Advait
UcraniaContenido
INSTRUCCIONES para uso médico del medicamento ADVAYT (ADVATE)
Composición:
Principio activo: factor de coagulación VIII (octocog alfa);
1 vial contiene:
250 UI* de factor de coagulación sanguínea humana VIII recombinante (ADNr)**, octocog alfa, que tras la reconstitución equivale aproximadamente a 50 UI/ml de factor de coagulación sanguínea humana VIII recombinante, octocog alfa;
500 UI* de factor de coagulación sanguínea humana VIII recombinante (ADNr)**, octocog alfa, que tras la reconstitución equivale aproximadamente a 100 UI/ml de factor de coagulación sanguínea humana VIII recombinante, octocog alfa;
1000 UI* de factor de coagulación sanguínea humana VIII recombinante (ADNr)**, octocog alfa, que tras la reconstitución equivale aproximadamente a 200 UI/ml de factor de coagulación sanguínea humana VIII recombinante, octocog alfa;
1500 UI* de factor de coagulación sanguínea humana VIII recombinante (ADNr)**, octocog alfa, que tras la reconstitución equivale aproximadamente a 300 UI/ml de factor de coagulación sanguínea humana VIII recombinante, octocog alfa;
2000 UI* de factor de coagulación sanguínea humana VIII recombinante (ADNr)**, octocog alfa, que tras la reconstitución equivale aproximadamente a 400 UI/ml de factor de coagulación sanguínea humana VIII recombinante, octocog alfa;
3000 UI* de factor de coagulación sanguínea humana VIII recombinante (ADNr)**, octocog alfa, que tras la reconstitución equivale aproximadamente a 600 UI/ml de factor de coagulación sanguínea humana VIII recombinante, octocog alfa;
Excipientes: trehalosa, histidina, tris(hidroximetil)aminometano, cloruro de sodio, cloruro de calcio, glutatión (reducido), polisorbato 80, manitol.
* La actividad (en unidades internacionales) se determina mediante un ensayo cromogénico cuantitativo comparado con un estándar interno, que corresponde al estándar de la OMS. La actividad específica es aproximadamente de 4000 a 10000 UI/mg de proteína.
** El factor de coagulación sanguínea humana VIII se produce mediante tecnología de ADN recombinante a partir de células de ovario de hámster chino. Durante el cultivo celular, así como en las etapas de purificación y formulación final, no se añaden proteínas exógenas de origen humano o animal.
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Principales propiedades físico-químicas: polvo granular blanco o casi blanco; tras la reconstitución: solución transparente e incolora, libre de partículas extrañas.
Grupo farmacoterapéutico. Sangre y órganos hematopoyéticos. Agentes antihemorrágicos. Vitamina K y otros agentes hemostáticos. Factores de coagulación sanguínea. Factor de coagulación sanguínea VIII.
Código ATC B02B D02.
Propiedades farmacológicas.
Farmacodinámica.
El complejo del factor VIII/factor de von Willebrand está compuesto por dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas diferentes. ADYNOVATE contiene factor de coagulación recombinante humano VIII (octocog alfa), una glicoproteína biológicamente equivalente a la glicoproteína del factor VIII presente en el plasma humano.
Octocog alfa es una glicoproteína compuesta por 2332 aminoácidos con una masa molecular aproximada de 280 kDa. Tras la administración por infusión a pacientes con hemofilia, octocog alfa se une al factor de von Willebrand endógeno en la circulación sanguínea del paciente. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la transformación del factor X en factor X activado. El factor X activado convierte la protrombina en trombina. Posteriormente, la trombina transforma el fibrinógeno en fibrina, formándose así un coágulo de fibrina. La hemofilia A es un trastorno hereditario ligado al sexo del sistema de coagulación sanguínea debido a niveles reducidos de actividad del factor VIII, lo que provoca hemorragias abundantes en articulaciones, músculos o órganos internos, que pueden aparecer espontáneamente o tras traumatismos accidentales o quirúrgicos. Los niveles plasmáticos de factor VIII aumentan mediante terapia sustitutiva, lo que permite una corrección temporal de la deficiencia del factor VIII, reduciendo así la hemorragia y la tendencia a presentar episodios hemorrágicos.
Se han recopilado datos sobre la inducción de tolerancia inmune (ITI) en pacientes con inhibidores frente al factor VIII. En un subestudio 060103 con niños que no habían recibido tratamiento previo antes de su inclusión en el estudio, se documentaron procedimientos de ITI en 11 de estos pacientes. Se realizó un análisis retrospectivo en 30 pacientes pediátricos en tratamiento ITI (en el estudio 060703). En un registro prospectivo no intervencional (PASS-INT-004) se documentó ITI en 44 pacientes pediátricos y adultos, de los cuales 36 completaron la terapia ITI. Los datos muestran que puede alcanzarse la tolerancia inmune.
En el estudio 060201 se compararon dos regímenes profilácticos a largo plazo en 53 pacientes previamente tratados: un régimen de dosificación basado en farmacocinética individual (entre 20 y 80 UI de factor VIII por kg de peso corporal cada 72 ± 6 horas, n = 23) y un régimen profiláctico estándar (entre 20 y 40 UI/kg cada 48 ± 6 horas, n = 30). El régimen basado en farmacocinética (según una fórmula específica) tiene como objetivo mantener niveles mínimos de factor VIII ≥ 1 % durante el intervalo de dosificación de 72 horas. Los datos de este estudio indican que ambos regímenes de dosificación profiláctica son comparables en cuanto a la reducción de la frecuencia de hemorragias.
La Agencia Europea de Medicamentos ha eximido al medicamento ADYNOVATE de la obligación de presentar resultados de estudios en todas las subpoblaciones pediátricas con hemofilia A (déficit congénito del factor VIII): «Inducción de tolerancia inmune (ITI) en pacientes con hemofilia A (déficit congénito del factor VIII) que han desarrollado inhibidores frente al factor VIII» y «Tratamiento y prevención de hemorragias en pacientes con hemofilia A (déficit congénito del factor VIII)» (véase la sección «Posología y forma de administración» para obtener información sobre el uso en pacientes pediátricos).
Farmacocinética.
Todos los estudios farmacocinéticos del medicamento ADYNOVATE se realizaron con pacientes previamente tratados con hemofilia A grave y moderadamente grave (nivel basal de factor VIII ≤ 2 %). El análisis de las muestras de plasma se llevó a cabo en un laboratorio central mediante un ensayo de un solo nivel.
Un total de 195 pacientes con hemofilia A grave (nivel basal de factor VIII < 1 %) proporcionaron parámetros farmacocinéticos que se incluyeron en el conjunto de análisis farmacocinéticos según el protocolo. Las categorías de estos análisis para lactantes (de 1 mes a < 2 años), niños pequeños (de 2 a < 5 años), niños mayores (de 5 a < 12 años), adolescentes (de 12 a < 18 años) y adultos (de 18 años o más) se utilizaron para resumir los parámetros farmacocinéticos, siendo la edad definida como la edad en el momento de la infusión farmacocinética.
Tabla 1
| Datos agregados de los parámetros farmacocinéticos del medicamento ADEWATE en pacientes con hemofilia A grave (nivel inicial de factor VIII < 1 %) |
|||||
| Parámetro (valor medio ± desviación estándar) |
Lactantes (n = 5) |
Niños pequeños (n = 30) |
Niños mayores (n = 18) |
Adolescentes (n = 33) |
Adultos (n = 109) |
| AUC total (UI * h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
| Recuperación incremental corregida en Cmax (UI/dl por UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
| Período de semidesintegración (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
| Concentración máxima en plasma tras la infusión (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
| Período de semivida (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
| Volumen de distribución en estado de equilibrio (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
| Depuración (ml/kg * h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Se calcula como Cmáx – nivel basal del factor VIII, dividido por la dosis en UI/kg, donde Cmáx es el valor máximo postinfusión del factor VIII.
La seguridad y la eficacia hemostática del medicamento ADYNOVATE en la población pediátrica son similares a las de los pacientes adultos. La recuperación ajustada y el período de semivida terminal (t½) fueron aproximadamente un 20 % más bajos en los niños más pequeños (hasta 6 años) que en los adultos, lo cual podría deberse en parte al mayor volumen de plasma por kilogramo de peso corporal conocido en pacientes más jóvenes.
Hasta la fecha, no existen datos sobre la farmacocinética del medicamento ADYNOVATE en pacientes que no hayan recibido tratamiento previo.
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit congénito del factor VIII). ADVATE se administra a pacientes de todas las edades.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquier excipiente incluido en la sección «Composición», o a proteínas de ratón o hámster.
Interacción con otros medicamentos y otras formas de interacción.
No se han realizado estudios de interacción con el medicamento ADVATE.
Características de uso.
Seguimiento
Para mejorar el seguimiento de los medicamentos biológicos, se debe registrar el nombre y el número de lote del medicamento administrado.
Hipersensibilidad
Con el uso del medicamento ADYVITE se han notificado reacciones de hipersensibilidad, incluyendo anafilaxia. El medicamento contiene proteínas de ratón y hámster en cantidades traza. Si aparecen síntomas de hipersensibilidad, se debe recomendar a los pacientes que interrumpan inmediatamente la administración del medicamento y consulten a su médico. Los pacientes deben ser informados sobre los signos precoces de reacciones de hipersensibilidad, tales como urticaria, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de shock, deben iniciarse medidas generales contra el shock.
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, expresadas en unidades de Bethesda (BU) por 1 ml de plasma mediante un análisis modificado. El riesgo de formación de inhibidores depende de la gravedad de la enfermedad y del nivel de exposición al factor VIII, siendo máximo durante los primeros 20 días de tratamiento. En casos raros, los inhibidores pueden desarrollarse después de los primeros 100 días de tratamiento.
Se han observado casos de reaparición del desarrollo de inhibidores (con títulos bajos) después de más de 100 días, tras el cambio de un medicamento de factor VIII recombinante a otro, en pacientes previamente tratados y con antecedentes de desarrollo de inhibidores. Por ello, es necesario realizar una vigilancia cuidadosa de los pacientes respecto al desarrollo de inhibidores tras un cambio de medicamento.
La relevancia clínica de la formación de un inhibidor depende de su título. Así, los inhibidores de bajo título, que son transitorios o permanecen constantemente bajos, suponen un menor riesgo de respuesta clínica insuficiente que los inhibidores de alto título.
En general, todos los pacientes que reciben tratamiento con factor de coagulación VIII requieren una vigilancia cuidadosa sobre la formación de inhibidores mediante observación clínica y pruebas de laboratorio. Si no se alcanzan los niveles esperados de actividad del factor VIII en plasma o si la hemorragia no se controla con la dosis adecuada, se debe realizar una prueba para detectar la presencia de un inhibidor del factor VIII. En pacientes con niveles altos de inhibidor, la terapia con factor VIII puede ser ineficaz, por lo que deben considerarse otras opciones de tratamiento. El tratamiento de estos pacientes debe ser realizado por médicos con experiencia en el manejo de la hemofilia y la formación de inhibidores del factor VIII.
Complicaciones relacionadas con la cateterización
Si se requiere un dispositivo de acceso venoso central (CVC), existe riesgo de complicaciones asociadas al CVC, incluyendo infecciones locales, bacteriemia y trombosis en el sitio de cateterización.
Factores relacionados con los excipientes
Sodio
Este medicamento contiene 10 mg de sodio por vial, lo que equivale al 0,5 % de la dosis diaria máxima recomendada por la OMS de 2 g de sodio para un adulto.
Se recomienda encarecidamente registrar cada vez el nombre y el número de lote del medicamento ADYVITE administrado al paciente, con el fin de establecer una relación entre el estado del paciente y el lote del medicamento.
niños
Las precauciones y medidas de seguridad en niños no difieren de las de los adultos.
Uso durante el embarazo y la lactancia.
No se han realizado estudios sobre el efecto del factor VIII en la fertilidad de animales. Debido a la baja frecuencia de hemofilia A en mujeres, no existe experiencia sobre el uso del factor VIII durante el embarazo y la lactancia. Por lo tanto, el factor VIII debe usarse durante el embarazo y la lactancia solo si claramente está indicado.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
ADYVITE no afecta la velocidad de reacción al conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia; asimismo, deben estar disponibles medios para reanimación en caso de que se produzca anafilaxia.
Dosificación
La dosis y la duración de la terapia sustitutiva dependen del grado de déficit del factor VIII, del sitio y magnitud de la hemorragia, así como del estado clínico del paciente.
La cantidad del factor VIII se expresa en unidades internacionales (UI), que se relacionan con el estándar de la OMS para los productos del factor VIII. La actividad del factor VIII en el plasma se expresa bien en porcentaje (en relación con el plasma normal humano), bien en UI (en relación con el estándar internacional del factor VIII en plasma sanguíneo).
1 unidad internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
Tratamiento «a demanda»
El cálculo de la dosis necesaria de factor VIII se basa en la relación empíricamente demostrada: 1 UI de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en 2 UI/dl de plasma. La dosis requerida se determina mediante la fórmula siguiente:
Dosis requerida (UI) = peso corporal (kg) × incremento deseado del factor VIII (%) × 0,5
En caso de episodios hemorrágicos posteriores, la actividad del factor VIII no debe descender por debajo de un determinado nivel de actividad plasmática (en % del nivel normal o en UI/dl) durante el período correspondiente. La siguiente tabla 2 puede utilizarse como guía para la elección de la dosis en caso de hemorragias y de intervenciones quirúrgicas.
Tabla 2
| Instrucciones para la selección de la dosis en caso de hemorragias e intervenciones quirúrgicas |
||
| Grado de hemorragia/ tipo de intervención quirúrgica |
Nivel necesario del factor VIII (% o UI/dl) |
Frecuencia de administración (horas)/duración del tratamiento (días) |
| Hemorragia |
||
| Hemartrosis precoz, hemorragia muscular o hemorragia en la cavidad oral. |
20–40 |
Repetir las inyecciones cada 12–24 horas (cada 8 a 24 horas en pacientes menores de 6 años) durante al menos 1 día, hasta que cese el sangrado indicado por dolor o se logre la cicatrización. |
| Hemartrosis avanzada, hemorragia muscular o hematoma. |
30–60 |
Repetir las inyecciones cada 12–24 horas (cada 8 a 24 horas en pacientes menores de 6 años) durante 3–4 días o más, hasta que desaparezcan el dolor o la incapacidad funcional. |
| Hemorragia amenazante para la vida. |
60–100 |
Repetir las inyecciones cada 8–24 horas (cada 6 a 12 horas en pacientes menores de 6 años) hasta que desaparezca la amenaza para la vida. |
| Intervención quirúrgica |
||
| Leve, incluida la extracción dental |
30–60 |
Cada 24 horas (cada 12 a 24 horas en pacientes menores de 6 años) durante al menos 1 día, hasta lograr la cicatrización. |
| Importante |
80–100 (antes y después de la cirugía) |
Repetir las inyecciones cada 8–24 horas (cada 6 a 24 horas en pacientes menores de 6 años) hasta una cicatrización adecuada; posteriormente continuar el tratamiento al menos durante 7 días para mantener la actividad del factor VIII entre el 30 % y el 60 % (UI/dl). |
La dosis y frecuencia de administración deben ajustarse según la situación clínica de cada caso particular. En determinadas circunstancias (por ejemplo, presencia de un inhibidor con título bajo), puede ser necesaria una dosis mayor que la calculada mediante la fórmula.
Durante el tratamiento, es conveniente determinar los niveles de actividad del factor VIII en el plasma sanguíneo y utilizar estos valores para establecer la dosis necesaria y la frecuencia de las inyecciones repetidas. En caso de intervenciones quirúrgicas mayores, es esencial un control preciso de la terapia sustitutiva mediante una prueba cuantitativa de la actividad del factor VIII en el plasma sanguíneo. Diferentes pacientes pueden presentar respuestas distintas al factor VIII, alcanzando diferentes niveles de recuperación in vivo y mostrando distintas intensidades de semivida.
Profilaxis
Para la profilaxis a largo plazo de hemorragias en pacientes con hemofilia A grave, las dosis habituales son de 20 a 40 UI de factor VIII por kg de peso corporal, con intervalos de administración de 2 a 3 días.
Vía de administración
ADVATE debe administrarse por vía intravenosa. Si el medicamento lo administra una persona que no es personal sanitario, es necesaria una preparación adecuada.
La velocidad de administración debe ajustarse para que sea cómoda para el paciente y no debe exceder los 10 ml/min.
Después de la reconstitución, la solución debe ser transparente, incolora, libre de partículas extrañas y con un pH entre 6,7 y 7,3.
Instrucciones para la reconstitución del medicamento antes de la administración
ADVATE debe administrarse por vía intravenosa tras la reconstitución del medicamento.
Debe realizarse una inspección visual de la solución reconstituida para detectar la presencia de partículas extrañas y/o cambios de color.
Después de la reconstitución, la solución debe ser transparente, incolora y no contener inclusiones extrañas. No utilice soluciones que estén turbias o que contengan inclusiones.
- Para la administración, debe utilizarse una jeringa con conector tipo Luer.
- Utilizar dentro de las tres horas siguientes a la reconstitución.
- No se debe almacenar la solución reconstituida en nevera.
Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
Reconstitución con el dispositivo BAXJECT II
- Para la reconstitución, utilice exclusivamente agua estéril para inyecciones y el dispositivo de dilución incluido en el envase.
- No utilice si el dispositivo BAXJECT II, su sistema de barrera estéril o su envase están dañados o presentan signos de deterioro.
- Debe seguirse una técnica aséptica.
- Si el medicamento se ha almacenado en nevera, retire ambos frascos —el del polvo ADVATE y el del diluyente— y déjelos alcanzar la temperatura ambiente (aproximadamente 15-25 °C).
- Lávese bien las manos con agua tibia y jabón.
- Retire las tapas de los frascos del polvo y del diluyente.
- Desinfecte los tapones con compresas de alcohol. Coloque los frascos sobre una superficie plana y limpia.
- Abra el envase del dispositivo BAXJECT II retirando la tapa de papel, sin tocar el dispositivo en su interior (fig. a). No saque el dispositivo del envase. No utilice si el dispositivo BAXJECT II, su sistema de barrera estéril o su envase están dañados o presentan signos de deterioro.
- Gire el envase e inserte la púa de plástico transparente en el tapón del frasco del diluyente. Sujete el envase por el borde y retírelo del dispositivo BAXJECT II (fig. b). No retire la tapa azul del dispositivo BAXJECT II.
- Para la dilución, utilice exclusivamente agua estéril para inyecciones y el dispositivo de dilución incluidos en el envase. Gire el sistema con el dispositivo BAXJECT II conectado al frasco del diluyente, de modo que el frasco quede encima del dispositivo. Inserte la púa de plástico blanco en el tapón del frasco del medicamento ADVATE. El vacío transferirá el diluyente al frasco de ADVATE (fig. c).
- Agite suavemente hasta la completa disolución del medicamento. Asegúrese de que ADVATE se ha disuelto completamente, ya que de lo contrario no toda la solución reconstituida pasará a través del filtro del dispositivo. El medicamento se disuelve rápidamente (normalmente en menos de 1 minuto). Tras la reconstitución, la solución debe ser clara, transparente y libre de partículas extrañas.
Fig. a Fig. b Fig. c
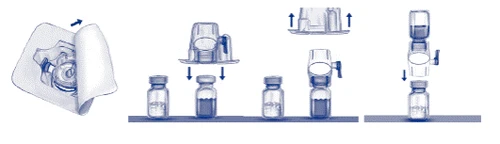
Administración
Siga las normas de asepsia.
Los medicamentos para administración parenteral deben inspeccionarse visualmente en busca de partículas extrañas inmediatamente antes de su uso, siempre que el frasco y la solución lo permitan. Solo debe utilizarse una solución transparente e incolora.
- Retire la tapa azul del BAXJECT II. No aspire aire a la jeringa. Conecte la jeringa al BAXJECT II.
- Gire el sistema (el frasco con la solución reconstituida debe quedar en la parte superior). Aspire lentamente la solución reconstituida a la jeringa tirando del émbolo.
- Desconecte la jeringa.
- Conecte una aguja-butterfly a la jeringa. Administre por vía intravenosa. La solución debe administrarse lentamente, a una velocidad determinada por el nivel de comodidad del paciente, sin exceder los 10 ml por minuto. Se debe controlar la frecuencia del pulso antes y durante la administración de ADVATE. Si se produce un aumento significativo de la frecuencia cardíaca, se debe reducir la velocidad de administración o interrumpir temporalmente la inyección, lo cual suele ser suficiente para que los síntomas desaparezcan rápidamente (véanse las secciones «Precauciones de uso» y «Reacciones adversas»).
Desde el punto de vista microbiológico, el medicamento debe administrarse inmediatamente después de la reconstitución. Sin embargo, se ha demostrado la estabilidad química y física del medicamento durante 3 horas a una temperatura de 25 °C tras la apertura del envase.
Durante el período de caducidad, el medicamento puede conservarse a temperatura ambiente (no superior a 25 °C) durante un único período no superior a 6 meses. La fecha de finalización del período de 6 meses de almacenamiento a temperatura ambiente debe indicarse en el envase del medicamento. No se debe volver a colocar el medicamento en nevera para su almacenamiento posterior.
Niños
En el tratamiento "a demanda", la dosificación en pacientes pediátricos (de 0 a 18 años) no difiere de la de los adultos. En pacientes menores de 6 años, para la terapia profiláctica, se recomienda una dosis de 20 a 50 UI de factor VIII por kg de peso corporal, 3 a 4 veces por semana.
Sobredosificación.
No se han notificado síntomas de sobredosificación con este medicamento.
Reacciones adversas
Los estudios clínicos del medicamento ADYVATE incluyeron 418 estudios con administración única del medicamento ADYVATE, en los que se observaron 93 reacciones adversas. Las reacciones adversas no deseadas frecuentes incluyeron la aparición de anticuerpos neutralizantes contra el factor VIII (inhibidores), cefalea y fiebre.
Se han observado raramente reacciones de hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, sensación de ardor y hormigueo en el sitio de infusión, escalofríos, hiperemia, urticaria generalizada, cefalea, urticaria, hipotensión, letargo, náuseas, inquietud, taquicardia, opresión en el pecho, hormigueo, vómitos, sibilancias), y en algunos casos estas reacciones han progresado hasta una anafilaxia aguda (incluyendo shock).
En pacientes que desarrollan anticuerpos contra proteínas de ratón y/o hámster, pueden presentarse reacciones de hipersensibilidad asociadas.
En pacientes con hemofilia A que reciben factor VIII, incluyendo ADYVATE, pueden desarrollarse anticuerpos neutralizantes (inhibidores). Si aparecen tales inhibidores, la respuesta clínica será insuficiente. En tales casos, se recomienda consultar a un centro especializado en el tratamiento de la hemofilia.
Lista de reacciones adversas en forma de tabla
En la tabla 3 que figura a continuación se indican las reacciones adversas notificadas con el medicamento en estudios clínicos y en notificaciones espontáneas, clasificadas por frecuencia de aparición. La tabla sigue la clasificación por sistemas de órganos de MedDRA (SOC) y términos de preferencia.
Las categorías de frecuencia se definen de la siguiente manera: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1 000 a < 1/100), raras (≥ 1/10 000 a < 1/1 000), muy raras (< 1/10 000), desconocidas (no se puede estimar con los datos disponibles). Dentro de cada categoría de frecuencia, las reacciones adversas se enumeran en orden decreciente de gravedad.
Tabla 3
Frecuencia de aparición de reacciones adversas notificadas en estudios clínicos y en notificaciones espontáneas
| Sistema de Clasificación Estándar de Clases de Órganos por MedDRA |
Reacciones adversas |
Frecuenciaa |
| Infecciones y parasitosis |
Gripe |
Infrecuente |
| Laringitis |
Infrecuente |
|
| Trastornos de la sangre y del sistema linfático |
Inhibición del factor VIII |
Infrecuente (POTL)d Muy frecuente (PBT)d |
| Linfangitis |
Infrecuente |
|
| Trastornos del sistema inmunitario |
Reacción anafiláctica |
No conocido |
| Hipersensibilidadc |
No conocido |
|
| Trastornos del sistema nervioso |
Cefalea |
Frecuente |
| Vertigo |
Infrecuente |
|
| Empeoramiento de la memoria |
Infrecuente |
|
| Pérdida de conciencia |
Infrecuente |
|
| Temblor |
Infrecuente |
|
| Migraña |
Infrecuente |
|
| Disgeusia |
Infrecuente |
|
| Trastornos oculares |
Inflamación ocular |
Infrecuente |
| Trastornos cardíacos |
Taquicardia |
Infrecuente |
| Trastornos vasculares |
Hematoma |
Infrecuente |
| Aflujo de sangre al rostro |
Infrecuente |
|
| Pálido |
Infrecuente |
|
| Trastornos respiratorios, torácicos y mediastínicos |
Disnea |
Infrecuente |
| Trastornos gastrointestinales |
Diárrhea |
Infrecuente |
| Dolor en la parte superior del abdomen |
Infrecuente |
|
| Náuseas |
Infrecuente |
|
| Vómitos |
Infrecuente |
|
| Lesiones de la piel y del tejido subcutáneo |
Picazón |
Infrecuente |
| Erupción cutánea |
Infrecuente |
|
| Hiperhidrosis |
Infrecuente |
|
| Urticaria |
Infrecuente |
|
| Trastornos generales y reacciones en el sitio de administración |
Hipertemia |
Frecuente |
| Edema periférico |
Infrecuente |
|
| Dolor en el pecho |
Infrecuente |
|
| Malestar en el pecho |
Infrecuente |
|
| Escalofríos |
Infrecuente |
|
| Mala sensación general |
Infrecuente |
|
| Hematoma en el sitio de punción vascular |
Infrecuente |
|
| Cansancio |
No conocido |
|
| Reacción en el sitio de inyección |
No conocido |
|
| Indisposición |
No conocido |
|
| Pruebas de laboratorio |
Nivel elevado de monocitos |
Infrecuente |
| Disminución del nivel del factor de coagulación sanguínea VIIIb |
Infrecuente |
|
| Disminución del hematocrito |
Infrecuente |
|
| Desviación de los valores normales en pruebas de laboratorio |
Infrecuente |
|
| Lesiones, envenenamientos y complicaciones procedimentales |
Complicaciones postprocedimiento |
Infrecuente |
| Hemorragia postprocedimiento |
Infrecuente |
|
| Reacción en el sitio del procedimiento |
Infrecuente |
a Calculado sobre la base del número total de pacientes que recibieron ADVATE (418).
b En un paciente, durante la infusión continua de ADVATE tras una intervención quirúrgica (entre 10 y 14 días después de la cirugía), se produjo una disminución inesperada del nivel del factor de coagulación VIII. Durante este período, se mantuvo un control hemostático constante, y los niveles plasmáticos del factor VIII y la velocidad de eliminación volvieron a niveles adecuados el día 15 tras la cirugía. Los análisis cuantitativos para detectar la presencia de inhibidores del factor VIII, realizados tras finalizar la infusión continua y tras finalizar el estudio, dieron resultados negativos.
c La explicación de esta reacción adversa se proporciona en la sección siguiente.
d La frecuencia se basa en datos de estudios con todos los medicamentos del factor de coagulación VIII que incluyeron pacientes con hemofilia A grave. PPT: pacientes previamente tratados; PBPT: pacientes previamente no tratados.
Descripción de reacciones adversas individuales
Reacciones adversas a residuos del proceso de fabricación
Se evaluó la presencia de anticuerpos frente a proteínas celulares del hámster chino (CHO) en 229 pacientes tratados: 3 pacientes mostraron una tendencia estadísticamente significativa en los títulos, 4 presentaron picos persistentes o potenciales picos transitorios y 1 paciente presentó ambos indicadores, sin manifestaciones clínicas. De los 229 pacientes evaluados para anticuerpos frente a IgG murina, 10 mostraron una tendencia estadísticamente significativa al aumento, 2 presentaron picos persistentes o potenciales picos transitorios y 1 paciente presentó ambos indicadores. Cuatro pacientes informaron casos aislados de urticaria, prurito, erupción cutánea y un ligero aumento en el recuento de eosinófilos tras aplicaciones repetidas del medicamento.
Hipersensibilidad
Las reacciones de tipo alérgico incluyen anafilaxia y se manifiestan como mareo, parestesias, erupción cutánea, enrojecimiento, edema facial, urticaria y prurito.
Pacientes pediátricos
Además de la formación de anticuerpos neutralizantes (inhibidores) en niños previamente no tratados y de las complicaciones relacionadas con la cateterización, no se han observado diferencias en las reacciones adversas notificadas en los estudios clínicos.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite continuar monitorizando la relación beneficio-riesgo del medicamento. Los profesionales sanitarios y farmacéuticos, así como los pacientes o sus representantes legales, deben informar sobre cualquier caso sospechoso de reacción adversa o de falta de eficacia del medicamento a través del sistema automatizado de farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Periodo de validez.
2 años.
Disolvente (agua para inyección): 5 años.
Condiciones de conservación.
Conservar entre 2 y 8 °C. ¡No congelar! Conservar en el envase original para protegerlo de la luz.
Mantener fuera del alcance de los niños.
Incompatibilidades
Dado que no se han realizado estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos ni con disolventes.
Envase.
1 vial con polvo para solución inyectable (250 UI, 500 UI, 1000 UI, 1500 UI, 2000 UI o 3000 UI) junto con 1 vial de disolvente (5 ml de agua para inyección) y 1 dispositivo de reconstitución BAXJECT II por caja.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
Baxalta Belgium Manufacturing SA, Bélgica / Baxalta Belgium Manufacturing SA, Belgium.
Dirección del fabricante y lugar de actividad.
Boulevard Rene Branquart 80, Lessines, 7860, Bélgica / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.