Inlita
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT INLYTA (INLYTA®)
Composition:
Active substance: axitinib;
1 film-coated tablet contains 1 mg or 5 mg of axitinib;
Excipients: microcrystalline cellulose; lactose monohydrate; sodium croscarmellose; magnesium stearate; Opadry® II red 32K15441 (HPMC 2910/hypromellose 15 cP; titanium dioxide (E 171); lactose monohydrate; triacetin; iron oxide red (E 172)).
Pharmaceutical form. Film-coated tablets.
Main physical and chemical properties:
Inlyta 1 mg tablets: red oval film-coated tablet, engraved with “Pfizer” on one side and “1” and “XNB” on the other;
Inlyta 5 mg tablets: red triangular film-coated tablet, engraved with “Pfizer” on one side and “5” and “XNB” on the other.
Pharmacotherapeutic group.
Antineoplastic agents. Protein kinase inhibitors. ATC code L01XE17.
Pharmacological Properties
Pharmacodynamics
Mechanism of Action
It has been established that axitinib at therapeutic plasma concentrations inhibits tyrosine kinase receptors, including vascular endothelial growth factor receptors VEGFR-1, VEGFR-2, and VEGFR-3. These receptors are involved in pathological angiogenesis, tumor growth, and progression of malignant neoplasms. Axitinib inhibited VEGF-induced proliferation and survival of endothelial cells in vitro and in mouse models. In mouse tumor xenograft models, axitinib was shown to inhibit tumor growth and phosphorylation of VEGFR-2.
To evaluate the effect of a single oral dose of Inlyta (5 mg) on QTc interval duration when administered with 400 mg ketoconazole and as monotherapy, a randomized, blinded, two-period crossover study was conducted involving 35 healthy volunteers. Within the first 3 hours after drug administration, no notable changes in mean QTc interval duration (i.e., more than 20 ms) were observed compared to the placebo group. However, a small increase in mean QTc interval duration (i.e., less than 10 ms) cannot be excluded.
Pharmacokinetics
Population pharmacokinetic analysis combined data from 17 studies involving healthy volunteers and oncology patients. The concentration-time profile of axitinib was adequately described by a two-compartment distribution model with first-order absorption and a lag time.
Absorption and Distribution. The median Tmax after a single 5 mg oral dose ranged from 2.5 to 4.1 hours. Based on the plasma elimination half-life, steady-state concentrations are expected to be reached within 2–3 days of dosing. Administration of axitinib at 5 mg twice daily resulted in approximately 1.4-fold greater drug accumulation compared to a single dose. The pharmacokinetics of axitinib at steady state are nearly linear within the dose range of 1 to 20 mg. The mean absolute bioavailability of axitinib after a single 5 mg oral dose is 58%.
Administration of Inlyta with a moderately fat-rich meal resulted in approximately a 10% decrease in AUC compared to administration in the fasted state in the morning. Administration with a high-fat, high-calorie meal increased AUC by 19% compared to administration in the fasted state in the morning. Inlyta may be taken with or without food (see "Dosage and Administration").
Axitinib is almost completely (>99%) bound to human plasma proteins, primarily to albumin, with moderate binding to α1-acid glycoprotein. In patients with advanced renal cell carcinoma (n = 20) who received the drug at a dose of 5 mg twice daily after a meal, geometric mean values of Cmax and AUC0–24 were 27.8 ng/mL (79%) and 265 ng·h/mL (77%), respectively. Clearance and apparent volume of distribution were 38 L/h (80%) and 160 L (105%), respectively (CV% values are given in parentheses).
Metabolism and Elimination. The elimination half-life of Inlyta from plasma ranges from 2.5 to 6.1 hours. Axitinib is primarily metabolized in the liver by CYP3A4/5. CYP1A2, CYP2C19, and UGT1A1 play a lesser role in axitinib metabolism. After oral administration of 5 mg radiolabeled axitinib, approximately 41% of radioactivity was excreted in feces and about 23% in urine. The main component detected in feces was unchanged axitinib, accounting for 12% of the administered dose. Unchanged axitinib was not detected in urine. The majority of radioactivity in urine was due to carboxylic acid and sulfoxide metabolites of the active substance. The main radioactive component in plasma is the N-glucuronide metabolite, which accounts for 50% of circulating plasma radioactivity. Unchanged axitinib and the sulfoxide metabolite each account for approximately 20% of circulating plasma radioactivity.
The in vitro affinity of the sulfoxide and N-glucuronide metabolites for VEGFR-2 is approximately ≥400-fold lower than that of axitinib.
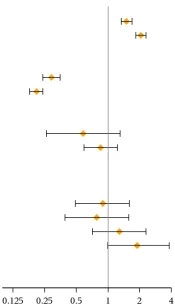
Effect of Other Medicinal Products on Inlyta. Axitinib is primarily metabolized in the liver by the CYP3A4/5 enzyme. In addition, the aqueous solubility of axitinib is pH-dependent: solubility decreases with increasing pH. The effects of a strong CYP3A4/5 inhibitor, a strong CYP3A4/5 inducer, and an antacid on the pharmacokinetics of axitinib are presented in Table 1 (see also sections "Dosage and Administration" and "Interaction with Medicinal Products and Other Types of Interactions").
Table 1
Effect on axitinib pharmacokinetics of concomitantly administered medicinal products and hepatic impairment
| Population characteristics |
Cmax |
Change range and 90% CI |
Recommendations |
| Strong CYP3A4/5 inhibitor: ketoconazole 400 mg once daily for 7 days |
Cmax AUC |
|
Reduce Inlyta dose* |
| Strong CYP3A4/5 inducer: rifampin 600 mg once daily for 9 days |
Cmax AUC |
Avoid coadministration |
|
| Antacid: rabeprozole 20 mg once daily for 5 days |
Cmax AUC |
No dose adjustment required |
|
| Impaired hepatic function |
|||
| Mild/normal |
Cmax AUC |
No dose adjustment required |
|
| Moderate/normal |
Cmax AUC |
Reduce Inlyta dose* |
|
| Severe/normal |
Experience not available |
||
AUC – area under the concentration-time curve.
Cmax – maximum concentration.
* See section "Dosage and administration".
PK – pharmacokinetics.
CI – confidence interval.
Pharmacokinetics in special patient populations
Children. Inlyta has not been studied in patients under 18 years of age.
Hepatic impairment. The effect of hepatic impairment on the pharmacokinetics of axitinib is shown in Table 1 (see also sections "Dosage and administration", "Special precautions").
Renal impairment. A population pharmacokinetic analysis was conducted involving 590 healthy volunteers and patients based on renal function parameters. Among the analyzed patients, five had severe renal impairment (creatinine clearance ranging from 15 to <29 ml/min), 64 had moderate renal impairment (30 to <59 ml/min), and 139 had mild renal impairment (creatinine clearance from 60 to <89 ml/min). Mild to severe renal impairment does not show a significant effect on the pharmacokinetics of axitinib. Data on the use of the drug in patients with end-stage renal disease are available for only one patient.
Other individual factors. Results of population pharmacokinetic analysis indicate no clinically significant effect of age, sex, race, body weight and body surface area, UGT1A1 genotype, or CYP2C19 genotype on axitinib clearance.
Clinical Characteristics
Indications.
Treatment of advanced renal cell carcinoma in cases where prior systemic therapy has proven ineffective.
Contraindications.
Hypersensitivity to axitinib or to any other component of the medicinal product.
Interaction with other medicinal products and other forms of interaction
Inhibitors of CYP3A4/5
Concomitant administration of ketoconazole (a strong CYP3A4/5 inhibitor) increased axitinib exposure levels in plasma of healthy volunteers. Concomitant use of Inlyta with strong CYP3A4/5 inhibitors should be avoided. Consumption of grapefruit or grapefruit juice may also increase axitinib plasma concentrations. Combination of these products with axitinib should also be avoided. It is recommended to select concomitant medications with no or minimal potential to inhibit CYP3A4/5 activity. If co-administration of Inlyta with a strong CYP3A4/5 inhibitor is necessary, the dose of axitinib should be reduced (see sections "Dosage and administration" and "Pharmacokinetics").
Inhibitors of CYP1A2 and CYP2C19
CYP1A2 and CYP2C19 play a minor role (< 10%) in axitinib metabolism. The effect of strong inhibitors of these isoenzymes on axitinib pharmacokinetics has not been studied. Due to the risk of increased axitinib plasma levels, caution should be exercised when administering strong inhibitors of the aforementioned isoenzymes concomitantly with axitinib.
Inducers of CYP3A4/5
Concomitant administration of rifampicin (a strong CYP3A4/5 inducer) decreased axitinib exposure levels in plasma of healthy volunteers. Concomitant use of Inlyta with strong CYP3A4/5 inducers (such as rifampicin, dexamethasone, phenytoin, carbamazepine, rifabutin, rifapentine, phenobarbital, St. John's wort) should be avoided. For concomitant use with Inlyta, it is recommended to select a medicinal product with no or minimal potential to induce CYP3A4/5 activity. Moderate CYP3A4/5 inducers (such as bosentan, efavirenz, etravirine, modafinil, and nafcillin) may also reduce axitinib plasma exposure levels. Use of these agents should also be avoided whenever possible.
In vitro studies of CYP and UGT inhibition and induction
In vitro studies have shown that axitinib does not inhibit CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, or UGT1A1 at therapeutic plasma concentrations.
In vitro studies demonstrated that axitinib may potentially inhibit CYP1A2. Therefore, concomitant administration of axitinib with CYP1A2 substrates may lead to increased plasma levels of CYP1A2 substrates (e.g., theophylline).
In vitro studies also showed that axitinib may potentially inhibit CYP2C8. However, concomitant administration of axitinib with paclitaxel, a known CYP2C8 substrate, did not result in increased paclitaxel plasma concentrations in patients with advanced cancer, indicating absence of clinically relevant CYP2C8 inhibition.
In vitro studies in human hepatocytes also demonstrated that axitinib does not induce CYP1A1, CYP1A2, or CYP3A4/5. Therefore, concomitant administration of axitinib is not expected to reduce plasma levels of CYP1A1, CYP1A2, and CYP3A4/5 substrates in vivo.
In vitro studies with P-glycoprotein
In vitro studies showed that axitinib inhibits P-glycoprotein. However, axitinib is not expected to exert an inhibitory effect on P-glycoprotein at therapeutic plasma concentrations. Therefore, concomitant administration of axitinib is not anticipated to increase plasma concentrations of digoxin or other P-glycoprotein substrates in vivo.
Special precautions for use.
Arterial hypertension
Adequate control of blood pressure should be ensured before initiating treatment with Inlyta. Patients should be monitored for signs of arterial hypertension, and if necessary, treated with standard antihypertensive medications. Depending on the severity of arterial hypertension, treatment with Inlyta should either be temporarily interrupted with subsequent dose reduction, or permanently discontinued (see section "Dosage and administration").
Arterial thromboembolism
The use of Inlyta in patients who have experienced arterial thromboembolism within the previous 12 months has not been studied. During clinical trials with Inlyta, arterial thromboembolic events (including transient ischemic attacks, cerebrovascular accidents, myocardial infarction, and retinal artery occlusion) occurred in 17 of 715 patients (2%), including two fatal cases following acute cerebrovascular events.
If arterial thromboembolism occurs during treatment, Inlyta should be permanently discontinued.
Venous thromboembolism
Venous thromboembolic events (VTE), including pulmonary embolism, deep vein thrombosis, retinal vein occlusion, and retinal vein thrombosis, some fatal, have been reported during clinical trials.
The use of Inlyta in patients who have experienced venous thromboembolism within the previous 6 months has not been studied. During clinical trials with Inlyta, venous thromboembolic events occurred in 22 of 715 patients (3%), including two fatal cases following pulmonary embolism.
Patients should be monitored for signs and symptoms of VTE and pulmonary embolism. Depending on the severity of VTE, treatment with Inlyta should either be temporarily interrupted with subsequent resumption without dose change, or permanently discontinued.
Increased hemoglobin or hematocrit
Treatment with axitinib may lead to increased hemoglobin or hematocrit, reflecting an increase in red blood cell mass. Increased red blood cell mass may elevate the risk of embolic and thrombotic events.
Hemoglobin or hematocrit should be monitored before starting and periodically during treatment with axitinib. If hemoglobin or hematocrit exceeds normal levels, patients should be managed according to standard medical practice to reduce hemoglobin or hematocrit to acceptable levels.
Bleeding
Bleeding events, including intracranial hemorrhage, hematuria, hemoptysis, lower gastrointestinal bleeding, melena, and fatal bleeding, have been reported.
The use of Inlyta in patients with signs of untreated brain metastases or recent active gastrointestinal bleeding has not been studied. These patients should not be treated with this medication. Depending on the severity and persistence of bleeding, treatment with Inlyta should either be temporarily interrupted with subsequent dose reduction, or permanently discontinued.
Heart failure
In a controlled clinical trial of Inlyta for the treatment of renal cell carcinoma, heart failure occurred in 6 of 359 patients (2%) receiving Inlyta and in 3 of 355 patients (1%) receiving sorafenib. Grade 3/4 heart failure occurred in 2 of 359 patients (1%) receiving Inlyta and in 1 of 355 patients (<1%) receiving sorafenib. Fatal heart failure occurred in 2 of 359 patients (1%) receiving Inlyta and in 1 of 355 patients (<1%) receiving sorafenib. Patients should be monitored throughout Inlyta treatment for symptoms of heart failure. Management of heart failure may require dose reduction, interruption, or permanent discontinuation of Inlyta (see section "Dosage and administration").
Gastrointestinal perforation and fistula formation
Gastrointestinal perforations, including one fatal case, have been reported during clinical trials. Cases of fistula formation have also been reported.
Patients should be periodically monitored during Inlyta treatment for signs of gastrointestinal perforation or fistula formation.
Thyroid dysfunction
Thyroid function should be monitored before starting and periodically during Inlyta treatment. Hypothyroidism and hyperthyroidism occurring during treatment should be managed according to standard medical practice to maintain normal thyroid function.
Impaired wound healing
Impaired wound healing may occur in patients receiving agents that inhibit the vascular endothelial growth factor (VEGF) signaling pathway. Therefore, Inlyta may negatively affect wound healing.
Treatment with Inlyta should be discontinued at least 2 days prior to planned surgery. Inlyta should not be used for at least 2 weeks after major surgery and until adequate wound healing has occurred. Depending on the severity and persistence of impaired wound healing, treatment with Inlyta may either be resumed at a reduced dose or permanently discontinued. The safety of resuming Inlyta after resolution of wound healing complications has not been established (see section "Dosage and administration").
Posterior reversible encephalopathy syndrome (PRES)
PRES is a neurological disorder that may present with headache, seizures, lethargy, confusion, blindness, visual disturbances, and other neurological disorders. Arterial hypertension of any degree (mild to severe) may also occur. MRI is required to confirm the diagnosis of PRES. Patients who develop PRES must permanently discontinue Inlyta. The safety of resuming Inlyta treatment in patients who previously experienced PRES is unknown (see section "Dosage and administration").
Proteinuria
Patients should be monitored for proteinuria before starting and periodically during Inlyta treatment. Patients who develop moderate or severe proteinuria should temporarily discontinue Inlyta, followed by dose reduction (see section "Dosage and administration").
Increased liver enzyme activity
Levels of ALT, AST, and bilirubin should be monitored before starting and periodically during Inlyta treatment.
Hepatic impairment
In a dedicated study of patients with hepatic impairment, systemic exposure levels after single doses of Inlyta in patients with mild hepatic impairment (Child-Pugh class A) were similar to those in patients with normal hepatic function, while systemic exposure to axitinib was higher in patients with moderate hepatic impairment (Child-Pugh class B) compared to patients with normal hepatic function. For patients with moderate hepatic impairment (Child-Pugh class B), a reduced starting dose of Inlyta is recommended. The use of Inlyta in patients with severe hepatic impairment (Child-Pugh class C) has not been studied (see sections "Dosage and administration" and "Pharmacokinetics").
Elderly patients and patients of different races
In a controlled clinical trial of Inlyta for the treatment of renal cell carcinoma, 34% of patients in the Inlyta treatment group were aged 65 years or older. The majority of patients were of Caucasian (77%) or Asian (21%) origin. Although increased sensitivity in elderly patients and Asians cannot be entirely ruled out, overall, no differences in safety and efficacy of Inlyta were observed between patients aged ≥65 years and younger patients, or between Caucasians and patients of other races.
Dose adjustment based on age or race is not required (see sections "Dosage and administration" and "Pharmacokinetics").
Lactose
This medicinal product contains lactose. It should not be administered to patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency, or glucose-galactose malabsorption syndrome.
Use during pregnancy or breastfeeding
There are insufficient data in humans to determine the risk of using the drug during pregnancy. Based on the mechanism of action and findings from animal studies, Inlyta may cause fetal harm when administered to pregnant women. In animal developmental toxicity studies, axitinib showed teratogenic, embryotoxic, and fetotoxic effects at exposure levels lower than those in humans receiving the recommended starting doses.
Women of reproductive potential should be informed of the potential risk to the fetus and the necessity of using effective contraception during treatment with Inlyta and for one week after the last dose. Men with female partners of reproductive potential should use effective contraception during treatment with Inlyta and for one week after the last dose. Animal studies have shown that Inlyta may impair fertility in both women and men of reproductive potential.
Currently, there are no data on the presence of axitinib in human breast milk or its effects on the breastfed infant or milk production. Due to the potential for serious adverse reactions in breastfed infants, breastfeeding women are advised to discontinue breastfeeding during treatment and for 2 weeks after the last dose.
Ability to affect the speed of reactions when driving or operating machinery
Patients should be warned about the possible occurrence of dizziness, somnolence, and visual disturbances during treatment with Inlyta and advised not to drive or operate machinery if these symptoms occur (see sections "Adverse reactions" and "Special precautions for use").
Administration and Dosage
The recommended initial oral dose of Inlita is 5 mg twice daily. The intervals between doses of Inlita should be approximately 12 hours; the drug can be administered independently of food intake (see section "Pharmacodynamics"). Tablets should be swallowed whole with a glass of water.
If vomiting occurs after taking a dose or if a dose is missed, an additional dose should not be taken. The next scheduled dose should be taken at the appropriate time.
Dosage Adjustment Recommendations.
Dosage increases or reductions should be based on individual safety and tolerability.
Recommendations for increasing or reducing the dose of Inlita are provided in Table 2.
The dose of Inlita may be increased in patients who have tolerated the drug well during treatment for at least two consecutive weeks, without experiencing adverse reactions of grade 2 or higher (according to the Common Terminology Criteria for Adverse Events), who have normal blood pressure, and who are not taking antihypertensive medications.
Table 2
Recommendations for Dose Increase or Reduction of Inlita
| Dose adjustment |
Dosing regimen |
| Recommended initial dose |
5 mg twice daily |
| Dose increase |
|
| First dose increase |
7 mg twice daily |
| Second dose increase |
10 mg twice daily |
| Dose reduction |
|
| First dose reductiona |
3 mg twice daily |
| Second dose reduction |
2 mg twice daily |
| a For management of adverse drug reactions. b From 5 mg twice daily. |
|
Dose modification recommendations for Inlyta adverse reactions are provided in Table 3.
Table 3
Dose modification recommendations for Inlyta in the event of adverse reactions
| Adverse reaction |
Severity |
Inlyta dosage modification |
| Arterial hypertension [see "Special precautions"] |
Systolic blood pressure > 150 mm Hg or diastolic blood pressure > 100 mm Hg despite antihypertensive therapy |
|
| Systolic blood pressure > 160 mm Hg or diastolic blood pressure > 105 mm Hg |
|
|
| Grade 4 or hypertensive crisis |
|
|
| Bleeding [see "Special precautions"] |
Grade 3 or 4 |
|
| Heart failure [see "Special precautions"] |
Asymptomatic cardiomyopathy (left ventricular ejection fraction decrease > 20% but < 50% below baseline or below lower limit of normal if baseline not measured) |
|
| Congestive heart failure with clinical symptoms |
|
|
| Impaired wound healing [see "Special precautions"] |
Any grade |
|
| Posterior reversible encephalopathy syndrome [see "Special precautions"] |
Any grade |
|
| Proteinuria [see "Special precautions"] |
≥ 2 g protein in 24 hours |
|
| Other adverse reactions |
Grade 3 |
|
| Grade 4 |
|
Dosage adjustments due to interactions with other medicinal products
Potent CYP3A4/5 inhibitors.
Concomitant use of Inlyta with potent CYP3A4/5 inhibitors (such as ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole) should be avoided. When concomitant use with Inlyta is required, an alternative agent with no or minimal potential to inhibit CYP3A4/5 activity should be considered. The possibility of adjusting the dosage of Inlyta in patients who are concomitantly receiving potent CYP3A4/5 inhibitors has not been studied. However, if co-administration of Inlyta with a potent CYP3A4/5 inhibitor is necessary, it is recommended to reduce the dose of Inlyta by approximately half, as such reduction is considered to restore AUC values to the range observed when Inlyta is administered without inhibitors. Subsequently, the dose of Inlyta may be increased or decreased based on individual safety and tolerability. After discontinuation of the potent inhibitor (after 3–5 half-lives of the inhibitor), the dose of Inlyta should be increased again to the level prescribed before initiation of the potent CYP3A4/5 inhibitor (see section "Interaction with other medicinal products and other forms of interaction").
Dosage adjustment in hepatic impairment.
In patients with mild hepatic impairment (Child-Pugh class A), no initial dose reduction of Inlyta is required. Based on pharmacokinetic data, patients with moderate hepatic impairment (Child-Pugh class B) should have the initial dose of Inlyta reduced by approximately half at the start of treatment. Subsequently, the dose of Inlyta may be increased or decreased based on individual safety and tolerability. The use of Inlyta in patients with severe hepatic impairment (Child-Pugh class C) has not been studied (see sections "Special warnings and precautions for use" and "Pharmacokinetics in special patient populations").
Renal impairment.
No dedicated studies have been conducted on the use of axitinib in patients with renal impairment. Population pharmacokinetic analyses showed no significant differences in axitinib clearance in patients with pre-existing mild, moderate, or severe renal impairment (creatinine clearance from ≤15 ml/min to <89 ml/min) (see section "Pharmacokinetics"). Patients with pre-existing mild, moderate, or severe renal impairment do not require adjustment of the initial dose. Inlyta should be used with caution in patients with end-stage renal disease (creatinine clearance <15 ml/min).
Use in elderly patients
Elderly patients do not require dose adjustment (see sections "Pharmacokinetics" and "Special warnings and precautions for use").
Children
The safety and efficacy of Inlyta in pediatric patients have not been established under real clinical conditions.
Safety and efficacy have been evaluated in clinical trials but not established in two open-label studies: a dose-finding study of Inlyta as monotherapy in 17 children aged 5 to <17 years with recurrent or refractory solid tumors (ADVL1315, NCT02164838), and a randomized study of Inlyta as monotherapy or in combination in 7 children aged 7 to <17 years (AREN1721, NCT03595124).
In these studies, no new safety concerns were observed in children.
Exposure in children receiving Inlyta at the maximum tolerated dose was lower than that previously observed in adults receiving the approved recommended starting dose.
Overdose
There is no specific antidote in case of Inlyta overdose.
In a controlled clinical trial of Inlyta for the treatment of renal cell carcinoma, one patient inadvertently received 20 mg twice daily for 4 days and experienced mild dizziness.
In a clinical dose-finding study of Inlyta, adverse reactions including arterial hypertension, seizures associated with arterial hypertension, and hemoptysis resulting in death occurred in participants receiving initial doses of 10 or 20 mg twice daily.
In case of suspected Inlyta overdose, administration of the drug should be discontinued and symptomatic treatment initiated.
Adverse Reactions
The following clinically significant adverse reactions are described in more detail in the "Use in Specific Populations" section:
- arterial hypertension;
- arterial thromboembolism;
- venous thromboembolism;
- hemorrhage;
- heart failure;
- gastrointestinal perforation and fistula formation;
- thyroid dysfunction;
- posterior reversible encephalopathy syndrome (PRES);
- proteinuria;
- elevated liver enzymes;
- hepatic dysfunction.
Because clinical trials are conducted under widely varying conditions, the frequency of adverse reactions observed in the clinical trials of a drug cannot be directly compared to the frequency in clinical trials of another drug; also, such frequencies may not reflect those observed in clinical practice.
The safety of Inlyta was evaluated in 715 patients in monotherapy clinical trials, of whom 537 had advanced renal cell carcinoma. The data presented in the "Adverse Reactions" section are from 359 patients with advanced renal cell carcinoma who participated in a randomized clinical trial comparing Inlyta to sorafenib.
Clinical Trial Experience
The median duration of treatment was 6.4 months (range: 0.03 to 22 months) for patients receiving Inlyta and 5 months (range: 0.03 to 20.1 months) for patients receiving sorafenib. Dose modifications or temporary treatment interruption due to adverse reactions were required in 199 of 359 patients (55%) receiving Inlyta and in 220 of 355 patients (62%) receiving sorafenib. Treatment was permanently discontinued due to adverse reactions in 34 of 359 patients (9%) receiving Inlyta and in 46 of 355 patients (13%) receiving sorafenib.
The most common adverse reactions (incidence ≥20%) observed with Inlyta were: diarrhea, arterial hypertension, fatigue, decreased appetite, nausea, dysphonia, palmar-plantar erythrodysesthesia syndrome, weight decrease, vomiting, asthenia, and constipation.
Table 4 lists adverse reactions reported in ≥10% of patients receiving Inlyta or sorafenib.
Table 4
| Adverse reactionsa |
Inlyta |
Sorafenib |
|||
| (N=359) |
(N=355) |
||||
| All gradesb |
Grade 3 or 4 |
All gradesb |
Grade 3 or 4 |
||
| % |
% |
% |
% |
||
| Diarrhea |
55 |
11 |
53 |
7 |
|
| Arterial hypertension |
40 |
16 |
29 |
11 |
|
| Fatigue |
39 |
11 |
32 |
5 |
|
| Decreased appetite |
34 |
5 |
29 |
4 |
|
| Nausea |
32 |
3 |
22 |
1 |
|
| Dysphonia |
31 |
0 |
14 |
0 |
|
| Palmar-plantar erythrodysesthesia syndrome |
27 |
5 |
51 |
16 |
|
| Weight decreased |
25 |
2 |
21 |
1 |
|
| Vomiting |
24 |
3 |
17 |
1 |
|
| Asthenia |
21 |
5 |
14 |
3 |
|
| Constipation |
20 |
1 |
20 |
1 |
|
| Hypothyroidism |
19 |
<1 |
8 |
0 |
|
| Cough |
15 |
1 |
17 |
1 |
|
| Mucosal inflammation |
15 |
1 |
12 |
1 |
|
| Arthralgia |
15 |
2 |
11 |
1 |
|
| Stomatitis |
15 |
1 |
12 |
<1 |
|
| Dyspnea |
15 |
3 |
12 |
3 |
|
| Abdominal pain |
14 |
2 |
11 |
1 |
|
| Headache |
14 |
1 |
11 |
0 |
|
| Limb pain |
13 |
1 |
14 |
1 |
|
| Rash |
13 |
<1 |
32 |
4 |
|
| Proteinuria |
11 |
3 |
7 |
2 |
|
| Dysgeusia |
11 |
0 |
8 |
0 |
|
| Skin dryness |
10 |
0 |
11 |
0 |
|
| Dyspepsia |
10 |
0 |
2 |
0 |
|
| Pruritus |
7 |
0 |
12 |
0 |
|
| Alopecia |
4 |
0 |
32 |
0 |
|
| Erythema |
2 |
0 |
10 |
<1 |
|
| a Percentage of all treatment-emergent reaction categories. b National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0. |
|||||
The adverse reactions (of all severity grades) observed in patients receiving Inlyta treatment listed below occurred at a frequency of <10%. These reactions included dizziness (9%), upper abdominal pain (8%), myalgia (7%), dehydration (6%), epistaxis (6%), anemia (4%), hemorrhoids (4%), hematuria (3%), tinnitus (3%), increased lipase levels (3%), glossodynia (3%), pulmonary embolism (2%), rectal hemorrhage (2%), hemoptysis (2%), deep vein thrombosis (1%), retinal vein occlusion/thrombosis (1%), polycythemia (1%), and transient ischemic attack (1%).
Table 5 lists the most common laboratory test abnormalities observed in ≥10% of patients receiving Inlyta or sorafenib.
Table 5
| Laboratory parameter abnormality |
N |
Inlyta |
N |
Sorafenib |
|||
| All gradesa |
Grade 3 or 4 |
All gradesa |
Grade 3 or 4 |
||||
| % |
% |
% |
% |
||||
| Clinical blood parameters |
|||||||
| Decreased hemoglobin level |
320 |
35 |
<1 |
316 |
52 |
4 |
|
| Decreased (absolute) lymphocyte count |
317 |
33 |
3 |
309 |
36 |
4 |
|
| Decreased platelet count |
312 |
15 |
<1 |
310 |
14 |
0 |
|
| Decreased white blood cell count |
320 |
11 |
0 |
315 |
16 |
<1 |
|
| Biochemical blood parameters |
|||||||
| Increased creatinine level |
336 |
55 |
0 |
318 |
41 |
<1 |
|
| Decreased bicarbonate level |
314 |
44 |
<1 |
291 |
43 |
0 |
|
| Hypocalcemia |
336 |
39 |
1 |
319 |
59 |
2 |
|
| Increased ALP level |
336 |
30 |
1 |
319 |
34 |
1 |
|
| Hyperglycemia |
336 |
28 |
2 |
319 |
23 |
2 |
|
| Increased lipase level |
338 |
27 |
5 |
319 |
46 |
15 |
|
| Increased amylase activity |
338 |
25 |
2 |
319 |
33 |
2 |
|
| Increased ALT level |
331 |
22 |
<1 |
313 |
22 |
2 |
|
| Increased AST level |
331 |
20 |
<1 |
311 |
25 |
1 |
|
| Hypernatremia |
338 |
17 |
1 |
319 |
13 |
1 |
|
| Hypoalbuminemia |
337 |
15 |
<1 |
319 |
18 |
1 |
|
| Hyperkalemia |
333 |
15 |
3 |
314 |
10 |
3 |
|
| Hypoglycemia |
336 |
11 |
<1 |
319 |
8 |
<1 |
|
| Hypnatremia |
338 |
13 |
4 |
319 |
11 |
2 |
|
| Hypophosphatemia |
336 |
13 |
2 |
318 |
49 |
16 |
|
and National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0:
ALP – alkaline phosphatase; ALT – alanine aminotransferase; AST – aspartate aminotransferase.
Changes in individual laboratory parameters (all grades) were observed in < 10% of patients receiving Inlyta, including increased hemoglobin levels above the upper normal limit (9% in the Inlyta group compared with 1% in the sorafenib group) and hypercalcemia (6% in the Inlyta group compared with 2% in the sorafenib group).
Post-marketing experience
The following adverse reactions have been identified during post-marketing use of Inlyta. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Vascular disorders: arterial aneurysms (including aortic), dissections, and ruptures.
Reporting suspected adverse reactions
Reporting suspected adverse reactions after medicine authorization is important. It allows ongoing monitoring of the benefit-risk balance of the medicine. Healthcare and pharmacy professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Information System for Pharmacovigilance at the following link: https://aisf.dec.gov.ua.
Shelf life. 3 years.
Storage conditions. Store below 30 °C in a place inaccessible to children.
Packaging. 14 tablets per blister. 2 or 4 blisters per cardboard box.
Prescription status. Prescription only.
Manufacturer. Pfizer Manufacturing Deutschland GmbH.
Manufacturer's address.
Mooswaldallee 1, 79108 Freiburg Im Breisgau, Germany.