Zarxio®
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU ZARXIO® (ZARXIO®)
Skład:
substancja czynna: filgrastym (rekombinacyjny ludzki czynnik stymulujący kolonie granulocytów);
1 ml roztworu zawiera 60 mln IU (600 µg) lub 96 mln IU (960 µg) filgrastymu;
szczypiora wstępnie wypełniona (szczypiora dawkowa) zawiera 30 mln IU (300 µg) lub 48 mln IU (480 µg) filgrastymu w 0,5 ml;
substancje pomocnicze: kwas glutaminowy, sorbitol (E 420), polisorbat 80, woda do wstrzykiwań.
Postać leku. Roztwór do wstrzykiwań lub do infuzji.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny lub lekko żółtawy roztwór.
Grupa farmakoterapeutyczna.
Imunostymulatory. Czynniki stymulujące wzrost kolonii. Filgrastym.
Kod ATC L03A A02.
Właściwości farmakologiczne.
Farmakodynamika.
Substancją czynną leku jest filgrastym – rekombinacyjny ludzki czynnik stymulujący kolonie granulocytów (G-CSF). Filgrastym wykazuje taką samą aktywność biologiczną jak endogenny ludzki G-CSF, różniąc się od niego jedynie tym, że jest białkiem nieglikozylowanym z dodatkowym N-końcowym resztkiem metioniny. Filgrastym otrzymywany technologią rekombinowanego DNA wydzielany jest z komórek bakterii Escherichia coli, do których aparatu genetycznego wprowadzono gen kodujący białko G-CSF.
Ludzki czynnik stymulujący kolonie granulocytów – glikoproteina – reguluje tworzenie funkcjonalnie aktywnych neutrofili oraz ich uwalnianie do krwi z szpiku kostnego. Filgrastym istotnie zwiększa liczbę neutrofili we krwi obwodowej już w ciągu pierwszych 24 godzin po podaniu, jednocześnie powodując nieznaczny wzrost liczby monocytów. Zwiększenie liczby neutrofili pod wpływem leku w zakresie dawek zalecanych zależy od wielkości dawki. Ich właściwości funkcjonalne są normalne lub wzmocnione, o czym świadczą wyniki badań chemotaksji i fagocytozy. Po zakończeniu leczenia lekiem liczba neutrofili we krwi obwodowej zmniejsza się o 50% w ciągu 1–2 dni, a do poziomu normalnego – w ciągu 1–7 dni.
Stosowanie filgrastymu istotnie zmniejsza częstość, ciężkość i czas trwania neutropenii u chorych po chemioterapii cytostatykami lub terapii mieloablatywnej z późniejszą przeszczepieniem szpiku kostnego. Stosowanie filgrastymu, zarówno pierwotne, jak i po chemioterapii, aktywuje komórki prekursorowe hematopoezy krwi obwodowej (KP-HKO). Te autologiczne KP-HKO można pobierać od chorego i podawać mu po leczeniu cytostatykami w wysokich dawkach, zamiast przeszczepu szpiku kostnego lub jako uzupełnienie do niego. Podanie KP-HKO przyspiesza regenerację hematopoezy, zmniejsza niebezpieczeństwo powikłań krwotocznych i potrzebę przetaczania masy płytkowej. U dzieci i dorosłych z wrodzoną neutropenią ciężką (TCH) filgrastym trwale zwiększa liczbę neutrofili we krwi obwodowej i zmniejsza częstość powikłań infekcyjnych.
Farmakokinetyka.
Po podaniu leku drogą dożylną i podskórnie obserwuje się pozytywną zależność liniową stężenia leku w osoczu od dawki. Po podaniu zalecanych dawek drogą podskórną stężenie leku w surowicy przekracza 10 ng/ml przez 8–16 godzin; objętość dystrybucji w krwi wynosi około 150 ml/kg. Tak po podaniu podskórnym, jak i dożylnym eliminacja leku z organizmu odpowiada kinetyce pierwszego rzędu. Średnia wartość okresu półtrwania filgrastymu w surowicy wynosi około 3,5 godziny, a szybkość klirensu wynosi około 0,6 ml/min na 1 kg masy ciała. Ciągłe podawanie leku przez infuzję przez 28 dni chorym po autologicznym przeszczepie szpiku kostnego nie wiązało się z objawami kumulacji ani wydłużenia okresu półtrwania leku.
Właściwości kliniczne.
Wskazania.
Skrócenie trwania i nasilenia neutropenii u pacjentów poddawanych intensywnej mielosupresyjnej chemioterapii lekami cytotoksycznymi z powodu nowotworów złośliwych (z wyłączeniem przewlekłej białaczki szpikowej i zespołu mielodysplastycznego) oraz skrócenie trwania neutropenii u pacjentów poddawanych chemioterapii wysokimi dawkami leków cytotoksycznych z następującą autologiczną lub allogeniczną transplantacją szpiku kostnego.
Bezpieczeństwo i skuteczność stosowania filgrastymu są podobne u dorosłych i dzieci otrzymujących chemioterapię lekami cytotoksycznymi.
-
Lek jest wskazany w celu mobilizacji krążących komórek macierzystych (KKM).
-
Długotrwałe stosowanie wskazane jest u dzieci i dorosłych z ciężką wrodzoną, cykliczną lub idiopatyczną neutropenią oraz neutropenią z bezwzględną liczbą neutrofili <0,5×10⁹/l w celu zwiększenia liczby neutrofili i zmniejszenia częstości infekcji.
-
Leczenie trwającej neutropenii (z bezwzględną liczbą neutrofili ≤1,0×10⁹/l) u pacjentów z zaawansowanym stadium infekcji HIV w celu zmniejszenia ryzyka infekcji bakteryjnych, gdy inne metody leczenia neutropenii są nieodpowiednie.
Przeciwwskazania.
- Nadwrażliwość na filgrastym, czynniki stymulujące kolonie, Escherichia coli lub dowolne substancje pomocnicze.
- Ciężka wrodzona neutropenia (zespoł Kostmanna) z zaburzeniami cytogenetycznymi oraz autoimmunologiczna neutropenia.
- Ostateczny etap przewlekłej niewydolności nerek (PNN).
- Przewlekła białaczka szpikowa i zespół mielodysplastyczny.
Interakcje z innymi lekami i inne rodzaje interakcji.
Bezpieczeństwo i skuteczność podawania leku Zarxio® w tym samym dniu co leki cytotoksyczne mielosupresyjne nie zostały ustalone. Ze względu na wrażliwość szybko dzielących się komórek mieloidalnych na mielosupresyjną chemioterapię cytotoksyczną, nie zaleca się podawania leku Zarxio® w ciągu 24 godzin przed lub po podaniu tych leków.
W przypadku jednoczesnego stosowania leku Zarxio® i 5-fluorouracylu nasilenie neutropenii może się nasilić. Interakcje z innymi czynnikami wzrostu hematopoetycznego i cytokinami są nieznane.
Ponieważ lit powoduje uwalnianie neutrofili, możliwe jest wzmocnienie działania leku Zarxio® przy podawaniu łącznie, jednak nie przeprowadzono takich badań.
Ze względu na niezgodność farmaceutyczną, nie wolno mieszać leku z 0,9 % roztworem chlorku sodu.
Szczególne środki ostrożności
Zjawiska nadwrażliwości
Zarejestrowano reakcje nadwrażliwości, w tym reakcje anafilaktyczne, pojawiające się na początku lub w trakcie leczenia u pacjentów przyjmujących filgrastym. Leczenie lekiem Zarxio® należy przerwać u pacjentów z klinicznie istotnymi reakcjami nadwrażliwości.
Nie należy stosować leku Zarxio® u pacjentów z podwyższoną wrażliwością na filgrastym lub pegfilgrastym w wywiadzie.
Zjawiska ze strony układu oddechowego
Istnieją doniesienia o rzadkich przypadkach niepożądanych działań na narządy oddechowe, w tym rozwoju zapalenia śródmiąższowego płuc w przebiegu stosowania G-CSF. Pacjenci, którzy niedawno chorowali na chorobę infiltracyjną płuc lub zapalenie płuc, mogą mieć zwiększone ryzyko. Pojawienie się takich objawów jak kaszel, podwyższona temperatura ciała i duszność w połączeniu z wykrytym obrazowo infiltratem płucnym oraz objawami postępującej niewydolności oddechowej pozwala podejrzewać zespół ostrej niewydolności oddechowej dorosłych (ARDS). W przypadku rozpoznania ARDS należy przerwać stosowanie filgrastymu i rozpocząć odpowiednie leczenie.
W okresie postmarketingowym zgłaszano bardzo rzadkie przypadki niepożądanych zjawisk ze strony płuc (krwioplucie, krwawienia z płuc, infiltracja płuc, duszność i niedotlenienie), w tym u zdrowych dawców. W przypadku podejrzenia lub potwierdzenia niepożądanego zjawiska ze strony płuc należy przerwać dalsze stosowanie filgrastymu i zapewnić pacjentowi odpowiednią pomoc medyczną.
Zapalenie kłębuszków nerek
Zapalenie kłębuszków nerek obserwowano u pacjentów otrzymujących filgrastym i pegfilgrastym. Zazwyczaj zapalenie kłębuszków nerek ustępuje po zmniejszeniu dawki lub odstawieniu filgrastymu i pegfilgrastymu. Zaleca się okresowe badanie moczu.
Zespół wycieku kapilarnego
Zgłaszano przypadki zespołu wycieku kapilarnego, który może zagrozić życiu, jeśli nie zostanie odpowiednio zdiagnozowany i leczony, po stosowaniu G-CSF, charakteryzującego się hipotensją tętniczą, hipoalbuminemią, obrzękami i hemokoncentracją. Pacjenci, u których pojawiają się objawy zespołu wycieku kapilarnego, wymagają starannego monitorowania i leczenia objawowego, w tym działań resuscytacyjnych.
Powiększenie śledziony i pęknięcie śledziony
Po podaniu filgrastymu u zdrowych dawców i pacjentów obserwowano przypadki bezobjawowego powiększenia śledziony i pęknięcia śledziony. Powiększenie śledziony jest bezpośrednią konsekwencją podania filgrastymu. U 31% pacjentów uczestniczących w badaniu stwierdzono powiększenie śledziony wykryte podczas badania palpacyjnego. Zarejestrowano kilka przypadków śmiertelnego pęknięcia śledziony. Dlatego konieczna jest staranna kontrola wielkości śledziony (badanie kliniczne i ultrasonograficzne). U dawców i/lub pacjentów skarżących się na ból w lewym górnym kwadrancie brzucha lub w lewym barku należy wykluczyć rozstrzygające rozpoznanie pęknięcia śledziony. Zmniejszenie dawki hamuje lub zatrzymuje postępowanie powiększenia śledziony, u 3% pacjentów konieczna była splenektomia.
Wzrost komórek nowotworowych
Ponieważ znane jest, że G-CSF wspomagają wzrost komórek mieloidalnych in vitro, podobne efekty mogą występować dla niektórych niemieloidalnych komórek in vitro.
Zespół mielodysplastyczny lub przewlekła białaczka szpikowa
Bezpieczeństwo i skuteczność stosowania filgrastymu u pacjentów z zespołem mielodysplastycznym lub przewlekłą białaczką szpikową nie zostały ustalone, dlatego filgrastym nie jest wskazany w tych stanach. Szczególną uwagę należy zwrócić na różnicowanie ostrej białaczki szpikowej i transformacji białaczkowej przewlekłej białaczki szpikowej.
Ostra białaczka mieloblastyczna (AML)
Ponieważ dane dotyczące bezpieczeństwa i skuteczności filgrastymu u pacjentów z wtórną ostrą białaczką mieloblastyczną (AML) są ograniczone, lek należy przepisywać ostrożnie.
Bezpieczeństwo i skuteczność podawania filgrastymu de novo pacjentom z AML w wieku < 55 lat z korzystnym rokowaniem cytogenetycznym [t(8;21), t(15;17) oraz inv(16)] nie zostały ustalone.
Trombocytopenia
Trombocytopenia była bardzo często obserwowana u pacjentów przyjmujących filgrastym. Konieczna jest staranna kontrola liczby płytek krwi, szczególnie w pierwszych tygodniach terapii z zastosowaniem filgrastymu. W przypadkach rozwoju trombocytopenii u pacjentów z ciężką przewlekłą neutropenią (CPN), tj. trwałego obniżenia liczby płytek krwi do poziomu < 100 × 10⁹/l, dalszą terapię filgrastymem należy tymczasowo odstawić lub zmniejszyć dawkę.
Leukocytoza
Liczba leukocytów we krwi osiąga lub przekracza 100 × 10⁹/l u mniej niż 5% chorych otrzymujących dawkę dzienną leku większą niż 0,3 mln IU/kg (3 μg/kg) masy ciała. Nie ma doniesień o jakichkolwiek niepożądanych działaniach bezpośrednio spowodowanych leukocytozą takiego stopnia ciężkości. Jednakże, biorąc pod uwagę możliwy ryzyko związane z ciężką leukocytozą, w trakcie leczenia filgrastymem należy regularnie kontrolować liczbę leukocytów. Jeśli liczba leukocytów przekroczy 50 × 10⁹/l po osiągnięciu oczekiwanego poziomu, należy natychmiast odstawić lek. W przypadku stosowania leku w celu mobilizacji PBSC należy go odstawić lub skorygować dawkę przy wzroście liczby leukocytów do > 70 × 10⁹/l.
Immunogenność
Tak jak w przypadku stosowania wszystkich leków białkowych, istnieje możliwość rozwoju immunogenności. Częstość występowania przeciwciał przeciwko filgrastymowi jest zazwyczaj niska. Wykryto przeciwciała wiążące, tak jak w przypadku stosowania wszystkich produktów biologicznych; obecnie nie są one jednak związane z działaniem neutralizującym.
Szczególne ostrzeżenia i środki ostrożności związane z chorobami współistniejącymi
Specjalne środki ostrożności u pacjentów z objawami lub chorobą sierpowatą
Zgłaszano przypadki kryzysu sierpowatego, w niektórych przypadkach śmiertelne, po stosowaniu filgrastymu u pacjentów z objawami sierpowatymi lub chorobą sierpowatą. Lekarze powinni zachować ostrożność przy przepisywaniu filgrastymu pacjentom z objawami sierpowatymi lub chorobą sierpowatą.
U chorych z anemią sierpowatą obserwowano przypadki rozwoju ostrego kryzysu hemolitycznego (zwiększenie liczby zmienionych komórek), czasem zakończonego śmiercią. Takim chorym należy przepisywać filgrastym ostrożnie i w trakcie terapii starannie monitorować odpowiednie wskaźniki kliniczne i laboratoryjne, zwracając szczególną uwagę na możliwe powiększenie śledziony i rozwój zakrzepicy naczyń krwionośnych.
Osteoporoza
U chorych z współistniejącą patologią kostną i osteoporozą w przypadku długotrwałego (ponad 6 miesięcy) stosowania filgrastymu zaleca się regularne kontrolowanie gęstości tkanki kostnej.
Szczególne środki ostrożności u pacjentów z chorobami nowotworowymi
Filgrastym nie powinien być stosowany w celu zwiększenia dawki chemioterapii cytotoksycznej ponad ustalone schematy dawkowania.
Ryzyko związane ze zwiększeniem dawki chemioterapii
Należy zachować szczególną ostrożność podczas leczenia chorych z nowotworami złośliwymi, którzy otrzymują wysokie dawki cytostatyków, ponieważ skuteczność takiego leczenia nie została ustalona. Wiadomo, że zwiększone dawki leków chemicznych wykazują większą toksyczność, prowadząc do rozwoju poważnych działań niepożądanych kardiologicznych, płucnych, neurologicznych i dermatologicznych (patrz instrukcje dotyczące stosowania współistniejących leków cytotoksycznych).
Wpływ chemioterapii na erytrocyty i trombocyty
Monoterapia filgrastymem nie zapobiega rozwojowi trombocytopenii i anemii spowodowanych mielosupresyjną chemioterapią. W przypadku stosowania wyższych dawek leków chemicznych (np. pełne dawki zgodnie z ustalonymi schematami) ryzyko ciężkiej trombocytopenii i anemii wzrasta.
Zaleca się regularne monitorowanie takich wskaźników morfologii krwi jak hematokryt i liczba płytek krwi. Należy zachować szczególną ostrożność przy stosowaniu pojedynczych lub kombinowanych leków chemioterapeutycznych, które mogą spowodować ciężką trombocytopenię.
W przypadku stosowania filgrastymu w celu mobilizacji PBSC stwierdzono zmniejszenie nasilenia i czasu trwania trombocytopenii spowodowanej mielosupresyjną lub mieloablatywną chemioterapią.
Inne specjalne środki ostrożności
Skuteczność leku u chorych z istotnie zmniejszoną liczbą komórek mieloidalnych prekursorowych nie była badana. Filgrastym zwiększa liczbę neutrofili poprzez działanie głównie na komórki prekursorowe neutrofili. Dlatego u chorych z zmniejszoną liczbą komórek prekursorowych (np. w wyniku intensywnej radioterapii, chemioterapii lub wskutek infiltracji szpiku kostnego komórkami nowotworowymi) liczba powstających neutrofili może być zmniejszona.
Czasem u chorych, którzy otrzymywali chemioterapię wysokich dawkach z następową transplantacją autologicznego szpiku kostnego, obserwowano zaburzenia naczyniowe, np. okluzję żył i zaburzenia gospodarki wodnej.
Istnieją dane dotyczące rozwoju reakcji „przeszczep przeciwko gospodarzowi” i przypadków śmiertelnych u chorych, którzy otrzymują G-CSF po allogenicznej transplantacji szpiku kostnego.
Wzmożony hematopoeza w szpiku kostnym w odpowiedzi na terapię czynnikami wzrostu wiąże się z pojawieniem się przemijających zmian patologicznych wykrywanych w scyntygrafii kości. Należy to uwzględnić przy interpretacji obrazów diagnostycznych kości.
Zgłaszano przypadki zapalenia aorty po podaniu filgrastymu, zarówno u zdrowych, jak i u chorych onkologicznych. Obserwowano następujące objawy: gorączkę, ból brzucha, niedowagę, ból pleców oraz podwyższone wskaźniki zapalenia (np. podwyższony poziom białka C-reaktywnego i leukocytozę). W większości przypadków zapalenie aorty rozpoznawano za pomocą tomografii komputerowej i zazwyczaj ustępowało po odstawieniu filgrastymu.
Szczególne środki ostrożności dla pacjentów wymagających mobilizacji PBSC
Mobilizacja
Nie istnieje żadne randomizowane porównanie dwóch zalecanych metod mobilizacji (filgrastym samodzielnie lub w połączeniu z mielosupresyjną chemioterapią) w obrębie tej samej populacji pacjentów. Stopień zmienności między poszczególnymi pacjentami oraz między laboratoriami analizującymi komórki CD34+ oznacza, że bezpośrednie porównanie różnych badań jest trudne. Dlatego trudno polecić optymalną metodę. Wybór metody mobilizacji należy rozważyć w kontekście ogólnych celów leczenia dla danego pacjenta.
Wcześniejszy wpływ substancji cytotoksycznych
U chorych, którzy wcześniej otrzymywali intensywną terapię mielosupresyjną, w trakcie stosowania filgrastymu w celu mobilizacji PBSC może nie dojść do zwiększenia liczby PBSC do zalecanego minimalnego poziomu (≥ 2,0 × 10⁶ komórek CD34+/kg) lub przyspieszenia odnowy płytek krwi.
Niektóre substancje cytotoksyczne wykazują szczególną toksyczność wobec komórek prekursorowych hematopoezy i negatywnie wpływają na ich mobilizację. Długotrwałe stosowanie melafalanu, karboplatyny lub karmustyny (BCNU) przed mobilizacją komórek prekursorowych może prowadzić do pogorszenia wyników. Jednak jednoczesne stosowanie melafalanu, karboplatyny lub BCNU z filgrastymem jest skuteczne w mobilizacji PBSC. Jeśli planowana jest transplantacja PBSC, zaleca się przeprowadzenie mobilizacji komórek macierzystych na wczesnym etapie leczenia chorego. Szczególną uwagę należy zwrócić na liczbę komórek prekursorowych aktywowanych u takich chorych przed podaniem leków chemioterapeutycznych w wysokich dawkach. Jeśli wyniki mobilizacji nie spełniają powyższych kryteriów, należy rozważyć zastosowanie alternatywnych metod leczenia, które nie wymagają stosowania komórek prekursorowych.
Ocena liczby komórek prekursorowych
Przy ocenie liczby PBSC zmobilizowanych u chorych, którzy otrzymali terapię z zastosowaniem filgrastymu, należy zwrócić szczególną uwagę na metodę ilościowego oznaczania. Wyniki analizy cytofluorymetrycznej liczby komórek CD34+ różnią się znacznie w zależności od zastosowanej metodyki oznaczania, dlatego należy ostrożnie interpretować wyniki oznaczeń uzyskane w innych laboratoriach.
Wyniki analizy statystycznej związku między liczbą podanych komórek CD34+ a szybkością normalizacji liczby płytek krwi po przeprowadzeniu chemioterapii wysokich dawkach wskazują na złożoną, ale stałą zależność. Rekomendacje dotyczące konieczności zapewnienia minimalnej liczby na poziomie ≥ 2,0 × 10⁶ komórek CD34+/kg opierają się na opublikowanych danych dotyczących doświadczenia z wystarczającej regeneracji wskaźników hematologicznych. Przy poziomie wyższym niż zalecany minimalny obserwuje się szybszą normalizację, przy poziomie niższym niż zalecany – dłuższy czas trwania.
Szczególne środki ostrożności dla zdrowych dawców poddawanych mobilizacji PBSC
Mobilizacja PBSC zdrowych dawców wpływa na ich stan zdrowia i stosowana jest wyłącznie w celu uzyskania allogennych komórek macierzystych do transplantacji.
Dawcy, u których przeprowadza się mobilizację PBSC w celu transplantacji, powinni spełniać standardowe wymagania dotyczące wskaźników klinicznych i wyników badań laboratoryjnych stawiane dawcom komórek macierzystych. Szczególną uwagę należy zwrócić na wskaźniki badań krwi i obecność chorób zakaźnych. Bezpieczeństwo i skuteczność podawania filgrastymu zdrowym dawcom w wieku do 16 lat oraz powyżej 60 lat nie były oceniane.
Przejściowa trombocytopenia (liczba płytek krwi < 100 × 10⁹/l) po podaniu filgrastymu i leukaferezie obserwowano u 35% pacjentów. Wśród nich zgłoszono dwa przypadki obniżenia liczby płytek krwi < 50 × 10⁹/l, które przypisano procedurze leukaferezy.
W przypadku konieczności przeprowadzenia więcej niż jednej procedury leukaferezy należy zwrócić szczególną uwagę na dawców, u których zawartość płytek krwi przed rozpoczęciem leukaferezy wynosi < 100 × 10⁹/l; zazwyczaj przeprowadzenie leukaferezy nie jest zalecane przy zawartości płytek krwi < 75 × 10⁹/l.
Leukaferezy nie należy przeprowadzać dawcom wymagającym terapii przeciwzakrzepowej lub z zaburzeniami hemostazy.
Monitorowanie stanu zdrowia dawców otrzymujących G-CSF w celu mobilizacji PBSC należy kontynuować aż do normalizacji wskaźników hematologicznych.
U zdrowych dawców po podaniu G-CSF obserwowano przemijające zmiany cytogennicze. Znaczenie tych zmian nie jest ustalone.
Trwają badania dotyczące oceny długoterminowego bezpieczeństwa leku u dawców. Ryzyko sprzyjania powstawaniu złośliwych klonów komórek mieloidalnych nie jest wykluczone. Centrom aferetycznym zaleca się przeprowadzanie systematycznych badań dawców komórek macierzystych przez okres co najmniej 10 lat w celu monitorowania wskaźników długoterminowego bezpieczeństwa.
Szczególne środki ostrożności dla biorców poddawanych mobilizacji PBSC za pomocą filgrastymu
Dane wskazują, że immunologicznej interakcji allogennych PBSC i biorcy towarzyszy wyższe ryzyko rozwoju ostrej i przewlekłej reakcji „przeszczep przeciwko gospodarzowi” w porównaniu z transplantacją szpiku kostnego.
Szczególne środki ostrożności dla pacjentów z CPN
Filgrastym nie należy stosować pacjentom z ciężką wrodzoną neutropenią, u których rozwinęła się białaczka, oraz chorym z objawami transformacji białaczkowej.
Badania składu krwi
Możliwe są inne zmiany morfologii krwi, w tym anemia i tymczasowy wzrost zawartości komórek mieloidalnych prekursorowych; konieczna jest staranna kontrola morfologii krwi.
Transformacja w białaczkę lub przedbiałaczkę
Szczególna ostrożność jest wymagana przy diagnozowaniu CPN, aby odróżnić ją od innych chorób hematologicznych, takich jak anemia aplastyczna, mielodysplazja i białaczka szpikowa. Przed rozpoczęciem leczenia należy przeprowadzić rozwiniętą analizę krwi z określeniem formuły leukocytów i liczby płytek krwi, a także określić obraz morfologiczny szpiku kostnego i karotyp.
Rozwój zespołu mielodysplastycznego (MDS) lub białaczki u pacjentów z ciężką przewlekłą neutropenią, którzy uczestniczyli w badaniach klinicznych dotyczących stosowania filgrastymu, obserwowany jest rzadko (w przybliżeniu u 3% przypadków). Te zaburzenia obserwowano wyłącznie u pacjentów z wrodzoną neutropenią. MDS i białaczka są częstymi powikłaniami choroby, ich związek z terapią filgrastymem jest niepewny. U około 12% pacjentów (bez zaburzeń cytogenetycznych przed rozpoczęciem terapii) w kolejnych badaniach obserwowano odchylenia, w tym monosomię 7. W przypadkach wystąpienia u pacjenta z ciężką przewlekłą neutropenią zaburzeń cytogenetycznych należy dokładnie rozważyć stosunek korzyści do ryzyka dalszego stosowania filgrastymu. Dalsze podawanie filgrastymu pacjentom w przypadku rozwoju MDS lub białaczki należy odstawić. Nie wiadomo, czy długotrwała terapia z zastosowaniem filgrastymu u pacjentów z ciężką przewlekłą neutropenią zwiększa ryzyko zaburzeń cytogenetycznych, MDS lub transformacji choroby w białaczkę. Badania morfologiczne i cytogenetyczne szpiku kostnego pacjentów należy przeprowadzać regularnie, w odstępach około co 12 miesięcy.
Inne szczególne środki ostrożności
Należy wykluczyć inne przyczyny wystąpienia neutropenii, np. infekcje wirusowe.
Hematuria występowała często, natomiast proteinuria obserwowana była u niewielkiej liczby pacjentów. Aby wczesnie wykryć te zjawiska, konieczne jest regularne przeprowadzanie badania moczu.
Wskaźniki bezpieczeństwa i skuteczności stosowania u noworodków i pacjentów z autoimmunologiczną neutropenią nie zostały ustalone.
Szczególne środki ostrożności dla pacjentów zakażonych HIV
Morfologia krwi
Konieczna jest staranna kontrola wartości bezwzględnej liczby neutrofili (ANC), szczególnie w pierwszych tygodniach terapii z zastosowaniem filgrastymu. U niektórych pacjentów obserwuje się bardzo szybką reakcję ze znacznym wzrostem liczby neutrofili w odpowiedzi na pierwszą dawkę filgrastymu. Zaleca się codzienne oznaczanie ANC w pierwszych 2-3 dniach podawania filgrastymu. Następnie, w pierwszych 2 tygodniach, oznaczanie ANC zaleca się przeprowadzać co najmniej 2 razy w tygodniu, później – 1 raz w tygodniu oraz co 2 tygodnie w okresie prowadzenia terapii wspomagającej. W okresie kolejnego podania filgrastymu w dawce indywidualnie ustalonej i dawce 30 mln IU/dobę (300 μg/dobę) możliwe są większe wahania wartości ANC pacjenta. W celu określenia wartości minimalnych (najniższych wartości ANC) zaleca się pobieranie próbek krwi pacjenta do analizy zawartości ANC bezpośrednio przed zaplanowanym podaniem filgrastymu.
Ryzyko związane ze stosowaniem leków o działaniu mielosupresyjnym w zwiększonych dawkach
Podanie filgrastymu samodzielnego nie wyklucza możliwości wystąpienia trombocytopenii i anemii spowodowanych mielosupresyjną chemioterapią. W wyniku stosowania leków chemicznych w większych dawkach lub większej liczby takich leków w połączeniu z filgrastymem ryzyko rozwoju trombocytopenii i anemii u pacjenta może wzrosnąć. Zaleca się regularne monitorowanie wskaźników morfologii krwi.
Choroby zakaźne i nowotworowe powodujące mielosupresję
Rozwój neutropenii może być konsekwencją infiltracji szpiku kostnego przez patogeny infekcji oportunistycznych, takie jak bakterie Mycobacterium avium, lub uszkodzenia przez nowotwory złośliwe, np. chłoniaki. W przypadku obecności u pacjenta chorób zakaźnych lub nowotworów złośliwych uszkadzających szpik kostny, oprócz podania filgrastymu, konieczne jest przeprowadzenie odpowiedniego leczenia choroby w celu wyeliminowania neutropenii. Wpływ filgrastymu na wyeliminowanie neutropenii spowodowanej chorobami zakaźnymi lub nowotworami złośliwymi uszkadzającymi szpik kostny nie został określony.
Substancje pomocnicze
Lek Zarxio® zawiera sorbitol, dlatego pacjentom z rzadką dziedziczną nietolerancją fruktozy nie należy stosować tego leku.
Kapselka ochronna zawiera pochodne naturalnego lateksu gumowego. Choć sam lek nie zawiera naturalnego lateksu gumowego. Bezpieczeństwo stosowania produktu u pacjentów wrażliwych na lateks nie zostało ocenione.
W celu poprawy śledzenia G-CSF, nazwa leku, który jest podawany, powinna być wyraźnie zaznaczona w karcie pacjenta.
Ten lek zawiera mniej niż 1 mmol (23 mg)/dawkę sodu, tj. jest praktycznie pozbawiony sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Bezpieczeństwo filgrastymu u kobiet w ciąży nie zostało ustalone. Istnieją dane o przenikaniu filgrastymu przez barierę łożyskową. Nie otrzymano danych o teratogenności filgrastymu w badaniach na zwierzętach. W badaniach na zwierzętach stwierdzono toksyczność reprodukcyjną. U zwierząt otrzymujących filgrastym obserwowano zwiększoną częstość poronień.
Lek Zarxio® nie jest zalecany do stosowania w czasie ciąży.
Nie wiadomo, czy filgrastym i jego metabolity przenikają do mleka matki, dlatego nie można wykluczyć ryzyka dla niemowląt. Dlatego należy podjąć decyzję o zaprzestaniu karmienia piersią lub stosowania filgrastymu, biorąc pod uwagę korzyści z karmienia piersią dla dziecka i korzyści z terapii dla kobiety.
W badaniach na zwierzętach stwierdzono, że filgrastym nie wpływa na układ rozrodczy ani płodność zwierząt.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwanie maszyn.
Filgrastym może mieć nieznaczny wpływ na reakcje podczas prowadzenia pojazdów lub obsługiwanie maszyn, a mianowicie – możliwe zawroty głowy.
Sposób stosowania i dawki.
Terapię lekiem Zarxio® można prowadzić w placówkach medycznych, w których istnieje dostęp do niezbędnego wyposażenia diagnostycznego. Lekarze powinni mieć doświadczenie w stosowaniu leków zawierających czynnik stymulujący kolonie granulocytów (G-CSF) oraz w leczeniu pacjentów z chorobami hematologicznymi.
Procedury mobilizacji oraz aferezy należy przeprowadzać we współpracy z lekarzami posiadającymi odpowiednie doświadczenie oraz możliwość niezbędnego monitorowania komórek prekursorowych hematopoezy.
Neutropenia u pacjentów otrzymujących cytotoksyczną chemioterapię z powodu nowotworów złośliwych.
Zalecana dawka dobową leku wynosi 0,5 mln IU/kg (5 μg/kg) masy ciała, podawana 1 raz na dobę. Pierwszą dawkę leku należy podać nie wcześniej niż po upływie 24 godzin od zakończenia cyklu cytotoksycznej chemioterapii. Lek stosuje się do momentu, gdy całkowita liczba neutrofili w morfologii krwi przekroczy oczekiwany poziom i osiągnie normę. Po chemioterapii przeciwnowotworowej z powodu nowotworów litych, chłoniaków oraz limfoleukoz, czas trwania leczenia do osiągnięcia wskazanych wartości wynosi do 14 dni. Po terapii indukcyjnej i konsolidacyjnej w ostrym białaczce szpikowej czas trwania leczenia może być znacznie wydłużony (do 38 dni), w zależności od rodzaju, dawki oraz schematu zastosowanej cytotoksycznej chemioterapii.
U pacjentów otrzymujących cytotoksyczną chemioterapię, przejściowy wzrost liczby neutrofili obserwuje się zazwyczaj w ciągu 1–2 dni od rozpoczęcia leczenia lekiem Zarxio®. Jednakże, aby osiągnąć stabilny efekt terapeutyczny, leczenie należy kontynuować aż do momentu, gdy liczba neutrofili przekroczy oczekiwany minimum i osiągnie normę. Nie zaleca się przedwczesnego odstawiania leku przed osiągnięciem przez liczbę neutrofili poziomu oczekiwanego minimum.
Sposób podania
Lek Zarxio® stosuje się w postaci wstrzyknięć podskórnych lub dożylnej infuzji (rozcieńczony w 5% roztworze glukozy) przez 30 minut, 1 raz na dobę. W większości przypadków preferowaną drogą podania jest droga podskórna. Przy dożylnej aplikacji pojedynczej dawki czas trwania działania leku może być skrócony. Kliniczne znaczenie tych danych w odniesieniu do stosowania wielokrotnych dawek leku nie zostało ustalone. Wybór sposobu podania zależy od indywidualnych cech danej sytuacji klinicznej i jest określany indywidualnie dla każdego pacjenta.
Pacjenci otrzymujący terapię mieloablatywną z kolejną przeszczepieniem szpiku kostnego.
Zalecana początkowa dawka leku Zarxio® wynosi 1 mln IU/kg (10 μg/kg) masy ciała na dobę. Pierwszą dawkę należy podać nie wcześniej niż po upływie 24 godzin od zakończenia cytotoksycznej chemioterapii i nie wcześniej niż po upływie 24 godzin od przeszczepienia szpiku kostnego.
Po osiągnięciu maksymalnego spadku liczby neutrofili (nadiru) należy dostosować dawkę dobową leku Zarxio® w zależności od zmian liczby neutrofili (patrz tabela).
Dobór dawki leku Zarxio® w odpowiedzi na osiągnięcie nadiru.
| Bezwzględna liczba neutrofili (ALN) |
Korekta dawki leku Zarxio® |
| ALN > 1 × 109/l przez 3 dni z rzędu |
Obniżenie dawki do 0,5 mln J/ED/kg (5 μg/kg) masy ciała na dobę |
| ALN > 1 × 109/l przez kolejne 3 dni z rzędu |
Przeciągnięcie leku |
| Jeśli podczas leczenia ALN spada do poziomu < 1 × 109/l, dawkę leku Zarxio® należy zwiększać zgodnie z powyższym schematem. |
|
Sposób podania
Lekarstwo należy rozpuścić w 20 ml 5% roztworu glukozy i stosować w postaci krótkotrwałej infuzji wewnątrzżylnej trwającej 30 minut lub długotrwałej podskórnej lub wewnątrzżylnej infuzji trwającej 24 godziny.
Mobilizacja krwiotwórczych komórek macierzystych krwi (KKMK) u pacjentów otrzymujących terapię mielosupresyjną lub mieloablacyjną z późniejszą autologiczną transfuzją KKMK
Chorzy otrzymujący terapię mielosupresyjną lub mieloablacyjną z późniejszą autologiczną transplantacją KKMK.
W celu mobilizacji KKMK przy stosowaniu leku Zarxio® jako monoterapii zalecana dawka wynosi 1 mln J/ kg (10 μg/kg) masy ciała na dobę przez 5–7 kolejnych dni w postaci długotrwałej infuzji podskórnej trwającej 24 godziny. Zazwyczaj wystarczają 1–2 sesje leukaferezy w dniu 5. i 6. W niektórych przypadkach należy przeprowadzić dodatkowo 1 sesję leukaferezy. Nie należy zmieniać dawki leku do ostatniej leukaferezy.
W celu mobilizacji KKMK po terapii mielosupresyjnej chemioterapią zalecana dawka leku Zarxio® wynosi 0,5 mln J/ kg (5 μg/kg) masy ciała na dobę codziennie, począwszy od pierwszego dnia po zakończeniu cyklu chemioterapii, aż do momentu, gdy liczba neutrofili przekroczy oczekiwany minimum i osiągnie normę. Leukaferezę należy przeprowadzać w okresie wzrostu ANC z < 0,5 × 10⁹/l do > 5 × 10⁹/l. Chorym, którzy nie otrzymali intensywnej chemioterapii, zazwyczaj wystarcza 1 sesja leukaferezy. W poszczególnych przypadkach zaleca się przeprowadzenie dodatkowych sesji leukaferezy.
Sposób podania
Filgrastym do mobilizacji KKMK przy stosowaniu oddzielnie można podawać w postaci ciągłej infuzji podskórnej trwającej 24 godziny lub iniekcji podskórnej. Do infuzji filgrastym należy rozpuścić w 20 ml 5% roztworu glukozy.
Filgrastym do mobilizacji KKMK po terapii mielosupresyjnej chemioterapią należy podawać podskórnie.
Mobilizacja KKMK u zdrowych dawców przed allogeniczną transplantacją KKMK.
W celu mobilizacji KKMK przed allogeniczną transplantacją KKMK u zdrowych dawców zalecana dawka leku Zarxio® wynosi 1 mln J/ kg (10 μg/kg) masy ciała na dobę przez 4–5 kolejnych dni. Leukaferezę przeprowadza się od 5. dnia, a w razie potrzeby kontynuuje do 6. dnia w celu uzyskania 4 × 10⁶ komórek CD34+/kg masy ciała biorcy.
Sposób podania
Filgrastym należy podawać w postaci iniekcji podskórnej.
U pacjentów z ciężką przewlekłą neutropenią (CPN)
Neutropenia dziedziczna
Zalecana dawka początkowa – 1,2 mln J/ kg (12 μg/kg) masy ciała na dobę w formie jednorazowej iniekcji podskórnej lub dawkach podzielonych.
Neutropenia idiopatyczna i okresowa
Zalecana dawka początkowa – 0,5 mln J/ kg (5 μg/kg) masy ciała na dobę jednorazowo lub dawkach podzielonych.
Dobór dawki
Lek Zarxio® należy podawać codziennie w formie iniekcji podskórnej do osiągnięcia i trwałego przekroczenia poziomu liczby neutrofili 1,5 × 10⁹/l. Po osiągnięciu efektu terapeutycznego należy określić minimalną dawkę skuteczną w celu utrzymania tego poziomu. W celu utrzymania wymaganej liczby neutrofili konieczne jest długotrwałe codzienne podawanie leku. Po 1–2 tygodniach leczenia początkową dawkę można podwoić lub zmniejszyć o połowę w zależności od skuteczności terapii. Następnie co 1–2 tygodnie należy przeprowadzać indywidualną korektę dawki w celu ustabilizowania średniej liczby neutrofili w zakresie od 1,5 × 10⁹/l do 10 × 10⁹/l. Chorym z ciężkimi infekcjami można zastosować schemat z szybszym zwiększeniem dawki. Bezpieczeństwo stosowania filgrastymu u chorych leczonych długotrwale dawkami Zarxio® wyższymi niż 2,4 mln J (24 μg/kg) na dobę nie zostało ustalone.
Sposób podania
Filgrastym należy podawać w postaci iniekcji podskórnej.
U pacjentów zakażonych HIV
Odzyskanie liczby neutrofili
Zalecana dawka początkowa leku – 0,1 mln J/ kg (1 μg/kg) masy ciała na dobę z zwiększeniem dawki do 0,4 mln J (4 μg/kg) masy ciała na dobę w formie jednorazowej iniekcji podskórnej do normalizacji liczby neutrofili (ANC > 2,0 × 10⁹/l). Normalizacja liczby neutrofili następuje zazwyczaj po 2 dniach. W niewielkiej liczbie przypadków (< 10 % pacjentów) w celu odzyskania liczby neutrofili dawkę leku można zwiększyć do 1 mln J/kg (10 μg/kg masy ciała na dobę).
Utrzymanie normalnej liczby neutrofili
Gdy neutropenia ustąpi, należy ustalić minimalną dawkę skuteczną w celu utrzymania normalnego poziomu neutrofili. Po osiągnięciu efektu terapeutycznego dawka utrzymująca wynosi 300 μg/ dobę 2–3 razy w tygodniu według schematu naprzemiennego (co drugi dzień). Następnie może być konieczna indywidualna korekta dawki i długotrwałe stosowanie leku w celu utrzymania średniej liczby neutrofili > 2,0 × 10⁹/l.
Sposób podania
Filgrastym należy podawać w postaci iniekcji podskórnej.
Osobliwe kategorie chorych
Korekty dawki nie trzeba wprowadzać u pacjentów z ciężką niewydolnością wątroby lub nerek, ponieważ parametry farmakokinetyczne i farmakodynamiczne okazały się podobne do tych u zdrowych ochotników.
Nie ma szczególnych zaleceń dotyczących stosowania leku Zarxio® u pacjentów w podeszłym wieku.
Dzieci.
W praktyce pediatrycznej profil bezpieczeństwa Zarxio® u chorych dzieci z CPN i chorobami nowotworowymi nie różnił się od profilu u dorosłych. Bezpieczeństwo i skuteczność stosowania leku u noworodków nie zostały ustalone.
Zalecenia dotyczące dawkowania dla chorych w wieku dziecięcym są takie same jak dla dorosłych otrzymujących mielosupresyjną cytotoksyczną chemioterapię.
Zalecenia przed zastosowaniem
Przed zastosowaniem leku należy przeprowadzić kontrolę wzrokową zawartości wstępnie wypełnionego strzykawki. Roztwór powinien być przejrzysty, bez cząstek. Krótkotrwałe działanie niskich temperatur nie wpływa negatywnie na stabilność leku. Lek nie zawiera substancji konserwujących. Aby uniknąć zakażenia mikrobiologicznego, należy pamiętać, że lek Zarxio® w wstępnie wypełnionej strzykawce przeznaczony jest tylko do jednorazowego użytku. W trakcie przechowywania i do użytku ambulatoryjnego można wyjąć lek z lodówki i przechowywać w temperaturze pokojowej (nie wyższej niż 25 °C) jednorazowo przez 8 dni. Po upływie tego czasu nie należy ponownie stawiać do lodówki, a należy zniszczyć.
Zalecenia dotyczące rozcieńczania leku.
Lek Zarxio® można podawać w rozcieńczonej postaci w 5% roztworze glukozy. Rozcieńczanie do stężenia niższego niż 0,2 mln J/ml (2 μg/ml) nie jest zalecane. Przy rozcieńczaniu do stężenia < 1,5 mln J/ml (15 μg/ml) należy dodatkowo podać albuminę ludzką do osiągnięcia stężenia 2 mg/ml. Na przykład, aby osiągnąć objętość roztworu 20 ml i całkowitą dawkę leku Zarxio® 30 mln J (300 μg) należy dodatkowo podać roztwór albuminy w objętości 0,2 ml (20% roztwór).
Przy rozcieńczaniu w roztworze glukozy lek jest pochłaniany przez szkło i inne materiały stosowane do podania infuzyjnego. Zabronione jest stosowanie roztworu chlorku sodu do rozcieńczania leku.
Stabilność chemiczną i fizyczną rozcieńczonego roztworu do infuzji po otwarciu opakowania potwierdzono przez 24 godziny w warunkach przechowywania w temperaturze od 2 do 8 °C. Z mikrobiologicznego punktu widzenia lek należy użyć natychmiast. Jeżeli lek nie zostanie użyty natychmiast, czas i warunki przechowywania po otwarciu opakowania do momentu zastosowania są odpowiedzialnością użytkownika. Rozcieńczanie powinno odbywać się w kontrolowanych i zatwierdzonych warunkach bezpyłowych.
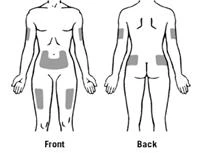
Preferowane obszary ciała do podania podskórnego Zarxio® przedstawiono na rysunku:
Instrukcje dotyczące samodzielnego podania iniekcji leku przez pacjenta
W tym rozdziale zawarte są informacje dotyczące samodzielnego podania iniekcji leku Zarxio® przez pacjenta. Ważne! Nie próbuj samodzielnie wykonywać iniekcji leku, jeśli nie został nauczony techniki podania przez lekarza lub pielęgniarkę. Lek Zarxio® jest dostarczany z urządzeniem ochronnym zapobiegającym urazowi igłą po zastosowaniu, a Twój lekarz lub pielęgniarka pokażą Ci, jak korzystać ze strzykawki. Jeżeli nie jesteś pewien, czy możesz wykonać iniekcję, lub masz pytania, skontaktuj się z lekarzem lub pielęgniarką o pomoc.
Uwaga! Nie używaj strzykawki, jeśli upadła na twardą powierzchnię lub jeśli upadła po zdjęciu osłonki igły.
- Umij ręce.
- Wyjmij jedną strzykawkę z opakowania i usuń urządzenie ochronne zapobiegające urazowi igłą po zastosowaniu z igły iniekcyjnej. Na strzykawkach znajdują się reliefowe podziałki na wypadek, gdy strzykawka ma być napełniona lekiem nie w pełni. Każda podziałka odpowiada objętości 0,1 ml. Jeśli strzykawka ma być napełniona nie w pełni, usuń nadmiar roztworu przed podaniem.
- Przetrzyj skórę w miejscu podania chusteczką alkoholową.
- Utwórz fałd skórny, zaciskając skórę kciukiem i palcem wskazującym.
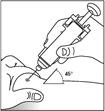
- Wprowadź igłę w fałd skórny szybkim pewnym ruchem. Wprowadź roztwór leku Zarxio® w sposób, w jaki pokazał to lekarz. Jeśli nie jesteś pewien, powinieneś skontaktować się z lekarzem lub farmaceutą.
|
|
Poniższa informacja przeznaczona jest wyłącznie dla personelu medycznego.
Urządzenie ochronne zapobiegające urazom igłą po zastosowaniu zakrywa igłę po iniekcji, aby zapobiec urazom spowodowanym ukłuciem igłą. Nie wpływa to na normalne działanie strzykawki. Powoli i równomiernie naciskaj na tłoczek, aż cała dawka zostanie wstrzyknięta i tłoczek przestanie się przesuwać. Kontynuując naciskanie na tłoczek, wyciągnij igłę z skóry. Urządzenie ochronne zapobiegające urazom igłą po zastosowaniu zakryje igłę po zwolnieniu tłoczka.
Utylizacja
Nie wykorzystany lek lub odpady należy zutylizować zgodnie z lokalnymi przepisami.
Przedawkowanie.
Objawy przedawkowania Zarxio® są nieznane. W ciągu 1–2 dni po zakończeniu leczenia lekiem liczba krążących neutrofilowych granulocytów zazwyczaj spada o 50%, a w ciągu 1–7 dni wraca do normy.
Efekty uboczne.
Częstość występowania efektów ubocznych sklasyfikowano w następujący sposób: bardzo często (≥ 1/10); często (≥ 1/100, < 1/10); rzadko (≥ 1/1000, < 1/100); pojedyncze (≥ 1/10000, < 1/1000); rzadkie (< 1/10000).
Najpoważniejsze efekty uboczne, które mogą wystąpić podczas leczenia filgrastymem, to: reakcje anafilaktyczne, poważne działania niepożądane ze strony płuc (w tym zapalenie śródmiąższowe płuc i ostre zaburzenia oddechowe), zespół przeciekania przez naczynia włosowate, ciężka splenomegalia/rozerwanie śledziony, zespół mielodysplastyczny lub białaczka u pacjentów z ciężką wrodzoną neutropenią, odrzucenie przeszczepu u pacjentów otrzymujących allogeniczne przeszczepienie szpiku kostnego lub przeszczepienie komórek prekursorowych krwi obwodowej oraz krystaliczny zespół wywołany przez komórki sierpowate u pacjentów z chorobą sierpowatą.
Najczęstsze działania niepożądane podczas terapii z zastosowaniem filgrastymu to: gorączka, ból kości i mięśni (w tym ból kości, ból pleców, ból stawów, mięśniowy ból, ból kończyn, ból mięśniowo-kościowy, ból mięśniowo-kościowy w klatce piersiowej, ból szyi), anemia, nudności i wymioty. U pacjentów z chorobami nowotworowymi ból mięśniowo-kościowy o niskim do średniego stopnia nasilenia obserwowano u 10 % pacjentów, o wysokim stopniu nasilenia – u 3 %. Ból kości i mięśni zazwyczaj ustępuje po podaniu standardowych środków przeciwbólowych.
Podczas mobilizacji PBSC u zdrowych dawców najpowszechniejszym efektem ubocznym był ból mięśniowo-szkieletowy.
U pacjentów z ciężką wrodzoną neutropenią (CWN) najczęstszymi efektami ubocznymi podczas terapii z zastosowaniem filgrastymu były: ból kości, ogólny ból mięśniowo-szkieletowy, powiększenie i rozerwanie śledziony. Zespół mielodysplastyczny (ZMD) lub białaczka obserwowano u pacjentów z wrodzoną neutropenią, którzy stosowali filgrastym.
Zespół przeciekania przez naczynia włosowate, stan zagrożony życia w przypadku braku natychmiastowego leczenia, obserwowano rzadko (≥1/1000 do <1/100) u pacjentów z nowotworami złośliwymi poddawanych chemioterapii, oraz u zdrowych dawców poddawanych procedurze mobilizacji komórek macierzystych krwi obwodowej po zastosowaniu ludzkich czynników stymulujących wzrost kolonii granulocytów.
W badaniach klinicznych u pacjentów zakażonych HIV efektami ubocznymi uznawanymi za związane ze stosowaniem filgrastymu były wyłącznie ból mięśniowo-szkieletowy, ból kości i mięśniowy ból.
Podawanie filgrastymu nie zwiększało częstości działań niepożądanych spowodowanych cytotoksyczną chemioterapią. Do działań niepożądanych obserwowanych z taką samą częstością u pacjentów otrzymujących filgrastym/chemioterapię oraz u pacjentów otrzymujących placebo/chemioterapię należą: nudności i wymioty, łysienie, biegunka, zmęczenie, brak apetytu, stan zapalny błon śluzowych, ból głowy, kaszel, wysypka skórna, ból w klatce piersiowej, osłabienie, ból gardła, zaparcia oraz nieokreślony ból.
U pacjentów poddawanych chemioterapii z podawaniem wysokich dawek leków z następową autologiczną transplantacją szpiku kostnego obserwowano zaburzenia ze strony układu naczyniowego.
Poniżej przedstawiono listę efektów ubocznych opisanych w trakcie prowadzenia badań klinicznych oraz uzyskanych jako spontaniczne zgłoszenia.
Infekcje i inwazje
Częste: sepsa, zapalenie oskrzeli, infekcje górnych dróg oddechowych, infekcje dróg moczowych.
Zaburzenia układu krwi i chłonnego
Bardzo często: trombocytopenia, anemia¹.
Częste: splenomegalia¹, obniżenie stężenia hemoglobiny⁵.
Rzadkie: leukocytoza¹, zaburzenia funkcji śledziony.
Pojedyncze: rozerwanie śledziony¹, krystaliczny zespół wywołany przez komórki sierpowate.
Zaburzenia układu odpornościowego
Częste: reakcje alergiczne, wysypka skórna, pokrzywka, obrzęk naczynioruchowy.
Rzadkie: reakcje nadwrażliwości, reakcje nadwrażliwości na lek¹, reakcje „przeszczep przeciwko gospodarzowi”².
Pojedyncze: reakcje anafilaktyczne.
Zaburzenia przemiany materii i odżywiania
Częste: wzrost stężenia dehydrogenazy mleczanowej (LDH) we krwi, zmniejszenie apetytu⁵.
Rzadkie: wzrost stężenia kwasu moczowego we krwi, hiperurykemia.
Pojedyncze: fałszywe zapalenie stawów¹, obniżenie poziomu glukozy we krwi, zaburzenia równowagi wodnej organizmu.
Zaburzenia psychiczne
Częste: bezsenność.
Zaburzenia układu nerwowego
Bardzo często: ból głowy¹.
Częste: zawroty głowy, hipestezja, parestezje.
Zaburzenia układu naczyniowego
Częste: nadciśnienie tętnicze, niedociśnienie tętnicze.
Rzadkie: choroba zatorowa żył wątroby⁴.
Pojedyncze: zespół przeciekania przez naczynia włosowate¹, zapalenie aorty.
Zaburzenia układu oddechowego, narządów klatki piersiowej i przestrzeni międzyłukowej
Częste: krwawienie z dróg oddechowych, duszność, kaszel¹, ból gardła¹,⁵, krwawienia z nosa, brak tchu.
Rzadkie: zespół ostrej niewydolności oddechowej¹, niewydolność oddechowa¹, obrzęk płuc¹, choroba śródmiąższowa płuc¹, powstawanie infiltratów w płucach¹, krwawienie płucne, niedotlenienie.
Zaburzenia przewodu pokarmowego
Bardzo często: biegunka¹,⁵, wymioty¹,⁵, nudności¹.
Częste: ból w jamie ustnej, zaparcia.
Zaburzenia układu wątrobowo-pęcherzykowego
Częste: hepatomegalia, wzrost stężenia fosfatazy alkalicznej we krwi.
Rzadkie: wzrost stężenia aminotransferazy asparaginianowej (AST), wzrost stężenia transferazy gamma-glutamylowej (GGT).
Zaburzenia skóry i tkanek podskórnych
Bardzo często: łysienie¹.
Częste: wysypka¹, zaczerwienienie.
Rzadkie: wysypka makularna i plamnicza, zespół Sweeta, zapalenie naczyń skóry¹.
Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej
Bardzo często: ból mięśni i kości³.
Częste: skurcze mięśni.
Rzadkie: osteoporoza.
Pojedyncze: nasilenie reumatoidalnego zapalenia stawów i objawów zapalenia stawów, zmniejszenie gęstości kości.
Zaburzenia nerek i dróg moczowych
Częste: dysuria, hematuria.
Rzadkie: białkomocz.
Pojedyncze: patologiczne zmiany w badaniu moczu, kłębuszkowe zapalenie nerek.
Zaburzenia ogólne i reakcje w miejscu podania
Bardzo często: zmęczenie¹, osłabienie, stan zapalny błon śluzowych¹, gorączka.
Częste: ból w klatce piersiowej¹, ból¹, osłabienie¹, niedowaga⁵, obrzęki obwodowe⁵, ból w miejscu podania.
Rzadkie: reakcje w miejscu iniekcji.
Zaburzenia, zatrucia i komplikacje po zabiegach
Częste: reakcja przetoczna⁵.
¹ Zobacz opis poszczególnych efektów ubocznych.
² Napływają doniesienia o reakcjach „przeszczep przeciwko gospodarzowi” i przypadkach śmiertelnych wśród pacjentów stosujących G-CSF po alogenicznym przeszczepieniu szpiku kostnego (zobacz opis poszczególnych efektów ubocznych).
³ W tym ból kości, ból pleców, ból stawów, mięśniowy ból, ból kończyn, ból mięśniowo-kościowy, ból mięśniowo-kościowy w klatce piersiowej, ból szyi.
⁴ O przypadkach informowano w okresie pozarejestrowym u pacjentów poddawanych alogenicznemu przeszczepieniu szpiku kostnego lub mobilizacji PBSC.
⁵ Niepożądane zjawiska występowały częściej u pacjentów otrzymujących filgrastym w porównaniu z grupą placebo i są związane z konsekwencjami podstawowego nowotworu lub cytotoksycznej chemioterapii.
Opis poszczególnych efektów ubocznych
Reakcje nadwrażliwości
W badaniach klinicznych oraz w okresie pozarejestrowym odnotowano reakcje typu nadwrażliwościowego, w tym anafilaksję, wysypkę, pokrzywkę, obrzęk Quinckego, duszność i niedociśnienie tętnicze, które pojawiają się na początku lub w dalszych etapach leczenia. Ogólnie rzecz biorąc, zgłoszenia napływały częściej po wstrzyknięciu dożylnym. W niektórych przypadkach objawy pojawiały się ponownie podczas próby prowokacyjnej, co wskazuje na związek przyczynowo-skutkowy. W przypadku poważnych reakcji alergicznych w dalszym ciągu nie należy stosować filgrastymu pacjentowi.
Zaburzenia ze strony płuc
W badaniach klinicznych oraz w okresie pozarejestrowym obserwowano działania niepożądane ze strony płuc, w tym krwawienie z dróg oddechowych, krwawienia płucne, infiltrowanie płuc, duszność i niedotlenienie, co prowadziło do niewydolności oddechowej lub zespołu ostrej niewydolności oddechowej dorosłych (ARDS), czasem zakończonego śmiercią. Napływają doniesienia o bardzo rzadkich przypadkach działań niepożądanych ze strony płuc u zdrowych dawców.
Splenomegalia i rozerwanie śledziony
Po podaniu filgrastymu zgłaszano powiększenie śledziony i rozerwanie śledziony. Niektóre przypadki rozerwania śledziony były śmiertelne.
W przypadkach wszystkich stopień powiększenia śledziony u pacjentów zakażonych HIV był niski do średniego według wyników badania lekarskiego, przebieg zaburzenia miał charakter łagodny; u żadnego z pacjentów nie zdiagnozowano nadczynności śledziony, żadnemu pacjentowi nie wykonano splenektomii. Ponieważ powiększenie śledziony jest częstym powikłaniem u pacjentów zakażonych HIV i występuje w różnym stopniu nasilenia u większości pacjentów z AIDS, związek przyczynowo-skutkowy ze stosowaniem filgrastymu pozostaje niejasny.
Zespół przeciekania przez naczynia włosowate
Zgłaszano przypadki zespołu przeciekania przez naczynia włosowate podczas przyjmowania ludzkich czynników stymulujących wzrost kolonii granulocytów. Zazwyczaj występowały one u chorych w zaawansowanym stadium choroby, z sepsą, u pacjentów otrzymujących wieloskładnikową chemioterapię lub poddawanych zabiegowi aferezy.
Zapalenie naczyń skóry
Zgłaszano zapalenie naczyń skóry u pacjentów otrzymujących filgrastym. Mechanizm zapalenia naczyń u pacjentów przyjmujących filgrastym nie jest znany. Podczas długotrwałego stosowania zapalenie naczyń skóry odnotowano u 2% chorych z CWN.
Leukocytoza
Leukocytoza (bezwzględna liczba leukocytów > 50 × 10⁹/l) występowała u 41% dawców. Ponadto po podaniu filgrastymu i leukaferezie u 35% dawców stwierdzono przemijającą trombocytopenię (bezwzględna liczba trombocytów < 100 × 10⁹/l).
Zespół Sweeta
Istnieją doniesienia o przypadkach zespołu Sweeta (ostry gorączkowy dermatoz niecharakterystyczny) u pacjentów z chorobami nowotworowymi stosujących filgrastym. Jednakże biorąc pod uwagę, że większość z tych pacjentów chorowała na białaczkę, chorobę, która często prowadzi do zespołu Sweeta, związek przyczynowo-skutkowy ze stosowaniem filgrastymu nie został potwierdzony.
Fałszywe zapalenie stawów (pirofosfatazowe chondrokalcynozę)
Istnieją doniesienia o przypadkach fałszywego zapalenia stawów u pacjentów z chorobami nowotworowymi stosujących filgrastym.
Reakcje „przeszczep przeciwko gospodarzowi”
Napływają doniesienia o reakcjach „przeszczep przeciwko gospodarzowi” i przypadkach śmiertelnych wśród pacjentów stosujących G-CSF po alogenicznym przeszczepieniu szpiku kostnego.
Imunogenność.
Zgodnie z danymi czterech badań klinicznych z udziałem zdrowych ochotników i pacjentów z chorobami nowotworowymi, u żadnego z nich w wyniku podania filgrastymu nie zaobserwowano powstawania przeciwciał anty-rG-CSF.
Dzieci.
Dane badań klinicznych u pacjentów pediatrycznych wskazują, że bezpieczeństwo i skuteczność stosowania filgrastymu są takie same u dorosłych i dzieci otrzymujących cytotoksyczną chemioterapię, co świadczy o braku różnic wiekowych w farmakokinetyce filgrastymu. Jedynym efektem ubocznym, który był systematycznie zgłaszany, był ból układu mięśniowo-szkieletowego, który nie różnił się od tego występującego u dorosłych.
Brak wystarczających danych do dalszej oceny stosowania filgrastymu u dzieci.
Inne specjalne kategorie pacjentów.
Pacjenci w podeszłym wieku
Nie zaobserwowano ogólnych różnic w bezpieczeństwie i skuteczności u osób powyżej 65 roku życia w porównaniu z młodymi dorosłymi (od 18 roku życia) otrzymującymi cytotoksyczną chemioterapię, a doświadczenie kliniczne nie wykazało różnic w odpowiedziach między pacjentami starszymi a młodymi dorosłymi. Brak wystarczających danych do oceny stosowania filgrastymu u pacjentów w podeszłym wieku w odniesieniu do innych zatwierdzonych wskazań filgrastymu.
Dzieci z CWN
Zgłaszano przypadki zmniejszenia gęstości mineralnej tkanki kostnej i osteoporozy u dzieci z ciężką przewlekłą neutropenią, które otrzymywały długotrwałe leczenie filgrastymem. Częstość występowania tego niepożądanego zjawiska w ramach badań klinicznych oceniano jako powszechną.
Zgłaszanie podejrzewanych działań niepożądanych.
Zgłaszanie podejrzewanych działań niepożądanych w okresie po rejestracji leku jest ważnym działaniem. Pozwala ono na kontynuowanie monitorowania stosunku korzyści do ryzyka stosowania danego leku.
Personel medyczny powinien zgłaszać przypadki wszelkich działań niepożądanych za pomocą systemu nadzoru farmakologicznego Ukrainy.
Okres ważności. 3 lata.
Warunki przechowywania.
Przechowywać w temperaturze 2-8 °C w oryginalnym opakowaniu. Nie zamrażać.
Przechowywać w miejscu niedostępnym dla dzieci.
Po rozcieńczeniu roztwór jest stabilny przez 24 godziny w temperaturze 2-8 °C.
Z mikrobiologicznego punktu widzenia roztwór należy użyć natychmiast.
Opakowanie.
0,5 ml roztworu w strzykawce wstępnie napełnionej z przezroczystego, bezbarwnego szkła, wyposażonej w tłok z szarym gumowym uszczelnieniem, igłą do wstrzykiwań, szarym gumowym osłonowym kapturkiem, zewnętrznym kapturkiem z polipropylenu oraz urządzeniem ochronnym zapobiegającym urazom igłą po zastosowaniu, w opakowaniu blisterowym.
1 lub 5 opakowań blisterowych w pudełku kartonowym.
Kategoria wydania. Na receptę.
Producent.
Novartis Pharmaceuticals Manufacturing GmbH
lub
Sandoz GmbH – Oddział Produkcyjny Aseptyczne Leki (Aseptyczne Leki)
Siedziba producenta i adres miejsca prowadzenia działalności.
Biochemiestrasse 10, Unterlangkampfen, Langkampfen, 6336, Austria
lub
Biochemiestrasse 10, 6336 Langkampfen, Austria