up more than the required amount of Hemlibra® for the prescribed dose. After completing these steps, leave the transfer needle in the vial and return to Step 6. Continue with the remaining steps (Steps 7–17). | | | | | | | | | |
Children.
Hemlibra® should be administered to children according to the instructions provided in the section "Dosage and Administration".
Overdose.
Experience with Hemlibra® overdose is limited. Accidental overdose may lead to hypercoagulability.
In case of accidental overdose, seek immediate medical advice and remain under medical supervision.
Adverse Reactions
The following serious adverse reactions are described in other sections of the prescribing information:
- Thrombotic microangiopathy associated with Hemlibra® and activated prothrombin complex concentrate (aPCC) (see section "Special Warnings and Precautions for Use").
- Thromboembolism associated with Hemlibra® and aPCC (see section "Special Warnings and Precautions for Use").
Clinical Trial Data
Because clinical trials are conducted under widely varying conditions, the frequency of adverse reactions observed in clinical trials of one product may not be directly compared to the frequency in trials of another product, and may not reflect the frequency observed in clinical practice.
Overall, adverse reactions in children treated with Hemlibra® were similar in type to those observed in adult patients with hemophilia A.
The adverse reactions listed below are based on pooled data from two randomized trials in adults and adolescents (HAVEN 1 and HAVEN 3), one single-arm trial in adults and adolescents (HAVEN 4), one single-arm trial in children (HAVEN 2), and a dose-finding trial, in which a total of 391 male patients with hemophilia A received at least one dose of Hemlibra® for routine prophylaxis. Of these, 281 patients (72%) were adults aged 18 years and older, 50 (13%) were adolescents aged 12 to 18 years, 55 (14%) were children aged 2 to 12 years, and 5 patients (1%) were children aged 1 month to 2 years. The median duration of treatment across all studies was 34.1 weeks (range: 0.1–224.4 weeks).
The most commonly reported adverse reactions observed in ≥ 10% of patients treated with Hemlibra® were injection site reactions, headache, and arthralgia.
Four patients (1%) in the clinical trials receiving Hemlibra® prophylaxis discontinued treatment due to adverse reactions, including thrombotic microangiopathy, skin necrosis, superficial venous thrombophlebitis, headache, and injection site reactions.
Table 3
Adverse reactions reported in ≥ 5% of patients based on pooled clinical trial data for Hemlibra®
| System organ class |
Adverse reaction |
Number of patients n (%) (N = 391) |
| General disorders and administration site conditions |
Injection site reactions * |
85 (22 %) |
| Pyrexia |
23 (6 %) |
|
| Nervous system disorders |
Headache |
57 (15 %) |
| Gastrointestinal disorders |
Diarrhea |
22 (6 %) |
| Musculoskeletal and connective tissue disorders |
Arthralgia |
59 (15 %) |
* Include bruising at injection site, discomfort at injection site, erythema at injection site, hematoma at injection site, induration at injection site, pain at injection site, pruritus at injection site, rash at injection site, injection site reactions, swelling at injection site, urticaria at injection site, and sensation of warmth at injection site.
Characteristics of aPCC treatment in all clinical studies
During aPCC treatment, 130 cases were observed in 36 patients, of which in 37 cases (10%) aPCC was administered at a mean cumulative dose >100 IU/kg/24 hours for 24 hours or longer; two out of 13 cases were associated with thrombotic events and three out of 13 cases were associated with the occurrence of TMA (Table 5). In the other cases, no thrombotic events or TMA were observed during aPCC treatment.
Table 4
Characteristics of aPCC treatment* in all clinical studies
| Duration of AKPK treatment |
Average cumulative dose of CAPK within 24 hours (IU/kg/24 h) |
||
| < 50 |
50–100 |
> 100 |
|
| < 24 hours |
11 |
76 |
18 |
| 24–48 hours |
0 |
6 |
3a |
| > 48 hours |
1 |
5 |
10a,b,b,b |
* A case of FVIII exposure is defined as all FVIII doses received by a patient for any reason and prior to a 36-hour treatment interruption.
a Thrombotic event.
b Thrombotic microangiopathy.
Description of selected adverse reactions
Injection site reactions
Overall, injection site reactions (ISRs) were reported in 85 patients (22%). All ISRs observed during clinical studies with Hemlibra® were mild to moderate in intensity, and 93% resolved without treatment. The most frequently reported ISRs were injection site erythema (11%), injection site pruritus (4%), and injection site pain (4%).
Other less common (< 1%) reactions
Rhabdomyolysis
Rhabdomyolysis was reported in two adult patients with asymptomatic elevations in serum creatine kinase levels without associated renal or musculoskeletal symptoms. In both cases, rhabdomyolysis occurred following increased physical activity.
Post-marketing experience
The following adverse reactions have been identified during post-marketing use of Hemlibra®. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and subcutaneous tissue disorders: rash, urticaria, angioedema.
Reporting of adverse reactions after marketing authorization of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare and pharmaceutical professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the automated pharmacovigilance information system at the following link: https://aisf.dec.gov.ua».
Shelf life.
24 months.
Storage conditions.
Store at 2 to 8 °C in the original packaging to protect from light. Do not freeze. Do not shake. Keep out of reach of children.
Incompatibilities.
No incompatibility of Hemlibra® with the recommended syringes and needles has been observed.
Packaging.
12 mg/0.4 mL or 30 mg/1 mL or 60 mg/0.4 mL or 105 mg/0.7 mL or 150 mg/1 mL in a vial.
1 vial per carton.
Prescription category.
Prescription only.
Manufacturer.
F. Hoffmann-La Roche Ltd
Manufacturer's address and place of business.
Wurmisweg, 4303 Kaiseraugst, Switzerland