Vegovі FlexTech
UkrainaSpis treści
INSTRUKCJA stosowania leku do celów medycznych Vegovі FlexTech (wegovy flextouch)
Skład:
substancja czynna: semaglutyd;
0,25 mg
każda wstępnie napełniona dawka strzykawki-pióra zawiera 1 mg semaglutydu* w 1,5 ml roztworu, 1 ml roztworu zawiera 0,68 mg semaglutydu*, jedna wstępnie napełniona strzykawka-pióro zawiera 4 dawki po 0,25 mg;
0,5 mg
każda wstępnie napełniona strzykawka-pióro zawiera 2 mg semaglutydu* w 1,5 ml roztworu, 1 ml roztworu zawiera 1,34 mg semaglutydu*, jedna wstępnie napełniona strzykawka-pióro zawiera 4 dawki po 0,5 mg;
1 mg
każda wstępnie napełniona strzykawka-pióro zawiera 4 mg semaglutydu* w 3 ml roztworu, 1 ml roztworu zawiera 1,34 mg semaglutydu*, jedna wstępnie napełniona strzykawka-pióro zawiera 4 dawki po 1 mg;
1,7 mg
każda wstępnie napełniona strzykawka-pióro zawiera 6,8 mg semaglutydu* w 3 ml roztworu, 1 ml roztworu zawiera 2,27 mg semaglutydu*, jedna wstępnie napełniona strzykawka-pióro zawiera 4 dawki po 1,7 mg;
2,4 mg
każda wstępnie napełniona strzykawka-pióro zawiera 9,6 mg semaglutydu* w 3 ml roztworu, 1 ml roztworu zawiera 3,2 mg semaglutydu*, jedna wstępnie napełniona strzykawka-pióro zawiera 4 dawki po 2,4 mg;
substancje pomocnicze: fosforan sodu dwu sodowy dihydorat; propylenoglikol; fenol; kwas chlorowodorowy (do regulacji pH); wodorotlenek sodu (do regulacji pH); woda do wstrzykiwań.
* Analog ludzkiego peptydu podobnego do glukagonu-1 (GLP-1), wytwarzany metodą rekombinowanego DNA w Saccharomyces cerevisiae.
Postać leku. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny roztwór izotoniczny; pH = 7,4.
Grupa farmakoterapeutyczna. Leki hipoglikemizujące inne niż insulina. Analogi peptydu podobnego do glukagonu-1 (GLP-1). Kod ATC A10BJ06.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Semaglutyd jest analogiem GLP-1 o 94% homologii do ludzkiego GLP-1. Działa jako agonista receptorów GLP-1, wiążąc się i aktywując selektywnie receptory GLP-1, które są celami działania naturalnego GLP-1.
GLP-1 jest fizjologicznym regulatorem apetytu i spożycia kalorii, a receptory GLP-1 występują w kilku obszarach mózgu biorących udział w regulacji apetytu.
Badania na zwierzętach wykazały, że semaglutyd działa w mózgu poprzez receptory GLP-1. Ma bezpośredni wpływ na obszary mózgu biorące udział w homeostatycznej regulacji spożycia pokarmu w podwzgórzu i rdzeniu przedłużonym. Semaglutyd może wpływać na hedoniczny układ nagrody poprzez bezpośredni i pośredni wpływ na obszary mózgu, w tym na przegrodę, podstawę kresu i migdałek.
Badania kliniczne wykazały, że semaglutyd zmniejsza spożycie energii, zwiększa uczucie sytości, wypełnienia i kontroli nad spożyciem pokarmu, zmniejsza uczucie głodu oraz częstotliwość i intensywność popędu do jedzenia. Ponadto semaglutyd zmniejsza popęd do spożycia pokarmów o wysokiej zawartości tłuszczu.
Semaglutyd reguluje homeostatyczne i hedoniczne wpływy z udziałem funkcji wykonawczych w celu regulacji spożycia kalorii, apetytu, nagrody i wyboru pokarmu.
Dodatkowo badania kliniczne wykazały, że semaglutyd obniża poziom glukozy we krwi działając w sposób zależny od stężenia glukozy, poprzez stymulację sekrecji insuliny i hamowanie sekrecji glukagonu przy podwyższonym stężeniu glukozy we krwi. Mechanizm obniżania poziomu glukozy we krwi towarzyszy także nieznaczne opóźnienie opróżniania żołądka w wczesnej fazie pokarmowej. W przypadku hipoglikemii semaglutyd zmniejsza sekrecję insuliny i nie zakłóca sekrecji glukagonu.
Ekspresja receptorów GLP-1 zachodzi również w sercu, układzie naczyniowym, układzie odpornościowym i nerkach.
W trakcie badań klinicznych semaglutyd wykazywał pozytywny wpływ na poziom lipidów w osoczu krwi, obniżał ciśnienie tętnicze skurczowe i zmniejszał stan zapalny. Ponadto badania na zwierzętach wykazały, że semaglutyd hamuje rozwój miażdżycy i wykazuje działanie przeciwzapalne na układ sercowo-naczyniowy.
Skutki farmakodynamiczne
Apetyt, spożycie energii i wybór pokarmu
Semaglutyd zmniejsza apetyt, nasilając uczucie sytości i nasyconia, jednocześnie zmniejszając uczucie głodu i potencjalne spożycie pokarmu. W badaniu fazy 1 spożycie energii podczas jedzenia ad libitum było o 35% niższe przy stosowaniu semaglutydzu w porównaniu z placebo po 20 tygodniach leczenia. Wspomagało to poprawę kontroli nad spożyciem pokarmu, osłabienie popędu do jedzenia i względnie mniejszy popęd do spożycia pokarmów o wysokiej zawartości tłuszczu. Popęd do jedzenia był dodatkowo oceniany w badaniu STEP 5 za pomocą Kwestionariusza Kontroli Nadżywiania (CoEQ). W 104. tygodniu obliczona różnica terapii była na korzyść semaglutydzu zarówno w zakresie kontroli popędu do jedzenia, jak i popędu do pokarmów słonych, natomiast nie zaobserwowano wyraźnego wpływu na popęd do pokarmów słodkich.
Poziomy lipidów na czczo i w okresie postprandialnym
W porównaniu z placebo semaglutyd w dawce 1 mg zmniejszał stężenia trójglicerydów na czczo i lipoprotein o bardzo niskiej gęstości (VLDL) na czczo o 12% i 21% odpowiednio. Poziomy trójglicerydów i VLDL w okresie postprandialnym w odpowiedzi na spożycie pokarmu o wysokiej zawartości tłuszczu zmniejszały się o ponad 40%.
Skuteczność i bezpieczeństwo kliniczne
Skuteczność i bezpieczeństwo stosowania semaglutydzu w celu kontroli masy ciała jako uzupełnienie diety o obniżonej wartości kalorycznej i zwiększonej aktywności fizycznej oceniano w czterech podwójnie ślepych, randomizowanych badaniach kontrolowanych placebo, trwających 68 tygodni (STEP 1–4). Ogółem 4684 pacjentów (2652 randomizowanych do leczenia semaglutydem) zostało włączonych do tych badań. Ponadto dwuletnią skuteczność i bezpieczeństwo stosowania semaglutydzu w porównaniu z placebo oceniano w podwójnie ślepych, randomizowanych badaniach kontrolowanych placebo (STEP 5), w których wzięło udział 304 pacjentów (152 pacjentów otrzymywało leczenie semaglutydem).
Leczenie semaglutydem wykazało lepsze, klinicznie istotne i trwałe zmniejszenie masy ciała w porównaniu z placebo u pacjentów z otyłością (BMI ≥ 30 kg/m²) lub nadmierną masą ciała (BMI ≥ 27 kg/m² do < 30 kg/m²) i co najmniej jedną chorobą współistniejącą związaną z masą ciała. Ponadto w trakcie badań większa część pacjentów osiągnęła utratę masy ciała ≥ 5%, ≥ 10%, ≥ 15% oraz ≥ 20% przy stosowaniu semaglutydzu w porównaniu z placebo. Redukcja masy ciała następowała niezależnie od występowania objawów przewodu pokarmowego, takich jak nudności, wymioty lub biegunka.
Leczenie semaglutydem wykazało również istotne statystycznie poprawy w zakresie obwodu talii, ciśnienia tętniczego skurczowego i poziomu aktywności fizycznej w porównaniu z placebo.
Skuteczność została wykazana niezależnie od wieku pacjenta, płci, rasy, pochodzenia etnicznego, wyjściowego poziomu masy ciała, BMI, obecności cukrzycy typu 2 oraz stopnia zaburzenia funkcji nerek. Występowały różnice w skuteczności we wszystkich podgrupach pacjentów. Względnie większa utrata masy ciała obserwowana była u kobiet i u pacjentów bez cukrzycy typu 2, a także u pacjentów z niższym wyjściowym poziomem masy ciała w porównaniu z wyższym.
Badanie STEP 1. Kontrola masy ciała
W podwójnie ślepych badaniach klinicznych trwających 68 tygodni 1961 pacjentów z otyłością (BMI ≥ 30 kg/m²) lub z nadmierną masą ciała (BMI ≥ 27 kg/m² do < 30 kg/m²) i co najmniej jedną chorobą współistniejącą związaną z masą ciała został randomizowany do grupy przyjmującej semaglutyd lub placebo. Wszyscy pacjenci przestrzegali diety o obniżonej wartości kalorycznej i zwiększonej aktywności fizycznej przez cały okres badania.
Redukcja masy ciała następowała we wczesnym etapie i trwała przez cały okres badania. Na końcu leczenia (tydzień 68) zmniejszenie masy ciała było lepsze i klinicznie istotne w porównaniu z placebo (patrz tabela 1 i rysunek 1). Ponadto większa część pacjentów osiągnęła utratę masy ciała ≥ 5%, ≥ 10%, ≥ 15% oraz ≥ 20% przy stosowaniu semaglutydzu w porównaniu z placebo (patrz tabela 1). Wśród pacjentów z wstępnie stwierdzonym stanem przedcukrzycowym większa część pacjentów osiągnęła normalny status glikemiczny na końcu leczenia semaglutydem w porównaniu z placebo (84,1% vs 47,8%).
Tabela 1. Badanie STEP 1: wyniki w 68. tygodniu
| Wskaźnik |
Vegovі FlexTech |
Placebo |
| Liczba pacjentów (N) |
1306 |
655 |
| Masa ciała (kg) |
||
| Poziom wyjściowy (kg) |
105,4 |
105,2 |
| Zmiana (%) od poziomu wyjściowego1,2 |
-14,9 |
-2,4 |
| Różnica (%) w porównaniu z placebo1 [95 % CI] |
-12,4 [-13,4; -11,5]* |
- |
| Zmiana (kg) od poziomu wyjściowego |
-15,3 |
-2,6 |
| Różnica (kg) w porównaniu z placebo1 [95 % CI] |
-12,7 [-13,7; -11,7] |
- |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 5 %3 |
83,5* |
31,1 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 10 %3 |
66,1* |
12,0 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 15 %3 |
47,9* |
4,8 |
| Obwód talii (cm) |
||
| Poziom wyjściowy |
114,6 |
114,8 |
| Zmiana od poziomu wyjściowego1 |
-13,5 |
-4,1 |
| Różnica w porównaniu z placebo1 [95 % CI] |
-9,4 [-10,3; -8,5]* |
- |
| Ciśnienie skurczowe (mmHg) |
||
| Poziom wyjściowy |
126 |
127 |
| Zmiana od poziomu wyjściowego1 |
-6,2 |
-1,1 |
| Różnica z placebo1 [95 % CI] |
-5,1 [-6,3; -3,9]* |
- |
* p < 0,0001 (niepoprawione, dwustronne) na rzecz.
1 Ocena wykonana przy użyciu modelu ANCOVA z wielokrotną korektą opartą na wszystkich danych, niezależnie od przerwania leczenia randomizowanego lub rozpoczęcia stosowania innych leków przeciwołżeniu lub chirurgii bariatrycznej.
2 Podczas badania leczenie zostało ostatecznie przerwane u 17,1 % i 22,4 % pacjentów zrandomizowanych do stosowania semaglutydu 2,4 mg i placebo, odpowiednio. Zakładając, że wszyscy pacjenci kontynuowali leczenie i nie otrzymywali dodatkowej terapii przeciwko otyłości, oszacowane zmiany masy ciała od randomizacji do 68. tygodnia na podstawie modelu mieszanego dla powtarzanych pomiarów, w tym wszystkie obserwacje do pierwszego przerwania, wyniosły -16,9 % i -2,4 % dla semaglutydu 2,4 mg i placebo, odpowiednio.
3 Ocena wykonana według modelu regresji binarnej na podstawie tej samej procedury imputacji, co w analizie pierwotnej.
HTMLIMG0END
Wartości obserwowane u pacjentów, którzy ukończyli każdą planowaną wizytę, oraz oszacowania z wielokrotną korektą (WK) danych uzyskanych od pacjentów, którzy wycofali się.
Rys. 1. STEP 1: Średnia zmiana masy ciała (%) od wartości wyjściowej do 68. tygodnia
Po 68-tygodniowym badaniu przeprowadzono 52-tygodniowy okres bez leczenia obejmujący 327 pacjentów, którzy ukończyli główny okres badania na dawce podtrzymującej semaglutydu lub placebo. W okresie bez leczenia od 68. do 120. tygodnia średnia masa ciała wzrosła w obu grupach leczenia. Jednak u pacjentów, którzy otrzymywali semaglutyd w trakcie głównego okresu badania, masa ciała pozostała o 5,6 % niższa niż wartość wyjściowa, w porównaniu do 0,1 % w grupie placebo.
Badanie STEP 2. Kontrola masy ciała u pacjentów z cukrzycą typu 2
W podwójnie ślepej, klinicznej próbie trwającej 68 tygodni 1210 pacjentów z nadmierną masą ciała lub otyłością (BMI ≥ 27 kg/m²) i zdiagnozowaną cukrzycą typu 2 zostało zrandomizowanych do grup otrzymujących raz w tygodniu semaglutyd 2,4 mg, semaglutyd 1 mg lub placebo. Pacjenci biorący udział w badaniu mieli niedostatecznie kontrolowaną cukrzycę (HbA1c 7–10 %) i otrzymywali albo jedynie dietę i aktywność fizyczną, albo 1–3 doustne leki przeciwcukrzycowe. Wszyscy pacjenci przestrzegali diety o obniżonej kaloryczności i zwiększonej aktywności fizycznej przez cały okres badania.
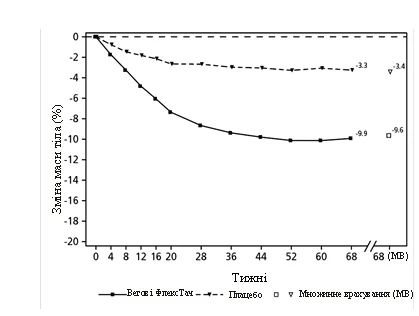
Leczenie semaglutydem przez 68 tygodni doprowadziło do istotnej i klinicznie znaczącej redukcji masy ciała oraz HbA1c w porównaniu do placebo (patrz tabela 2 i rysunek 2).
Tabela 2. Badanie STEP 2: wyniki na 68. tydzień
| Wskaźnik |
Vegovі FlexTech |
Placebo |
| Liczba pacjentów (N) |
404 |
403 |
| Masa ciała (kg) |
||
| Poziom wyjściowy (kg) |
99,9 |
100,5 |
| Zmiana (%) od poziomu wyjściowego1,2 |
-9,6 |
-3,4 |
| Różnica (%) w porównaniu z placebo1 [95 % CI] |
-6,2 [-7,3;-5,2]* |
- |
| Zmiana (kg) od poziomu wyjściowego |
-9,7 |
-3,5 |
| Różnica (kg) w porównaniu z placebo1 [95 % CI] |
-6,1 [-7,2;-5,0] |
- |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 5 %3 |
67,4* |
30,2 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 10 %3 |
44,5* |
10,2 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 15 %3 |
25,0* |
4,3 |
| Obwód talii (cm) |
||
| Poziom wyjściowy |
114,5 |
115,5 |
| Zmiana od poziomu wyjściowego1 |
-9,4 |
-4,5 |
| Różnica w porównaniu z placebo1 [95 % CI] |
-4,9 [-6,0; -3,8]* |
- |
| Ciśnienie tętnicze skurczowe (mm Hg) |
||
| Poziom wyjściowy |
130 |
130 |
| Zmiana od poziomu wyjściowego1 |
-3,9 |
-0,5 |
| Różnica z placebo1 [95 % CI] |
-3,4 [-5,6; -1,3]** |
- |
| HbA1c (mmol/mol (%)) |
||
| Poziom wyjściowy |
65,3 (8,1) |
65,3 (8,1) |
| Zmiana od poziomu wyjściowego1 |
-17,5 (-1,6) |
-4,1 (-0,4) |
| Różnica z placebo1 [95 % CI] |
-13,5 [-15,5; -11,4] (-1,2 [-1,4; -1,1])* |
- - |
* p < 0,0001 (niepoprawione dwustronne) na korzyść; ** p < 0,05 (niepoprawione dwustronne) na korzyść.
1 Ocena wykonana przy użyciu modelu ANCOVA z wielokrotną korektą opartą na wszystkich danych, niezależnie od przerwania leczenia randomizowanego lub rozpoczęcia stosowania innych leków przeciwożrzeniowych lub chirurgii bariatrycznej.
2 Podczas badania leczenie zostało ostatecznie przerwane u 11,6 % i 13,9 % pacjentów randomizowanych do stosowania semaglutydu 2,4 mg i placebo, odpowiednio. Zakładając, że wszyscy pacjenci kontynuowali leczenie i nie otrzymywali dodatkowego leczenia otyłości, oszacowane zmiany od randomizacji do 68. tygodnia dla masy ciała na podstawie mieszanego modelu dla pomiarów powtarzalnych, obejmującego wszystkie obserwacje do pierwszego przerwania, wynosiły -10,6 % i -3,1 % dla semaglutydu 2,4 mg i placebo, odpowiednio.
3 Ocena wykonana według modelu regresji binarnej na podstawie tej samej procedury korekty co w analizie pierwotnej.
**
**
Wartości obserwowane u pacjentów, którzy ukończyli każdą planowaną wizytę, oraz oszacowania z wielokrotną korektą (WK) danych uzyskanych od pacjentów, którzy wycofali się z badania.
Rys. 2. STEP 2: Średnia zmiana masy ciała (%) od wartości wyjściowej do 68. tygodnia
Badanie STEP 3. Kontrola masy ciała za pomocą intensywnej terapii behawioralnej
W podwójnie ślepej próbie klinicznej trwającej 68 tygodni 611 pacjentów z otyłością (BMI ≥ 30 kg/m²) lub z nadmierną masą ciała (BMI ≥ 27 kg/m² do < 30 kg/m²) oraz z co najmniej jednym współistniejącym schorzeniem związanym z masą ciała, zostało randomizowanych do grupy semaglutydu lub placebo. Wszyscy pacjenci otrzymywali intensywną terapię behawioralną (ITB), składającą się z bardzo restrykcyjnej diety, zwiększonej aktywności fizycznej i konsultacji behawioralnych przez cały okres badania.
Leczenie semaglutydem i ITB przez 68 tygodni doprowadziło do istotnego i klinicznie znaczącego zmniejszenia masy ciała w porównaniu z placebo (patrz tabela 3).
Tabela 3. Badanie STEP 3: wyniki w 68. tygodniu
| Wskaźnik |
Vegovі FlexTech |
Placebo |
| Liczba pacjentów (N) |
407 |
204 |
| Masa ciała (kg) |
||
| Poziom wyjściowy (kg) |
106,9 |
103,7 |
| Zmiana (%) od poziomu wyjściowego1,2 |
-16,0 |
-5,7 |
| Różnica (%) w porównaniu z placebo1 [95 % CI] |
-10,3 [-12,0;-8,6]* |
- |
| Zmiana (kg) od poziomu wyjściowego |
-16,8 |
-6,2 |
| Różnica (kg) w porównaniu z placebo1 [95 % CI] |
-10,6 [-12,5;-8,8] |
- |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 5 %3 |
84,8* |
47,8 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 10 %3 |
73,0* |
27,1 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 15 %3 |
53,5* |
13,2 |
| Obwód talii (cm) |
||
| Poziom wyjściowy |
113,6 |
111,8 |
| Zmiana od poziomu wyjściowego1 |
-14,6 |
-6,3 |
| Różnica w porównaniu z placebo1 [95 % CI] |
-8,3 [-10,1; -6,6]* |
- |
| Ciśnienie tętnicze skurczowe (mm Hg) |
||
| Poziom wyjściowy |
124 |
124 |
| Zmiana od poziomu wyjściowego1 |
-5,6 |
-1,6 |
| Różnica z placebo1 [95 % CI] |
-3,9 [-6,4; -1,5]* |
- |
*p < 0,0001 (niepoprawione dwustronnie) na korzyść.
1 Ocena przeprowadzona przy użyciu modelu ANCOVA z wielokrotną korektą na podstawie wszystkich danych, niezależnie od przerwania leczenia randomizowanego lub rozpoczęcia stosowania innych leków przeciwożyciowych lub chirurgii bariatrycznej.
2 Podczas badania leczenie zostało ostatecznie przerwane u 16,7 % i 18,6 % pacjentów randomizowanych do stosowania semaglutyd 2,4 mg i placebo, odpowiednio. Zakładając, że wszyscy pacjenci randomizowani kontynuowali leczenie i nie otrzymywali dodatkowego leczenia otyłości, oszacowane zmiany od randomizacji do 68. tygodnia dla masy ciała, na podstawie mieszanego modelu dla pomiarów powtarzalnych, w tym wszystkie obserwacje do pierwszego przerwania, wynosiły -17,6 % i -5,0 % dla semaglutyd 2,4 mg i placebo, odpowiednio.
3 Ocena przeprowadzona według modelu regresji binarnej na podstawie tej samej procedury korekty, co w analizie pierwotnej.
Badanie STEP 4. Utrzymywanie kontroli masy ciała
W podwójnie ślepej próbie klinicznej trwającej 68 tygodni włączone zostało 902 pacjentów z otyłością (BMI ≥ 30 kg/m²) lub z nadmierną masą ciała (BMI ≥ 27 kg/m² do < 30 kg/m²) i co najmniej jednym współistniejącym schorzeniem związanym z masą ciała. Wszyscy pacjenci przestrzegali diety o obniżonej wartości kalorycznej i zwiększali aktywność fizyczną przez cały okres badania. Od tygodnia 0 do tygodnia 20 (okres wprowadzania) wszyscy pacjenci otrzymywali semaglutyd. W 20. tygodniu (poziom wyjściowy) pacjenci, którzy osiągnęli dawkę utrzymującą 2,4 mg, zostali randomizowani do kontynuowania leczenia lub przejścia na placebo. W tygodniu 0 (początek okresu wprowadzania) pacjenci mieli średnią masę ciała 107,2 kg i średni BMI 38,4 kg/m².
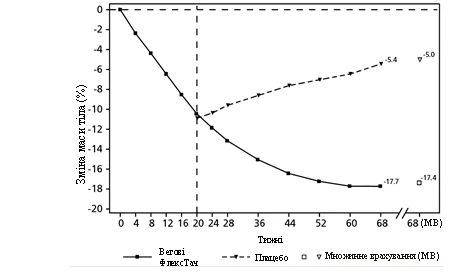
Pacjenci, którzy osiągnęli dawkę utrzymującą 2,4 mg w 20. tygodniu (poziom wyjściowy) i kontynuowali leczenie semaglutydem przez 48 tygodni (od 20. do 68. tygodnia), nadal tracili masę ciała i osiągali większe oraz klinicznie istotne zmniejszenie masy ciała w porównaniu do pacjentów, którzy przeszli na placebo. (patrz tabela 4 i rysunek 3). Masa ciała systematycznie wzrastała od 20. do 68. tygodnia u pacjentów, którzy przeszli na placebo w 20. tygodniu (poziom wyjściowy). Jednak obserwowana średnia masa ciała była niższa w 68. tygodniu niż na początku okresu wprowadzania (tydzień 0) (patrz rysunek 3). Pacjenci, którzy otrzymywali semaglutyd od tygodnia 0 (wprowadzenie) do tygodnia 68 (koniec leczenia), osiągnęli średnią zmianę masy ciała o -17,4 %, przy czym utratę masy ciała ≥ 5 % osiągnęło 87,8 %, ≥ 10 % osiągnęło 78,0 %, ≥ 15 % osiągnęło 62,2 % i ≥ 20 % osiągnęło 38,6 % tych pacjentów.
Tabela 4. Badanie STEP 4: wyniki od tygodnia 20 do tygodnia 68
| Wskaźnik |
Vegovі FlexTech |
Płacebo |
| Liczba pacjentów (N) |
535 |
268 |
| Masa ciała (kg) |
||
| Poziom wyjściowy1 (kg) |
96,5 |
95,4 |
| Zmiana (%) od poziomu wyjściowego1,2,3 |
-7,9 |
6,9 |
| Różnica (%) w porównaniu z płacebem2 [95 % CI] |
-14,8 [-16,0; -13,5]* |
- |
| Zmiana (kg) od poziomu wyjściowego |
-7,1 |
6,1 |
| Różnica (kg) w porównaniu z płacebem2 [95 % CI] |
-13,2 [-14,3; -12,0] |
- |
| Obwód talii (cm) |
||
| Poziom wyjściowy |
105,5 |
104,7 |
| Zmiana od poziomu wyjściowego1 |
-6,4 |
3,3 |
| Różnica w porównaniu z płacebem2 [95 % CI] |
-9,7 [-10,9; -8,5]* |
- |
| Ciśnienie tętnicze skurczowe (mmHg) |
||
| Poziom wyjściowy |
121 |
121 |
| Zmiana od poziomu wyjściowego1,2 |
0,5 |
4,4 |
| Różnica z płacebem2 [95 % CI] |
-3,9 [-5,8; -2,0]* |
- |
* p < 0,0001 (niepoprawione dwustronne) na rzecz lepszego wyniku.
1 Poziom wyjściowy = tydzień 20.
2 Ocena przeprowadzona przy użyciu modelu ANCOVA z wielokrotnym uwzględnieniem na podstawie wszystkich danych, niezależnie od przerwania randomizowanego leczenia lub rozpoczęcia stosowania innych leków przeciwożyciowych lub chirurgii bariatrycznej.
3 Podczas badania leczenie zostało ostatecznie przerwane u 5,8 % i 11,6 % pacjentów randomizowanych do stosowania semaglutydu 2,4 mg i placebo, odpowiednio. Zakładając, że wszyscy randomizowani pacjenci kontynuowali leczenie i nie otrzymywali dodatkowego leczenia otyłości, oszacowane zmiany od randomizacji do 68. tygodnia dla masy ciała na podstawie mieszanego modelu dla powtarzanych pomiarów, obejmującego wszystkie obserwacje do pierwszego przerwania, wyniosły -8,1 % i 6,5 % dla semaglutydu 2,4 mg i placebo, odpowiednio.
**
**
Wartości obserwowane u pacjentów, którzy ukończyli każdą zaplanowaną wizytę, oraz oszacowania z wielokrotnym uwzględnieniem (WU) danych uzyskanych od pacjentów, którzy wycofali się z badania.
Rys. 3. STEP 4: Średnia zmiana masy ciała (%) od tygodnia 0 do 68. tygodnia
Badanie STEP 5. Dane dwuletnie
W podwójnie ślepej próbie klinicznej trwającej 104 tygodnie 304 pacjentów z otyłością (BMI ≥ 30 kg/m²) lub z nadmierną masą ciała (BMI ≥ 27 kg/m² do < 30 kg/m²) oraz z co najmniej jednym stowarzyszonym schorzeniem związanym z masą ciała, zostali randomizowani do grupy stosującej semaglutyd lub placebo. Wszyscy pacjenci przestrzegali diety o obniżonej zawartości kalorii oraz zwiększonej aktywności fizycznej przez cały okres badania. Na poziomie wyjściowym pacjenci mieli średni BMI 38,5 kg/m² oraz średnią masę ciała 106,0 kg.
Leczenie semaglutydem przez 104 tygodnie doprowadziło do istotnego i klinicznie znaczącego obniżenia masy ciała w porównaniu do placebo. Średnia masa ciała zmniejszała się od poziomu wyjściowego do 68. tygodnia podczas stosowania semaglutydu, po czym osiągnięto plateau. W przypadku stosowania placebo średnia masa ciała zmniejszyła się w mniejszym stopniu, a plateau osiągnięto po około 20 tygodniach leczenia (patrz tabela 5 i rysunek 4). Pacjenci otrzymujący semaglutyd osiągnęli średnią zmianę masy ciała wynoszącą -15,2 %, u 74,7 % tych pacjentów utrata masy ciała wyniosła ≥ 5 %, u 59,2 % – ≥ 10 % oraz u 49,7 % – ≥ 15 %. 80 % i 37 % pacjentów z wczesnymi objawami cukrzycy na początku leczenia osiągnęło normalny stan glikemiczny na końcu leczenia semaglutydem i placebo, odpowiednio.
Tabela 5. Badanie STEP 5: wyniki na 104. tydzień
| Wskaźnik |
Vegovі FlexTech |
Placebo |
| Liczba pacjentów (N) |
152 |
152 |
| Masa ciała (kg) |
||
| Poziom wyjściowy (kg) |
105,6 |
106,5 |
| Zmiana (%) od poziomu wyjściowego1,2 |
-15,2 |
-2,6 |
| Różnica (%) w porównaniu z placebo1 [95 % CI] |
-12,6 [-15,3; -9,8]* |
- |
| Zmiana (kg) od poziomu wyjściowego |
-16,1 |
-3,2 |
| Różnica (kg) w porównaniu z placebo1 [95 % CI] |
-12,9 [‑16,1; ‑9,8] |
- |
| Pacjenci (%), którzy osiągnęli redukcję masy ciała ≥ 5 %3 |
74,7* |
37,3 |
| Pacjenci (%), którzy osiągnęli redukcję masy ciała ≥ 10 %3 |
59,2* |
16,8 |
| Pacjenci (%), którzy osiągnęli redukcję masy ciała ≥ 15 %3 |
49,7* |
9,2 |
| Obwód talii (cm) |
||
| Poziom wyjściowy |
115,8 |
115,7 |
| Zmiana od poziomu wyjściowego1 |
-14,4 |
5,2 |
| Różnica w porównaniu z placebo1 [95 % CI] |
-9,2 [-12,2; -6,2]* |
- |
| Ciśnienie tętnicze skurczowe (mm Hg) |
||
| Poziom wyjściowy |
126 |
125 |
| Zmiana od poziomu wyjściowego1 |
-5,7 |
-1,6 |
| Różnica z placebo1 [95 % CI] |
-4,2 [-7,3; -1,0]* |
- |
* p < 0,0001 (nieprzetestowane dwustronnie) na rzecz semaglutydu.
1 Ocena wykonana przy użyciu modelu ANCOVA z wielokrotną korektą na podstawie wszystkich danych, niezależnie od przerwania leczenia randomizowanego lub rozpoczęcia stosowania innych leków przeciwołżucia lub chirurgii bariatrycznej.
2 Podczas badania leczenie zostało ostatecznie zakończone u 13,2 % i 27,0 % pacjentów randomizowanych do grupy semaglutydu i placebo, odpowiednio. Zakładając, że wszyscy pacjenci kontynuowali leczenie i nie otrzymywali dodatkowej terapii przeciwołżucia, oszacowane zmiany od randomizacji do 68. tygodnia masy ciała, na podstawie modelu mieszanego dla pomiarów powtarzalnych, w tym wszystkie obserwacje do pierwszego przerwania, wyniosły -16,7 % i -0,6 % dla semaglutydu i placebo, odpowiednio.
3 Ocena wykonana przy użyciu modelu regresji binarnej na podstawie tej samej procedury korekty, co w analizie pierwotnej.
HTMLIMG3END
Wartości obserwowane u pacjentów, którzy ukończyli każdą planowaną wizytę, oraz oszacowania z wielokrotną korektą (WK) danych od pacjentów, którzy wypadli.
Rys. 4. STEP 5: Średnia zmiana masy ciała (%) od tygodnia 0 do tygodnia 104
Badanie STEP 8. Porównanie semaglutydu z liraglutydem
W randomizowanym, otwartym, bezpośrednim, placebo-kontrolowanym badaniu trwającym 68 tygodni, 338 pacjentów z otyłością (BMI ≥ 30 kg/m²) lub nadmierną masą ciała (BMI ≥ 27 kg/m² do < 30 kg/m²) oraz z co najmniej jednym stowarzyszonym schorzeniem związanym z masą ciała, zostało zrandomizowanych do grupy stosującej semaglutyd raz w tygodniu, liraglutyd 3 mg raz dziennie lub placebo. Stosowanie semaglutydu raz w tygodniu i liraglutydu 3 mg było otwarte, ale każda grupa leczenia aktywnego była podwójnie ślepa w stosunku do placebo, podawanego z tą samą częstotliwością dawkowania. Na poziomie wyjściowym pacjenci mieli średni BMI 37,5 kg/m² i średnią masę ciała 104,5 kg.
Leczenie semaglutydem raz w tygodniu przez 68 tygodni skutkowało istotnym i klinicznie istotnym zmniejszeniem masy ciała w porównaniu z liraglutydem. Średnia masa ciała zmniejszała się od poziomu wyjściowego do 68. tygodnia podczas stosowania semaglutydu. Przy stosowaniu liraglutydu średnia masa ciała zmniejszała się w mniejszym stopniu (patrz tabela 6). 37,4 % pacjentów otrzymujących semaglutyd straciło ≥ 20 % masy ciała, w porównaniu z 7,0 % pacjentów otrzymujących leczenie liraglutydem. W tabeli 6 przedstawiono wyniki punktów końcowych potwierdzających: utratę masy ciała ≥ 10 %, ≥ 15 % oraz ≥ 20 %.
Tabela 6. Badanie STEP 8: wyniki w 68. tygodniu porównania semaglutydu i liraglutydu
| Wskaźnik |
Vegovі FlexTech |
Liraglutyd 3 mg |
| Liczba pacjentów (N) |
126 |
127 |
| Masa ciała (kg) |
||
| Początkowy poziom (kg) |
102,5 |
103,7 |
| Zmiana (%) od początkowego poziomu1,2 |
-15,8 |
-6,4 |
| Różnica (%) w porównaniu z liraglutydem1 [95 % CI] |
‑9,4 [‑12,0;‑6,8]* |
- |
| Zmiana (kg) od początkowego poziomu |
‑15,3 |
‑6,8 |
| Różnica (kg) w porównaniu z liraglutydem1 [95 % CI] |
‑8,5 [‑11,2;‑5,7] |
- |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 10 %3 |
69,4* |
27,2 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 15 %3 |
54,0* |
13,4 |
| Pacjenci (%), którzy osiągnęli utratę masy ciała ≥ 20 %3 |
37,4* |
7,0 |
* p < 0,0001 (niepoprawione, dwustronne) na rzecz semaglutydynu.
1 Ocena przeprowadzona przy użyciu modelu ANCOVA z korektą wielokrotną na podstawie wszystkich danych, niezależnie od przerwania leczenia randomizowanego lub rozpoczęcia stosowania innych leków przeciwożrzeniowych lub zabiegów bariatrycznych.
2 Podczas badania leczenie zostało ostatecznie przerwane u 13,5 % i 27,6 % pacjentów randomizowanych do stosowania semaglutydynu i liraglutynu odpowiednio. Zakładając, że wszyscy pacjenci kontynuowali leczenie i nie otrzymywali dodatkowego leczenia otyłości, oszacowane zmiany od randomizacji do 68 tygodnia dla masy ciała na podstawie mieszанego modelu dla pomiarów powtarzalnych, w tym wszystkie obserwacje do pierwszego przerwania, wyniosły -16,7 % i -6,7 % odpowiednio dla semaglutydynu i liraglutynu.
3 Ocena przeprowadzona przy użyciu modelu regresji binarnej na podstawie tej samej procedury korekty, co w analizie pierwotnej.
Wpływ na skład ciała
W podbadaniu STEP 1 (N = 140) skład ciała oceniano metodą podwójnej absorpcjometrii rentgenowskiej (DEXA). Wyniki oceny DEXA wykazały, że leczenie semaglutydyną wiązało się z większym zmniejszeniem masy tłuszczu w porównaniu z masą mięśniową, co prowadziło do poprawy składu ciała w porównaniu z placebo po 68 tygodniach. Ponadto zmniejszenie całkowitej masy tłuszczu wiązało się ze zmniejszeniem tłuszczu wisceralnego. Wyniki te wskazują, że większa część całkowitej utraty masy ciała była związana ze zmniejszeniem tkanki tłuszczowej, w tym tłuszczu wisceralnego.
Poprawa funkcjonowania fizycznego
Semaglutydyna wykazała niewielką poprawę wskaźników funkcjonowania fizycznego. Funkcje fizyczne oceniano zarówno za pomocą ogólnego kwestionariusza jakości życia związanego ze zdrowiem, Short Form-36v2 Health Survey, Acute Version (SF-36), jak i kwestionariusza wpływu masy ciała na jakość życia Impact of Weight on Quality of Lite Clinical Trials Version (IWQOL-Lite)-CT.
Ocena układu sercowo-naczyniowego
W badaniu SUSTAIN 6 zakwalifikowano 3297 pacjentów z niedostatecznie kontrolowanym cukrzycą typu 2 i wysokim ryzykiem sercowo-naczyniowym, którzy zostali randomizowani do podskórnego stosowania semaglutydynu w dawce 0,5 mg lub 1 mg raz w tygodniu albo placebo w połączeniu ze standardowym leczeniem. Czas trwania leczenia wyniósł 104 tygodnie. Średni wiek pacjentów wynosił 65 lat, a średni wskaźnik BMI – 33 kg/m².
Pierwotnym punktem końcowym był czas od randomizacji do pierwszego wystąpienia dużego niekorzystnego zdarzenia sercowo-naczyniowego (MACE): zgon z przyczyn sercowo-naczyniowych, zawał mięśnia serca nieśmiertelny lub udar mózgu nieśmiertelny. Łączna liczba zdarzeń MACE wyniosła 254, w tym 108 (6,6 %) w grupie semaglutydynu i 146 (8,9 %) w grupie placebo.
Bezpieczeństwo sercowo-naczyniowe leczenia semaglutydyną w dawkach 0,5 lub 1 mg zostało potwierdzone, ponieważ stosunek ryzyka (HR) dla semaglutydynu w porównaniu z placebo wyniósł 0,74 [0,58; 0,95] [95% CI], co wynikało ze zmniejszenia częstości występowania nieśmiertelnego zawału mięśnia serca i nieśmiertelnego udaru mózgu bez różnicy w częstości zgonów z przyczyn sercowo-naczyniowych (patrz rysunek 5).
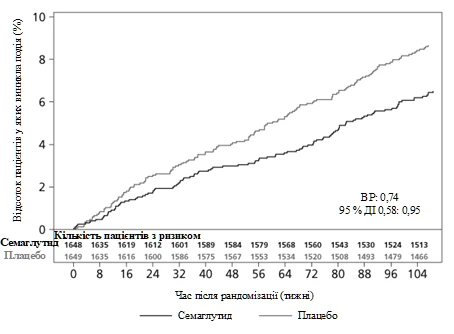
Rys. 5. Krzywa przeżycia Kaplana-Meiera dla czasu do pierwszego wystąpienia złożonego zdarzenia: zgon z przyczyn sercowo-naczyniowych, nieśmiertelny zawał mięśnia serca i nieśmiertelny udar mózgu (badanie kliniczne SUSTAIN 6)
Dzieci
Europejski Urząd ds. Leków odroczył obowiązek przedłożenia wyników badań leku Vegovі FlexTech w jednej lub kilku podgrupach populacji pediatrycznej w zakresie kontroli masy ciała (patrz sekcja „Sposób stosowania i dawki” w celu uzyskania informacji na temat stosowania u dzieci).
Badanie STEPTEENS: Kontrola masy ciała u pacjentów w wieku od 12 do 18 lat
W 68-tygodniowym badaniu podwójnie ślepych 201 dzieci w okresie dojrzewania w wieku od 12 do 18 lat z otyłością lub nadmierną masą ciała oraz co najmniej jednym współistniejącym schorzeniem związanym z masą ciała zostało randomizowanych w stosunku 2:1 do otrzymywania semaglutydynu lub placebo. Wszyscy pacjenci przestrzegali diety o ograniczonej kaloryczności i zwiększali aktywność fizyczną przez cały okres trwania badania.
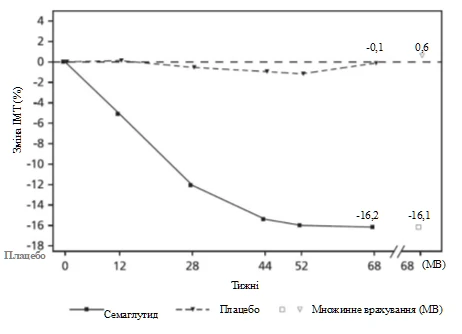
Na koniec leczenia (68 tydzień) poprawa wskaźnika BMI przy stosowaniu semaglutydynu była większa i klinicznie istotna w porównaniu z placebo (patrz tabela 6 i rysunek 6). Ponadto większy odsetek pacjentów osiągnął utratę masy ciała ≥ 5 %, ≥ 10 % oraz ≥ 15 % przy stosowaniu semaglutydynu w porównaniu z placebo (patrz tabela 6).
Tabela 6. STEPTEENS: Wyniki po 68 tygodniach
| Wskaźnik |
Vegovі FlexTech |
Placebo |
| Liczba pacjentów (N) |
134 |
67 |
| Wskaźnik masy ciała (BMI) |
||
| Poziom wyjściowy (BMI) |
37,7 |
35,7 |
| Zmiana (%) od poziomu wyjściowego1,2 |
-16,1 |
0,6 |
| Różnica (%) w porównaniu z placebo1 [95 % CI] |
-16,7 [-20,3; -13,2] |
- |
| Poziom wyjściowy (BMI SDS (indeks odchylenia standardowego)) |
3,4 |
3,1 |
| Zmiana od poziomu wyjściowego BMI SDS1 |
-1,1 |
-0,1 |
| Różnica w porównaniu z placebo1 [95 % CI] |
-1,0 [-1,3; -0,8] |
- |
| Masa ciała |
||
| Poziom wyjściowy (kg) |
109,9 |
102,6 |
| Zmiana (%) od poziomu wyjściowego1 |
-14,7 |
2,8 |
| Różnica (%) w porównaniu z placebo1 [95 % CI] |
-17,4 [-21,1; -13,8] |
- |
| Zmiana (kg) od poziomu wyjściowego1 |
-15,3 |
2,4 |
| Różnica (kg) w porównaniu z placebo1 [95 % CI] |
-17,7 [-21,8; -13,7] |
- |
| Pacjenci (%), którzy osiągnęli redukcję masy ciała ≥ 5 %3 |
72,5* |
17,7 |
| Liczba pacjentów (N) |
134 |
67 |
| Pacjenci (%), którzy osiągnęli redukcję masy ciała ≥ 10 %3 |
61,8 |
8,1 |
| Pacjenci (%), którzy osiągnęli redukcję masy ciała ≥ 15 %3 |
53,4 |
4,8 |
| Obwód talii (cm) |
||
| Poziom wyjściowy |
111,9 |
107,3 |
| Zmiana od poziomu wyjściowego1 |
-12,7 |
-0,6 |
| Różnica w porównaniu z placebo1 [95 % CI] |
-12,1 [-15,6; -8,7]* |
- |
| Ciśnienie tętnicze skurczowe (mmHg) |
||
| Poziom wyjściowy |
120 |
120 |
| Zmiana od poziomu wyjściowego1 |
-2,7 |
-0,8 |
| Różnica z placebo1 [95 % CI] |
-1,9 [-5,0; -1,1]* |
- |
*p < 0,0001 (niepoprawione, dwustronne) na korzyść leczenia.
1 Ocena przeprowadzona przy użyciu modelu ANCOVA z wielokrotnym uwzględnieniem na podstawie wszystkich danych, niezależnie od przerwania leczenia randomizowanego lub rozpoczęcia stosowania innych leków przeciwożrzeniowych lub chirurgii bariatrycznej.
2 Podczas badania leczenie zostało ostatecznie przerwane u 10,4 % i 10,4 % pacjentów randomizowanych do stosowania semaglutydu 2,4 mg i placebo, odpowiednio. Zakładając, że wszyscy pacjenci kontynuowali leczenie i nie otrzymywali dodatkowej terapii przeciwożrzeniowej, oszacowane zmiany masy ciała od randomizacji do 68. tygodnia na podstawie mieszанego modelu dla pomiarów powtarzalnych, obejmujące wszystkie obserwacje do pierwszego przerwania, wynosiły -17,9 % i -0,6 % dla semaglutydu 2,4 mg i placebo, odpowiednio.
3 Ocena przeprowadzona przy użyciu modelu regresji binarnej na podstawie tej samej procedury uwzględnienia, co w analizie pierwotnej.
Wartości obserwowane u pacjentów, którzy ukończyli każdą planowaną wizytę, oraz oszacowania z wielokrotnym uwzględnieniem (MВ) danych pochodzących od pacjentów, którzy wycofali się z badania.
Rys. 6. STEP TEENS: Średnia zmiana BMI (%) od wartości wyjściowej do 68. tygodnia
Farmakokinetyka.
W porównaniu z naturalnym GLP-1 semaglutyd charakteryzuje się wydłużonym okresem półtrwania wynoszącym około 1 tydzień, co umożliwia jego podawanie podskórne raz w tygodniu. Głównym mechanizmem tej długotrwałej aktywności jest wiązanie z albuminą, które prowadzi do zmniejszenia klirensu nerkowego i ochrony przed degradacją metaboliczną. Ponadto semaglutyd jest stabilny wobec degradacji przez enzym DPP-4.
Absorpcja
Średnie stężenie równowagowe semaglutydu po podaniu dawki utrzymującej podskórnie wynosiło około 75 nmol/l u pacjentów z nadwagą (BMI ≥ 27 kg/m² do < 30 kg/m²) lub otyłością (BMI ≥ 30 kg/m²) na podstawie danych z badań fazy 3a, w których 90 % pacjentów miało stężenia średnie w zakresie od 51 nmol/l do 110 nmol/l. W przypadku dawek od 0,25 mg do 2,4 mg podawanych raz w tygodniu ekspozycja równowagowa semaglutydu wzrastała w sposób zależny od dawki. Ekspozycja równowagowa była stabilna w czasie, jak określono do 68. tygodnia. Podobna ekspozycja była osiągana po podaniu podskórnej dawki semaglutydu w okolicy przedniej ściany brzucha, uda lub ramienia. Bezwzględna biodostępność semaglutydu po podaniu podskórnej dawki wynosiła 89 %.
Rozkład
Średni objętość rozkładu semaglutydu po podaniu podskórnym u pacjentów z nadwagą lub otyłością wynosił około 12,4 l. Semaglutyd silnie wiązał się z albuminą osocza krwi (> 99 %).
Metabolizm/biotransformacja
Przed wydaleniem semaglutyd jest aktywnie metabolizowany poprzez proteolityczne rozszczepienie peptydowej struktury białka i dalsze beta-oksydowanie bocznego łańcucha kwasu tłuszczowego. Enzym neutralnej endopeptydazy (NEP) został zidentyfikowany jako jeden z aktywnych enzymów metabolicznych.
Wydalanie
Substancje związane z semaglutydem są wydalane głównie z moczem i kałem. Około 3 % podanej dawki wydalało się z moczem w niezmienionej formie. Klirens semaglutydu u pacjentów z nadwagą (BMI ≥ 27 kg/m² do < 30 kg/m²) lub otyłością (BMI ≥ 30 kg/m²) wynosił około 0,05 l/h. Przy okresie półtrwania trwającym około 1 tydzień semaglutyd będzie obecny w krążeniu przez około 7 tygodni po podaniu ostatniej dawki 2,4 mg.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku
Wiek pacjenta nie wpływa na farmakokinetykę semaglutydu na podstawie danych z badań fazy 3 z udziałem pacjentów w wieku 18–86 lat.
Płeć, rasa i pochodzenie etniczne
Płeć, rasa (kaukaska, czarnoskóra lub azjatycka) oraz pochodzenie etniczne (hiszpańskie lub latynoamerykańskie, niehiszpańskie lub nielatynoamerykańskie) pacjenta nie wpływały na farmakokinetykę semaglutydu na podstawie danych z badań fazy 3a.
Masa ciała
Masa ciała pacjenta wpływała na ekspozycję na semaglutyd. Wyższa masa ciała prowadzi do niższej ekspozycji leku; różnica 20 % w masie ciała między pacjentami prowadzi do prawie 18 % różnicy w ekspozycji. Dawkowanie semaglutydu 2,4 mg raz w tygodniu zapewnia wystarczającą ekspozycję systemową przy masie ciała w zakresie 54,4–245,6 kg, ocenianą w kontekście odpowiedzi na ekspozycję podczas badań klinicznych.
Uszkodzenie funkcji nerek
Uszkodzenie funkcji nerek nie miało klinicznie istotnego wpływu na farmakokinetykę semaglutydu. Wykazano to po podaniu pojedynczej dawki 0,5 mg semaglutydu u pacjentów z uszkodzeniem funkcji nerek różnego stopnia nasilenia (łagodnego, umiarkowanego, ciężkiego oraz u pacjentów dializowanych) w porównaniu z pacjentami z normalną funkcją nerek. Potwierdzono to również danymi z badań klinicznych fazy 3a u pacjentów z nadwagą (BMI ≥ 27 kg/m² do < 30 kg/m²) lub otyłością (BMI ≥ 30 kg/m²) oraz z uszkodzeniem funkcji nerek od łagodnego do umiarkowanego stopnia nasilenia.
Uszkodzenie funkcji wątroby
Uszkodzenie funkcji wątroby nie miało żadnego wpływu na ekspozycję semaglutydu. Farmakokinetykę semaglutydu oceniano u pacjentów z różnym stopniem uszkodzenia funkcji wątroby (łagodnym, umiarkowanym, ciężkim) w porównaniu z pacjentami z normalną funkcją wątroby w badaniu z zastosowaniem pojedynczej dawki 0,5 mg semaglutydu.
Stan przedcukrzycowy i cukrzyca
Stan przedcukrzycowy i cukrzyca nie miały klinicznie istotnego wpływu na ekspozycję semaglutydu na podstawie danych z badań fazy 3.
Imunogenność
Wytwarzanie przeciwciał przeciwko semaglutydowi podczas leczenia semaglutydem występowało rzadko (patrz sekcja „Reakcje niepożądane”) i odpowiedź wydawała się nie wpływać na farmakokinetykę semaglutydu.
Dzieci
Właściwości farmakokinetyczne semaglutydu oceniano w badaniu klinicznym z udziałem pacjentów-adolescentów z otyłością lub nadwagą i co najmniej jednym współistniejącym schorzeniem związanym z masą ciała, w wieku od 12 do < 18 lat (124 pacjentów, masa ciała 61,6–211,9 kg). Ekspozycja na semaglutyd u dzieci była podobna do tej u dorosłych z otyłością lub nadwagą.
Bezpieczeństwo i skuteczność stosowania semaglutydu u dzieci poniżej 12. roku życia nie zostały zbadane.
Dane doksykliniczne dotyczące bezpieczeństwa
Dane doksykliniczne oparte na badaniach farmakologicznej bezpieczeństwa, toksyczności powtarzanych dawek oraz genotoksyczności nie wykazały żadnego ryzyka dla człowieka.
Nieśmiertelne guzy pochodzące z komórek C tarczycy, obserwowane u gryzoniów, należą do efektów charakterystycznych dla klasy agonistów receptorów GLP-1. W dwuletnim badaniu kancerogennym na szczurach i myszach semaglutyd powodował powstawanie guzów komórek C tarczycy przy klinicznie istotnych poziomach ekspozycji. Nie obserwowano żadnych innych nowotworów, których powstanie mogłoby być związane z leczeniem. Guzy komórek C u gryzoniów są spowodowane niegenotoksycznym, specyficznym, zależnym od receptora GLP-1 mechanizmem, na który gryzonie są częściowo wrażliwe. Znaczenie tego mechanizmu dla człowieka jest niskie, ale nie może być całkowicie wykluczone.
W badaniach dotyczących płodności u szczurów semaglutyd nie wpływał na skuteczność kojarzenia ani na płodność u samców. U samic szczurów obserwowano wydłużenie cyklu estrusowego i niewielkie zmniejszenie liczby ciał żółtych (owulacji) przy dawkach towarzyszących utracie masy ciała u samic.
W badaniach rozwoju embrionów i płodów u szczurów semaglutyd powodował działanie embriotoksyczne przy ekspozycji niższej niż klinicznie istotne poziomy. Semaglutyd powodował znaczne zmniejszenie masy ciała u samic oraz zmniejszenie przeżywalności i wzrostu embrionów. U płodów obserwowano liczne wady rozwojowe szkieletu i wady wewnętrzne, w tym zmiany w kościach długich, żebrach, kręgosłupie, kościach ogonowych, naczyniach krwionośnych i komorach mózgu. Ocena mechanistyczna wykazała, że efekt embriotoksyczny obejmuje zależne od receptorów GLP-1 zaburzenie dostarczania substancji odżywczych do embrionu poprzez worek żółtkowy u szczurów. Ze względu na różnice w budowie anatomicznej i funkcji worka żółtkowego u różnych gatunków szczurów oraz brak ekspresji receptorów GLP-1 w worku żółtkowym u małp naczłonków ten mechanizm uważa się za mało prawdopodobny u człowieka. Jednakże nie można wykluczyć bezpośredniego wpływu semaglutydu na płód.
W badaniach toksycznego wpływu leku na rozwój u królików i makaków jawajskich przy klinicznie istotnych poziomach ekspozycji obserwowano zwiększenie częstości poronień i pewne zwiększenie częstości wad płodu. Te wyniki korelowały ze znaczną utratą masy ciała u samic sięgającą 16 %. Nie wiadomo, czy te efekty są związane ze zmniejszeniem spożycia pokarmu przez samice z powodu bezpośredniego wpływu GLP-1.
Postnatalny wzrost i rozwój oceniano u makaków jawajskich. Młode były nieco mniejsze przy urodzeniu, ale ich masa ciała normalizowała się w okresie karmienia piersią.
U młodych szczurów semaglutyd powodował opóźnienie dojrzewania płciowego zarówno u samców, jak i u samic. Opóźnienie to nie wpływało ani na płodność i zdolność reprodukcyjną obu płci, ani na zdolność samic do utrzymania ciąży.
Charakterystyka kliniczna.
Wskazania .
Dorośli
Lek Vegovі FlexTech wskazany jest do kontroli masy ciała jako uzupełnienie diety o obniżonej wartości kalorycznej oraz zwiększonej aktywności fizycznej u dorosłych pacjentów z początkowym indeksem masy ciała (BMI)
- powyżej 30 kg/m² (otyłość) lub
- od 27 do 30 kg/m² (nadwaga) w przypadku co najmniej jednego współistniejącego schorzenia związanego z masą ciała, takiego jak: dysglikemia (przedcukrzycę lub cukrzycę typu 2), nadciśnienie tętnicze, dyslipidemia, obturacyjne bezdechy sennego lub chorobę serca i naczyń krwionośnych.
Dzieci ≥ 12 lat
Lek Vegovі FlexTech wskazany jest do kontroli masy ciała jako uzupełnienie diety o obniżonej wartości kalorycznej oraz zwiększonej aktywności fizycznej u dzieci w wieku od 12 lat z
- otyłością* oraz
- masą ciała powyżej 60 kg.
Należy przerwać stosowanie Vegovі FlexTech i ponownie ocenić wskazania, jeśli u dzieci nie stwierdzono co najmniej 5% spadku wskaźnika BMI po 12 tygodniach przyjmowania dawki 2,4 mg lub maksymalnej dawki dobrze tolerowanej.
* Otyłość (BMI ≥ 95. percentyl), zgodnie z wykresami wzrostu BMI według płci i wieku (CDC.gov) (patrz tabela 7).
Tabela 7. Progowe wartości BMI dla otyłości (≥ 95. percentyl) według płci i wieku dla dzieci od 12 roku życia (kryteria CDC)
| Wiek (lata) |
WSP (kg/m2) przy 95. centylu |
|
| Płeć męska |
Płeć żeńska |
|
| 12 |
24,2 |
25,2 |
| 12,5 |
24,7 |
25,7 |
| 13 |
25,1 |
26,3 |
| 13,5 |
25,6 |
26,8 |
| 14 |
26,0 |
27,2 |
| 14,5 |
26,4 |
27,7 |
| 15 |
26,8 |
28,1 |
| 15,5 |
27,2 |
28,5 |
| 16 |
27,5 |
28,9 |
| 16,5 |
27,9 |
29,3 |
| 17 |
28,2 |
29,6 |
| 17,5 |
28,6 |
30,0 |
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na substancje pomocnicze leku (patrz sekcja „Skład”).
Interakcje z innymi lekami i inne rodzaje interakcji.
Semaglutyd opóźnia opróżnianie żołądka i potencjalnie może wpływać na szybkość wchłaniania doustnych leków stosowanych jednocześnie. Nie zaobserwowano klinicznie istotnego wpływu na szybkość opróżniania żołądka po podaniu semaglutydu w dawce 2,4 mg, prawdopodobnie z powodu efektu tolerancji. Semaglutyd należy stosować z ostrożnością u pacjentów przyjmujących leki doustne, które wymagają szybkiego wchłaniania w przewodzie pokarmowym.
Paracetamol
Wyniki oceny farmakokinetyki paracetamolu w trakcie standaryzowanego testu z posiłkiem wykazały, że semaglutyd opóźnia szybkość opróżniania żołądka. Po jednoczesnym podaniu paracetamolu i semaglutydu w dawce 1 mg wartości AUC0-60min i Cmax paracetamolu zmniejszyły się odpowiednio o 27% i 23%. Całkowite narażenie na paracetamol (AUC0–5h) nie uległo zmianie. Nie zaobserwowano klinicznie istotnego wpływu semaglutydu na paracetamol. W przypadku jednoczesnego stosowania paracetamolu i semaglutydu nie jest wymagana korekta dawki.
Środki antykoncepcyjne doustne
Nie przewiduje się, że semaglutyd będzie obniżał skuteczność doustnych środków antykoncepcyjnych. Po jednoczesnym podaniu semaglutydu z doustnym kombinowanym lekiem antykoncepcyjnym (etynoestradiol 0,03 mg/lewonorgestrel 0,15 mg) semaglutyd nie wywierał klinicznie istotnego wpływu na całkowite narażenie na etynoestradiol i lewonorgestrel. Narażenie na etynoestradiol nie było zaburzone; narażenie na lewonorgestrel zwiększyło się o 20% w stanie stacjonarnym. Cmax żadnej z tych substancji nie uległo zmianie.
Atorwastatyna
Semaglutyd nie zmieniał całkowitego narażenia na atorwastatynę po podaniu pojedynczej dawki atorwastatyny (40 mg). Cmax atorwastatyny zmniejszyła się o 38%. Uznano to za klinicznie nieistotne.
Digoksyna
Semaglutyd nie zmieniał całkowitego narażenia ani Cmax digoksyny po podaniu pojedynczej dawki digoksyny (0,5 mg).
Metformina
Semaglutyd nie zmieniał całkowitego narażenia ani Cmax metforminy po podaniu metforminy w dawce 500 mg dwa razy dziennie przez 3,5 dnia.
Warfaryna
Semaglutyd nie zmieniał całkowitego narażenia ani Cmax warfaryny R- i S- po podaniu pojedynczej dawki warfaryny (25 mg), a farmakodynamiczne efekty warfaryny mierzone za pomocą międzynarodowego wskaźnika znormalizowanego (INR) nie ulegały klinicznie istotnym zmianom. Jednakże u pacjentów przyjmujących warfarynę lub inne pochodne kumaryny, na początku leczenia semaglutydem zaleca się częste monitorowanie INR.
Dzieci
Badania interakcji przeprowadzono wyłącznie na dorosłych pacjentach.
Szczególne środki ostrożności dotyczące stosowania.
Śledzenie
W celu poprawy śledzenia produktów biologicznych nazwa i numer serii podawanego leku powinny być wyraźnie zapisane na opakowaniu.
Odewodnienie
Stosowanie agonistów receptorów GLP-1 może wiązać się z reakcjami niepożądanymi ze strony przewodu pokarmowego, które mogą prowadzić do odwodnienia, co w rzadkich przypadkach może skutkować pogorszeniem funkcji nerek. Pacjentów należy poinformować o potencjalnym ryzyku odwodnienia związanym z reakcjami niepożadanymi ze strony przewodu pokarmowego oraz podjąć środki zapobiegające odwodnieniu.
Ostre zapalenie trzustki
Obserwowano przypadki ostrego zapalenia trzustki podczas stosowania agonistów receptorów GLP-1 (patrz dział «Działania niepożądane»). Pacjentów należy poinformować o charakterystycznych objawach ostrego zapalenia trzustki. W przypadku podejrzenia zapalenia trzustki należy odstawić leczenie semaglutydem; jeśli zapalenie trzustki zostanie potwierdzone, nie można ponownie wznawiać leczenia semaglutydem.
Semaglutyd należy stosować z ostrożnością u pacjentów z wywiadem zapalenia trzustki.
W przypadku braku innych objawów i oznak ostrego zapalenia trzustki samo podwyższenie stężenia enzymów trzustkowych nie jest objawem prognostycznym ostrego zapalenia trzustki.
Cukrzyca typu 2
Semaglutydu nie należy stosować jako zamiennika insuliny u pacjentów z cukrzycą typu 2.
Semaglutydu nie należy stosować w połączeniu z innymi agonistami receptorów GLP-1. Takiego połączenia nie oceniano, dlatego należy uznać za możliwe zwiększenie ryzyka działań niepożądanych związanych z przedawkowaniem.
Hipoglikemia u pacjentów z cukrzycą typu 2
Wiadomo, że insulina i pochodne sulfonowe powodują hipoglikemię. U pacjentów leczonych semaglutydem w połączeniu z pochodnymi sulfonowymi lub insuliną może występować zwiększone ryzyko wystąpienia hipoglikemii. Ryzyko hipoglikemii można zmniejszyć, obniżając dawkę pochodnych sulfonowych lub insuliny na początku leczenia agonistą receptora GLP-1. Nie oceniano dodawania leku VegoVі FlexTech do leczenia pacjentów otrzymujących insulinę.
Retinopatia cukrzycowa u pacjentów z cukrzycą typu 2
U pacjentów z retinopatią cukrzycową, którzy otrzymywali semaglutyd, obserwowano zwiększone ryzyko powikłań retinopatii cukrzycowej (patrz dział «Działania niepożądane»). Szybkie poprawienie kontroli stężenia glukozy było związane z tymczasowym pogorszeniem retinopatii cukrzycowej, jednak nie można wykluczyć wpływu innych mechanizmów. Pacjentów z retinopatią cukrzycową należy dokładnie monitorować i leczyć zgodnie z zaleceniami klinicznymi. Brak doświadczenia w stosowaniu leku VegoVі FlexTech u pacjentów z cukrzycą typu 2 z niekontrolowaną lub potencjalnie niestabilną retinopatią cukrzycową. Nie zaleca się stosowania leku VegoVі FlexTech u tych pacjentów.
Niebadane populacje
Nie badano bezpieczeństwa i skuteczności stosowania leku VegoVі FlexTech u pacjentów:
- stosujących inne leki do kontroli masy ciała,
– z cukrzycą typu 1,
- z ciężkim zaburzeniem funkcji nerek (patrz dział «Sposób stosowania i dawki»),
- z ciężką niewydolnością wątroby (patrz dział «Sposób stosowania i dawki»),
- z niewydolnością serca klasy IV według klasyfikacji New York Heart Association (NYHA).
Nie zaleca się stosowania u tych pacjentów.
Istnieje ograniczone doświadczenie w stosowaniu leku VegoVі FlexTech u pacjentów:
- w wieku 75 lat i starszych (patrz dział «Sposób stosowania i dawki»),
- z lekkim lub umiarkowanym zaburzeniem funkcji wątroby (patrz dział «Sposób stosowania i dawki»),
– z chorobami zapalnymi jelit,
- z gastroparezą cukrzycową.
U tych pacjentów lek VegoVі FlexTech należy stosować z ostrożnością.
Zawartość sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg)/dawkę, dlatego lek można uważać za pozbawiony sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym
Kobietom w wieku rozrodczym zaleca się stosowanie środków antykoncepcyjnych podczas leczenia semaglutydem (patrz dział «Interakcje z innymi lekami i inne formy interakcji»).
Ciąża
Badania na zwierzętach wykazały obecność toksyczności rozrodczej (patrz dział «Dane przedkliniczne dotyczące bezpieczeństwa»). Dane dotyczące stosowania semaglutydu u ciężarnych kobiet są ograniczone. Dlatego nie należy stosować semaglutydu w okresie ciąży. Jeśli pacjentka planuje zajście w ciążę lub jest w ciąży, należy przerwać stosowanie semaglutydu. Z uwagi na długi okres półtrwania semaglutydu należy przerwać jego stosowanie co najmniej 2 miesiące przed planowaną ciążą (patrz dział «Farmakokinetyka»).
Karmienie piersią
Podczas laktacji u szczurów semaglutyd wydzielany był z mlekiem. Nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią. Nie należy stosować semaglutydu w okresie karmienia piersią.
Płodność
Wpływ semaglutydu na płodność u ludzi jest nieznany. Stosowanie semaglutydu u samców szczurów nie wpływało na płodność. U samic szczurów obserwowano wydłużenie cyklu estralnego i zmniejszenie liczby owulacji przy dawkach związanych z utratą masy ciała.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Semaglutyd nie wpływa lub niemal nie wpływa na zdolność prowadzenia pojazdów i obsługiwania maszyn. Jednak może występować zawroty głowy, głównie w okresie zwiększania dawki. W przypadku wystąpienia zawrotów głowy należy zachować ostrożność podczas prowadzenia pojazdów lub pracy z innymi maszynami.
Pacjenci z cukrzycą typu 2
Pacjentom stosującym semaglutyd w połączeniu z pochodnymi sulfonowymi lub z insuliną należy zalecić środki ostrożności, aby uniknąć rozwoju hipoglikemii podczas prowadzenia pojazdów lub pracy z innymi maszynami (patrz dział «Szczególne środki ostrożności dotyczące stosowania»).
Sposób stosowania i dawki.
Dawkowanie
Dorośli
Dawkę utrzymującą semaglutydu 2,4 mg raz w tygodniu osiąga się poprzez rozpoczęcie od dawki 0,25 mg. Aby zmniejszyć ryzyko wystąpienia działań niepożądanych ze strony przewodu pokarmowego, dawkę należy zwiększać stopniowo w ciągu 16-tygodniowego okresu, aż do dawki utrzymującej 2,4 mg raz w tygodniu (patrz tabela 8). W przypadku wystąpienia znaczących działań ze strony przewodu pokarmowego należy rozważyć opóźnienie zwiększenia dawki lub jej zmniejszenie do poprzedniego poziomu, aż stan się poprawi. Dawek wyższych niż 2,4 mg na tydzień nie zaleca się.
Tabela 8. Harmonogram zwiększania dawki
| Podwyższenie dawki |
Dawka tygodniowa |
| Tydzień 1–4 |
0,25 mg |
| Tydzień 5–8 |
0,5 mg |
| Tydzień 9–12 |
1 mg |
| Tydzień 13–16 |
1,7 mg |
| Dawka utrzymaniowa |
2,4 mg |
Dzieci
U dzieci w wieku od 12 lat należy stosować ten sam schemat zwiększania dawki, co u dorosłych (patrz tabela 8). Dawka powinna być zwiększana do 2,4 mg (dawka utrzymaniowa) lub do osiągnięcia maksymalnej dawki dobrze tolerowanej. Nie zaleca się dawki tygodniowej wyższej niż 2,4 mg.
Pacjenci z cukrzycą typu 2
Na początku stosowania semaglutydu u pacjentów z cukrzycą typu 2 należy rozważyć możliwość zmniejszenia dawki współpodawanego insuliny lub stymulatorów sekrecji insuliny (takich jak pochodne sulfonowych moczników), aby zmniejszyć ryzyko hipoglikemii, patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”.
Pominięta dawka
Jeśli dawkę pominięto, należy ją podać jak najszybciej w ciągu 5 dni od pominięcia. Jeśli minęło już więcej niż 5 dni, pominiętą dawkę należy pominąć, a następną dawkę podać w zaplanowanym dniu. W ten sposób pacjenci mogą przywrócić regularny schemat stosowania leku raz w tygodniu. Jeśli pominięto więcej niż jedną dawkę, należy rozważyć możliwość zmniejszenia dawki początkowej przy ponownym rozpoczęciu leczenia.
Szczególne grupy pacjentów
Pacjenci w podeszłym wieku (powyżej 65 roku życia)
Nie jest wymagana korekta dawki w zależności od wieku pacjenta. Doświadczenie leczenia pacjentów w wieku ≥ 75 lat jest ograniczone, a nie można wykluczyć zwiększonej wrażliwości u niektórych osób starszych.
Naruszenie funkcji nerek
U pacjentów z łagodnym lub umiarkowanym naruszeniem funkcji nerek nie jest wymagana korekta dawki. Doświadczenie stosowania semaglutydu u pacjentów z ciężkim naruszeniem funkcji nerek jest ograniczone. Semaglutyd nie jest zalecany pacjentom z ciężkim naruszeniem funkcji nerek (eSzP < 30 ml/min/1,73 m²), w tym pacjentom z nerek w stadium końcowym (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”, „Efekty niepożądane” oraz „Farmakokinetyka”).
Naruszenie funkcji wątroby
U pacjentów z łagodnym lub umiarkowanym naruszeniem funkcji wątroby nie jest wymagana korekta dawki. Doświadczenie stosowania semaglutydu u pacjentów z ciężkim naruszeniem funkcji wątroby jest ograniczone. Semaglutyd nie jest zalecany pacjentom z ciężkim naruszeniem funkcji wątroby; u pacjentów z łagodnym lub umiarkowanym naruszeniem funkcji wątroby należy stosować z ostrożnością (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności podczas stosowania” oraz „Farmakokinetyka”).
Sposób podania
Podanie podskórne
Vegovі FlexTech stosuje się raz w tygodniu w dowolnym czasie dnia niezależnie od posiłku.
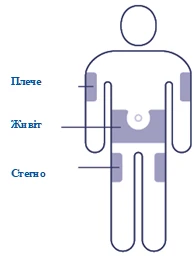
Lek podaje się podskórnie w okolicę przedniej ściany brzucha, uda lub barku. Miejsce wstrzyknięcia można zmieniać. Nie wolno podawać leku do wnętrza żyły ani do tkanki mięśniowej.
Jeśli konieczna jest zmiana dnia tygodniowego podania leku, można to zrobić, pod warunkiem że odstęp czasu między dwiema dawkami wynosi co najmniej 3 dni (> 72 godziny). Po wybraniu nowego dnia podania leku należy kontynuować jego stosowanie według schematu raz w tygodniu.
Podczas jednorazowego podania leku z wypełnionej wcześniej ruchomki strzykawki Vegovі FlexTech ruchomek należy mocno przycisnąć do skóry, aż do zatrzymania się żółtej wstążki. Iniekcja trwa około 5–10 sekund.
Przed zastosowaniem leku pacjentom należy zalecić dokładne zapoznanie się z instrukcją obsługi ruchomki strzykawki Vegovі FlexTech zawartą w ulotce do leku.
Aby uzyskać dodatkowe informacje przed zastosowaniem, patrz sekcja „Instrukcja obsługi ruchomki strzykawki”.
| Instrukcja obsługi długopisu strzykawki Vegovі FlexTech |
|||
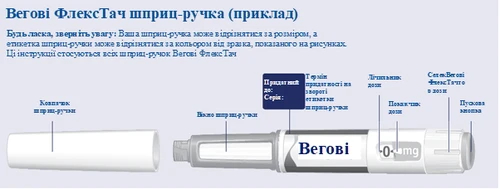
| Przed rozpoczęciem stosowania długopisu strzykawki Vegovі FlexTech raz w tygodniu zawsze uważnie przeczytaj niniejszą instrukcję i porozmawiaj ze swoim lekarzem, pielęgniarką lub farmaceutą na temat prawidłowego wstrzykiwania Vegovі FlexTech. Vegovі FlexTech to długopis strzykawka z zestawem dawek, który zawiera cztery przepisane dawki odpowiadające czterem zastosowaniom raz w tygodniu. Proszę korzystać z tabeli umieszczonej wewnątrz pokrywy tekturowego pudełka, aby śledzić, ile dawek zostało już wykorzystanych i ile dawek pozostało w długopisie strzykawce. Vegovі FlexTech jest dostarczany w pięciu różnych długopisach strzykawkach, z których każda zawiera jedną z następujących przepisanych dawek semaglutydu:
Zawsze zaczynaj od sprawdzenia etykiety długopisu strzykawki, aby upewnić się, że zawiera przepisaną Ci dawkę Vegovі FlexTech Twój długopis jest przeznaczony do użytku z jednorazowymi igłami 30G, 31G i 32G o długości do 8 mm. Opakowanie zawiera:
|
|||
|
|
|||
|
|||
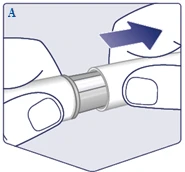
| Sprawdź nazwę i dawkę swojego długopisu strzykawki, aby upewnić się, że zawiera przepisaną Ci dawkę leku Vegovі FlexTech. Zdejmij osłonkę długopisu strzykawki. (Patrz rysunek A). |
|
||
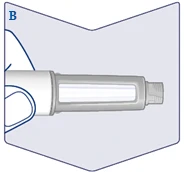
| Upewnij się, że roztwór w długopisie strzykawce jest przezroczysty i bezbarwny. Spójrz przez okienko długopisu strzykawki. Jeśli roztwór jest mętny lub zabarwiony, nie wolno używać długopisu strzykawki. (Patrz rysunek B). |
|
||
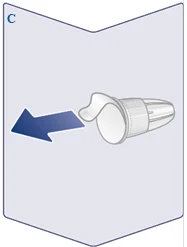
| Zawsze używaj nowej igły do każdego zastrzyku. Weź igłę, gdy będziesz gotowy do wykonania zastrzyku. Sprawdź, czy nie uszkodzono papierowej membrany ani zewnętrznej osłonki igły, ponieważ może to wpłynąć na sterylność. Jeśli występują jakiekolwiek uszkodzenia, użyj nowej igły. Zdejmij papierową membranę. (Patrz rysunek C). |
|
||
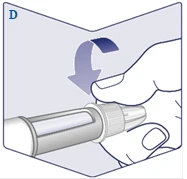
| Nakręć igłę na długopis strzykawki. Obróć ją, aby igła trzymała się mocno na długopisie strzykawki. (Patrz rysunek D). |
|
||
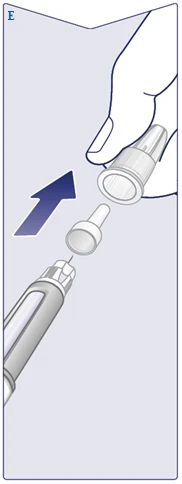
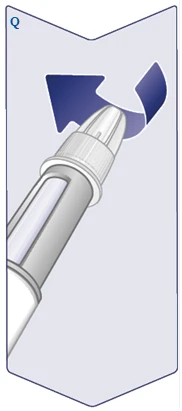
| Na igle znajdują się dwie osłonki. Musisz zdjąć obie osłonki. Jeśli zapomnisz zdjąć obie osłonki, nie będziesz mógł wstrzyknąć Vegovі FlexTech. Zdejmij zewnętrzną osłonkę igły i zachowaj ją. Będzie potrzebna po zakończeniu zastrzyku do bezpiecznego zdjęcia igły z długopisu strzykawki. Zdejmij wewnętrzną osłonkę igły i wyrzuć ją. Na końcu igły może pojawić się kropla roztworu. Jednakże należy sprawdzić przepływ roztworu przy pierwszym użyciu nowego długopisu strzykawki. Zobacz punkt „Sprawdzenie działania każdego nowego długopisu strzykawki”. Nigdy nie używaj wygiętej lub uszkodzonej igły. Aby uzyskać dodatkowe informacje dotyczące postępowania z igłami, zobacz punkt „O Twoich igłach” poniżej niniejszej instrukcji. (Patrz rysunek E). |
|
||
| Sprawdzenie działania każdego nowego długopisu strzykawki |
|||
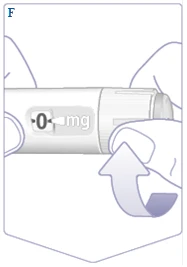
| Jeśli już korzystałeś z długopisu strzykawki Vegovі FlexTech, przejdź do punktu 2. „Ustawienie dawki”. Tylko przed pierwszym użyciem każdego nowego długopisu strzykawki Vegovі FlexTech sprawdź przepływ. Obracaj selektorem dawki, aż licznik dawki pokaże symbol sprawdzenia przepływu (). (Patrz rysunek F). |
|
||
| Upewnij się, że symbol sprawdzenia przepływu pokrywa się z wskaźnikiem dawki. (Patrz rysunek G). |
|
||
| Sprawdzenie przepływu |
|||
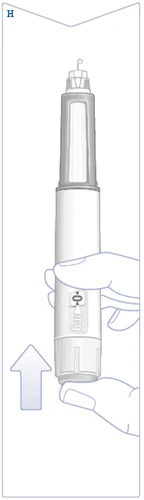
| Trzymaj długopis strzykawki igłą do góry. Naciśnij i przytrzymaj przycisk uruchamiający, aż licznik dawki powróci do symbolu . Symbol musi pokrywać się z wskaźnikiem dawki. Na końcu igły powinna pojawić się kropla roztworu. Ta kropla oznacza, że długopis strzykawki jest gotowy do użycia. Jeśli kropla się nie pojawi, sprawdź przepływ ponownie. Należy to zrobić tylko dwukrotnie. Jeśli kropla nadal się nie pojawia, zamień igłę i sprawdź przepływ ponownie. Nie używaj długopisu strzykawki, jeśli kropla roztworu nadal się nie pojawia. (Patrz rysunek H). |
|
||
|
|||
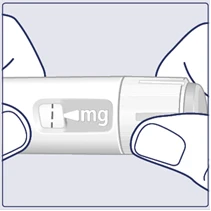
| Obracaj selektorem dawki, aż licznik dawki się zatrzyma i pokaże przepisaną Ci dawkę. (Patrz rysunek I). |
|
||
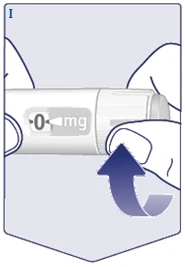
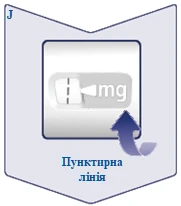
| Przerywana linia () na liczniku dawki pomoże określić dawkę. Selektor dawki kliknie inaczej, gdy obracasz go do przodu, do tyłu lub po dawce. Usłyszysz kliknięcie za każdym razem, gdy obracasz selektorem dawki. Nie ustawiaj dawki, licząc liczbę kliknięć, które słyszysz. (Patrz rysunek J). |
|
||
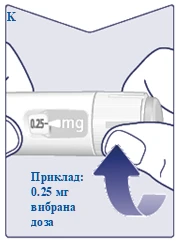
| Gdy przepisana dawka pokryje się ze wskaźnikiem dawki, wybrałeś swoją dawkę. Na tym rysunku dawka pokazana jest jako przykład. Jeśli licznik dawki zatrzyma się przed osiągnięciem przepisanej dawki, sprawdź punkt „Czy wystarczy Ci Vegovі FlexTech?” poniżej niniejszych instrukcji. (Patrz rysunek K). |
|
||
| Wybierz miejsce zastrzyku Wybierz miejsce na ramieniu, udzie lub brzuchu (utrzymuj odległość 5 cm od pępka). Możesz robić zastrzyk w to samo miejsce ciała co tydzień, ale upewnij się, że wstrzyknięcie nie będzie w tym samym punkcie, co ostatnim razem. |
|
||
|
|||
| Wprowadź igłę pod skórę. Upewnij się, że widzisz licznik dawki. Nie zakrywaj licznika dawki palcami. Może to przerwać zastrzyk. (Patrz rysunek L). |
|
||
| Naciśnij i przytrzymaj przycisk uruchamiający, aż licznik dawki pokaże . (Patrz rysunek M). Trzymaj igłę pod skórą po tym, jak licznik dawki powróci do 0, i licz powoli do 6. powinien pokrywać się z wskaźnikiem dawki. Następnie możesz usłyszeć lub poczuć kliknięcie, gdy licznik dawki powróci do . (Patrz rysunek N). |
|
||
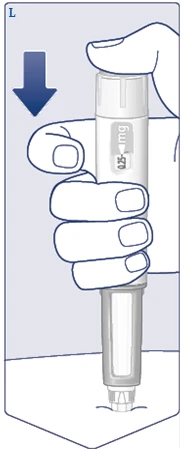
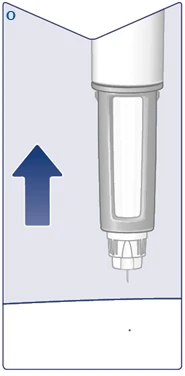
| Wyjmij igłę ze skóry. Jeśli wyciągniesz igłę zbyt wcześnie, możesz zobaczyć wyciek roztworu z końcówki igły i pełna dawka nie zostanie podana. Jeśli w miejscu zastrzyku pojawi się kropla krwi, delikatnie przyłóż do niego, aby zatrzymać krwawienie. Możesz zobaczyć kroplę roztworu na końcówce igły po wstrzyknięciu. Jest to normalne i nie wpływa na objętość podanej dawki. (Patrz rysunek O). |
|
||
|
|||
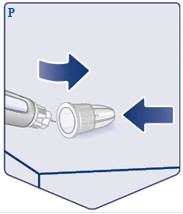
| Połóż zewnętrzną osłonkę igły na płaskiej powierzchni i załóż zewnętrzną osłonkę na igłę, nie dotykając igły ani zewnętrznej osłonki igły. Gdy tylko igła zostanie przykryta, ostrożnie całkowicie naciśnij na zewnętrzną osłonkę igły. (Patrz rysunek P). |
|
||
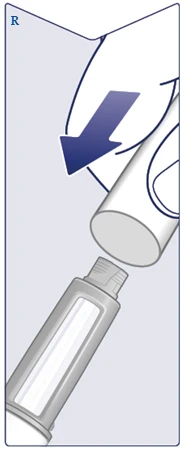
| Odkręć igłę i bezpiecznie ją zutylizuj zgodnie z instrukcjami lekarza, pielęgniarki, farmaceuty lub lokalnych władz. Nigdy nie próbuj ponownie założyć wewnętrznej osłonki igły na igłę. Możesz się poranić igłą. Zawsze zdejmuj igłę z długopisu bezpośrednio po każdym zastrzyku, aby zapobiec zablokowaniu igły, zanieczyszczeniu, zakażeniu, niedokładnemu dawkowaniu. Nigdy nie przechowuj długopisu z przykręconą igłą. (Patrz rysunek Q). |
|
||
| Zakryj długopis strzykawki osłonką po każdym użyciu, aby chronić roztwór przed działaniem światła. (Patrz rysunek R). |
|
||
| Gdy długopis strzykawki będzie pusty, zutylizuj długopis strzykawki bez igły zgodnie z instrukcjami otrzymanymi od lekarza, pielęgniarki, farmaceuty lub lokalnych władz. Osłonkę długopisu i pustą skrzynkę można wyrzucić razem z odpadami komunalnymi. |
|||
| O Twoich igłach |
|||
| Jak rozpoznać zablokowaną lub uszkodzoną igłę
Jak postępować z zablokowaną igłą
|
|||
| Opieka nad Twoim długopisem strzykawki |
|||
| Postępuj ostrożnie ze swoim długopisem strzykawki. Nieodpowiednie postępowanie lub niewłaściwe użycie może prowadzić do podania niewłaściwej dawki. Jeśli dojdzie do takiej sytuacji, możesz nie uzyskać oczekiwanego efektu działania tego leku.
|
|||
| Czy wystarczy Ci Vegovі FlexTech? |
|||
| Jeśli licznik dawki zatrzyma się przed przepisaną dawką, roztworu nie wystarczy do podania pełnej dawki. Zutylizuj ten długopis strzykawki i użyj nowego długopisu strzykawki Vegovі FlexTech. |
|
||
| Ważne informacje |
|||
|
|||








Dzieci
U dzieci w wieku od 12 lat nie jest wymagana korekta dawki. Nie ustalono bezpieczeństwa i skuteczności stosowania semaglutydu u dzieci w wieku poniżej 12 lat.
Przedawkowanie.
Przedawkowanie semaglutydu może być związane z zaburzeniami przewodu pokarmowego, które mogą prowadzić do odwodnienia organizmu. W przypadku przedawkowania należy obserwować pacjenta pod kątem objawów klinicznych i rozpocząć odpowiednie leczenie wspierające.
Niepożądane działania.
Streszczenie profilu bezpieczeństwa
W czterech badaniach klinicznych fazy 3 uczestniczyło 2650 pacjentów przyjmujących lek Vegovі FlexTech. Czas trwania badań wynosił 68 tygodni. Najczęściej zgłaszane niepożądane działania dotyczyły przewodu pokarmowego, w tym nudności, biegunki, zaparcia i wymioty.
Niepożądane działania
Poniżej przedstawiono niepożądane działania wykryte w badaniach klinicznych fazy 3. Częstość występowania niepożądanych działań określono na podstawie danych z badań klinicznych fazy 3.
Niepożądane działania sklasyfikowano według układów narządów i częstości występowania. Ocena częstości występowania niepożądanych działań przeprowadzona została według następującej skali: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1000 do < 1/100), niezbyt często (od ≥ 1/10000 do < 1/1000), bardzo rzadko (< 1/10000).
Zaburzenia układu odpornościowego: rzadko – reakcja anafilaktyczna.
Zaburzenia metabolizmu i trawienia: często – hipoglikemia u pacjentów z cukrzycą typu 2a.
Zaburzenia układu nerwowego: bardzo często – ból głowyb; często – zawroty głowyb.
Zaburzenia narządu wzroku: często – retinopatia cukrzycowa u pacjentów z cukrzycą typu 2a.
Zaburzenia układu sercowo-naczyniowego: niezbyt często – hipotensja, hipotensja ortostatyczna, zwiększona częstość akcji sercab,c.
Zaburzenia ze strony przewodu pokarmowego: bardzo często – wymiotyb, biegunkab, zaparcieb, nudnościab, ból brzuchab; często – zapalenie żołądka b,c, choroba refluksowa przełyku b, dyspepsja b, odbijanie b, wzdęcia b, brzuch b; niezbyt często – ostre zapalenie trzustki a, opóźnione opróżnianie żołądka.
Zaburzenia wątroby i dróg żółciowych: często – kamica żółciowa a.
Zaburzenia skóry i tkanek podskórnych: często – wypadanie włosów a; rzadko – obrzęk naczynioruchowy.
Ogólne zaburzenia i reakcje w miejscu wstrzyknięcia: bardzo często – zmęczenie b,c; często – reakcje w miejscu wstrzyknięcia c.
Badania laboratoryjne: niezbyt często – podwyższony poziom amylazy, podwyższony poziom lipazy c.
a Opis poszczególnych niepożądanych działań podano poniżej.
b Głównie obserwowane w okresie zwiększania dawki.
c Zgrupowane priorytetowe terminy.
Opis poszczególnych niepożądanych działań
Niepożądane działania ze strony przewodu pokarmowego
W trakcie badań trwających 68 tygodni nudności występowały u 43,9% pacjentów leczonych semaglutydem (u 16,1% w grupie placebo), biegunka – u 29,7% (u 15,9% w grupie placebo), a wymioty – u 24,5% (u 6,3% w grupie placebo). Większość przypadków miała charakter łagodny lub umiarkowany i była nietrwała. Zaparcia występowały u 24,2% pacjentów leczonych semaglutydem (u 11,1% w grupie placebo) i były łagodne lub umiarkowane, ale dłuższe trwanie. U pacjentów leczonych semaglutydem średnia długość trwania nudności wynosiła 8 dni, wymiotów – 2 dni, biegunki – 3 dni, a zaparcia – 47 dni.
Pacjenci z umiarkowanym zaburzeniem funkcji nerek (eGFR ≥ 30 ml/min/1,73 m²) mogą odczuwać silniejsze działania przewodu pokarmowego podczas leczenia semaglutydem.
Działania przewodu pokarmowego doprowadziły do całkowitego zakończenia leczenia u 4,3% pacjentów.
Ostre zapalenie trzustki
Częstość przypadków ostrego zapalenia trzustki potwierdzonych przez ekspertów w trakcie badań klinicznych fazy 3 wynosiła 0,2% przy stosowaniu semaglutydu i < 0,1% przy stosowaniu placebo.
Ostra choroba żółciowa / kamica żółciowa
Kamica żółciowa została zarejestrowana u 1,6% pacjentów i doprowadziła do zapalenia pęcherza żółciowego u 0,6% pacjentów leczonych semaglutydem. Kamica żółciowa i zapalenie pęcherza żółciowego występowały odpowiednio u 1,1% i 0,3% pacjentów leczonych placebo.
Wypadanie włosów
O wypadaniu włosów zgłaszano u 2,5% pacjentów leczonych semaglutydem i u 1,0% pacjentów leczonych placebo. Reakcja miała głównie charakter łagodny, a większość pacjentów wyzdrowiała podczas kontynuacji leczenia. Wypadanie włosów występowało częściej u pacjentów z większą utratą masy ciała (≥ 20%).
Zwiększenie częstości akcji serca
W badaniach klinicznych fazy 3 u pacjentów leczonych semaglutydem obserwowano średnie zwiększenie częstości akcji serca o 3 uderzenia na minutę (ud./min) od początkowej średniej wartości 72 ud./min. Odsetek pacjentów z zwiększeniem częstości akcji serca od poziomu wyjściowego ≥ 10 ud./min w dowolnym momencie w trakcie okresu leczenia wynosił 67,0% w grupie semaglutydu w porównaniu do 50,1% w grupie placebo.
Imunogenność
Z uwagi na potencjalne właściwości immunogenne leków zawierających białka lub peptydy, u pacjentów leczonych semaglutydem mogą powstawać przeciwciała. Odsetek pacjentów z pozytywnym wynikiem testu na obecność przeciwciał anty-semaglutydowych w dowolnym czasie po rozpoczęciu leczenia był niski (2,9%), a żaden z pacjentów nie miał przeciwciał neutralizujących semaglutyd ani przeciwciał z efektem neutralizującym GLP-1 do zakończenia badań klinicznych. W trakcie leczenia wysokie stężenia semaglutydu mogły obniżyć czułość testów, dlatego nie można wykluczyć ryzyka fałszywie negatywnych wyników. Jednakże u pacjentów z pozytywnym wynikiem testu na obecność przeciwciał obecność przeciwciał była tymczasowa i nie miała widocznego wpływu na skuteczność i bezpieczeństwo.
Hipoglikemia u pacjentów z cukrzycą typu 2
W badaniu STEP 2 hipoglikemia klinicznie istotna występowała u 6,2% (0,1 przypadku na pacjenta-rok) pacjentów leczonych semaglutydem w porównaniu do 2,5% (0,03 przypadku na pacjenta-rok) pacjentów leczonych placebo. Hipoglikemia przy stosowaniu semaglutydu występowała zarówno przy współrzutynowym stosowaniu sulfonamidów, jak i bez niego. Jeden epizod (0,2% pacjentów, 0,002 przypadku na pacjenta-rok) został zarejestrowany jako ciężki u pacjenta, który nie przyjmował jednocześnie sulfonamidu. Ryzyko hipoglikemii zwiększało się przy stosowaniu semaglutydu z sulfonamidami.
Zaostrzenie retinopatii cukrzycowej
W badaniu klinicznym trwającym 2 lata dotyczącego semaglutydu w dawkach 0,5 mg i 1 mg w porównaniu do placebo wzięło udział 3297 pacjentów z cukrzycą typu 2, wysokim ryzykiem sercowo-naczyniowym, długotrwałą cukrzycą i niewystarczającą kontrolą poziomu glukozy we krwi. W tym badaniu oceniano reakcje związane z zaostrzeniem retinopatii cukrzycowej, które występowały częściej u pacjentów leczonych semaglutydem (3,0%) niż u tych, którzy przyjmowali placebo (1,8%). Obserwacja ta dotyczyła pacjentów z potwierdzoną retinopatią cukrzycową, którzy przyjmowali insulinę. Różnica między wariantami leczenia pojawiła się wcześnie i utrzymywała się przez cały okres badania. W badaniu STEP 2 o chorobach siatkówki zgłosiło 6,9% pacjentów przyjmujących VegoFlexTech, 6,2% pacjentów przyjmujących semaglutyd 1 mg i 4,2% pacjentów przyjmujących placebo. Większość przypadków została zarejestrowana jako retinopatia cukrzycowa (4,0%, 2,7% i 2,7% odpowiednio) i retinopatia nieproliferacyjna (0,7%, 0% i 0% odpowiednio).
Dzieci
W badaniu klinicznym przeprowadzonym wśród dzieci w wieku od 12 do 18 lat z otyłością lub nadmierną masą ciała z co najmniej jednym towarzyszącym schorzeniem związanym z masą ciała, 133 pacjentów otrzymywało VegoFlexTech. Czas trwania badania wynosił 68 tygodni.
Ogólnie częstość, typ i nasilenie niepożądanych działań u dzieci były takie same jak u dorosłych. Kamica żółciowa została zarejestrowana u 3,8% pacjentów przyjmujących VegoFlexTech i u 0% pacjentów przyjmujących placebo.
Po 68 tygodniach leczenia nie stwierdzono wpływu na wzrost ani rozwój pokwitania.
Zgłaszanie niepożądanych działań
Zgłaszanie niepożądanych działań po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich przedstawiciele prawni powinni zgłaszać wszystkie przypadki podejrzanych niepożądanych działań i braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 3 lata.
Po pierwszym zastosowaniu: 6 tygodni. Przechowywać w temperaturze poniżej 30 °C lub w lodówce (przy temperaturze 2 °C – 8 °C).
Warunki przechowywania.
Przechowywać w lodówce (2 °C – 8 °C). Trzymać z dala od elementów mrozących. Nie zamrażać.
Przechowywać strzykawkę-pensetkę z założonym kapturkiem chroniącym przed działaniem światła.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Ten lek nie może być mieszany z innymi lekami, ponieważ nie przeprowadzono badań niezgodności.
Opakowanie.
0,25, 0,5 mg
Szkło kartusz (szkło typu I) o pojemności 1,5 ml, z jednej strony zamknięte gumowym (chlorobutyl) tłokiem, z drugiej strony – aluminiową pokrywką z laminowaną wkładką gumową (bromobutyl/polizopren). Kartusz umieszczony w jednorazowej wstępnie napełnionej strzykawce-pensetce wykonanej z polipropylenu, polioksymetylenu, poliwęglanu i akrylonitrylu butadienu styrenu.
1 wstępnie napełniona strzykawka-pensetka oraz 4 jednorazowe igły NovoFine® Plus w tekturowym pudełku.
1 mg, 1,7 mg
Szkło kartusz (szkło typu I) o pojemności 3 ml, z jednej strony zamknięte gumowym (chlorobutyl) tłokiem, z drugiej strony – aluminiową pokrywką z laminowaną wkładką gumową (bromobutyl/polizopren). Kartusz umieszczony w jednorazowej wstępnie napełnionej strzykawce-pensetce wykonanej z polipropylenu, polioksymetylenu, poliwęglanu i akrylonitrylu butadienu styrenu.
1 wstępnie napełniona strzykawka-pensetka oraz 4 jednorazowe igły NovoFine® Plus w tekturowym pudełku.
2,4 mg
Szkło kartusz (szkło typu I) o pojemności 3 ml, z jednej strony zamknięte gumowym (chlorobutyl) tłokiem, z drugiej strony – aluminiową pokrywką z laminowaną wkładką gumową (bromobutyl/polizopren). Kartusz umieszczony w jednorazowej wstępnie napełnionej strzykawce-pensetce wykonanej z polipropylenu, polioksymetylenu, poliwęglanu i akrylonitrylu butadienu styrenu.
1 lub 3 wstępnie napełnione strzykawki-pensetki oraz 4 lub 12 jednorazowych igieł NovoFine® Plus w tekturowym pudełku.
Kategoria wydania. Na receptę.
Wnioskodawca/Wytwórca.
A/S Novo Nordisk.
Miejsce zamieszkania wnioskodawcy/wytwórcy oraz adres miejsca prowadzenia działalności.
Novo Allé
2880 Bagsværd
Dania.