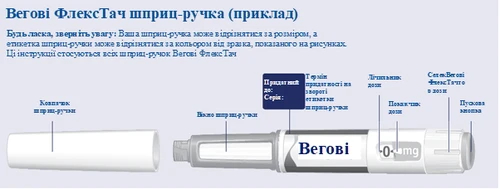
Vegovy Flexsta
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO Wegovy flextouch (wegovy flextouch)
Composición:
Principio activo: semaglutida;
0,25 mg
cada plumavitamina precargada contiene 1 mg de semaglutida* en 1,5 ml de solución, 1 ml de solución contiene 0,68 mg de semaglutida*, 1 plumavitamina precargada contiene 4 dosis de 0,25 mg;
0,5 mg
cada plumavitamina precargada contiene 2 mg de semaglutida* en 1,5 ml de solución, 1 ml de solución contiene 1,34 mg de semaglutida*, 1 plumavitamina precargada contiene 4 dosis de 0,5 mg;
1 mg
cada plumavitamina precargada contiene 4 mg de semaglutida* en 3 ml de solución, 1 ml de solución contiene 1,34 mg de semaglutida*, 1 plumavitamina precargada contiene 4 dosis de 1 mg;
1,7 mg
cada plumavitamina precargada contiene 6,8 mg de semaglutida* en 3 ml de solución, 1 ml de solución contiene 2,27 mg de semaglutida*, 1 plumavitamina precargada contiene 4 dosis de 1,7 mg;
2,4 mg
cada plumavitamina precargada contiene 9,6 mg de semaglutida* en 3 ml de solución, 1 ml de solución contiene 3,2 mg de semaglutida*, 1 plumavitamina precargada contiene 4 dosis de 2,4 mg;
Excipientes: fosfato disódico dihidrato; propilenglicol; fenol; ácido clorhídrico (para ajuste del pH); hidróxido sódico (para ajuste del pH); agua para preparaciones inyectables.
* Análogo del péptido 1 similar al glucagón humano (GLP-1), producido mediante tecnología de ADN recombinante en Saccharomyces cerevisiae.
Forma farmacéutica. Solución inyectable.
Características fisicoquímicas principales: solución isotónica, transparente e incolora; pH = 7,4.
Grupo farmacoterapéutico. Agentes hipoglucemiantes, excepto insulina. Análogos del péptido 1 similar al glucagón (GLP-1). Código ATC A10BJ06.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
El semaglutida es un análogo del GLP-1 con un 94 % de homología respecto al GLP-1 humano. El semaglutida actúa como agonista del receptor del GLP-1, uniendo y activando selectivamente los receptores del GLP-1, que son el blanco del GLP-1 nativo.
El GLP-1 es un regulador fisiológico del apetito y del consumo calórico, y los receptores del GLP-1 están presentes en varias regiones del cerebro implicadas en la regulación del apetito.
Estudios en animales muestran que el semaglutida actúa en el cerebro a través del receptor del GLP-1. El semaglutida tiene un efecto directo sobre las regiones del cerebro implicadas en la regulación homeostática de la ingesta alimentaria en el hipotálamo y el tronco encefálico. El semaglutida puede influir en el sistema hedónico de recompensa mediante un efecto directo e indirecto sobre regiones cerebrales, incluyendo el septum, el tálamo y la amígdala.
Los estudios clínicos muestran que el semaglutida reduce el consumo energético, aumenta la sensación de saciedad, plenitud y control sobre la ingesta de alimentos, disminuye la sensación de hambre, así como la frecuencia e intensidad del deseo de comer. Además, el semaglutida reduce el deseo por alimentos ricos en grasas.
El semaglutida regula las aportaciones homeostáticas y hedónicas con función ejecutiva para controlar el consumo calórico, el apetito, la recompensa y la elección de alimentos.
Además, los estudios clínicos han demostrado que el semaglutida reduce la glucemia actuando de forma dependiente de la glucosa, estimulando la secreción de insulina y suprimiendo la secreción de glucagón cuando la concentración de glucosa en sangre está elevada. El mecanismo de reducción de la glucemia también se asocia con un ligero retraso en el vaciamiento gástrico durante la fase postprandial temprana. En caso de hipoglucemia, el semaglutida disminuye la secreción de insulina y no interfiere con la secreción de glucagón.
La expresión del receptor del GLP-1 también se produce en el corazón, el sistema vascular, el sistema inmunitario y los riñones.
En los estudios clínicos, el semaglutida tuvo un efecto positivo sobre los niveles de lípidos en plasma, redujo la presión arterial sistólica y disminuyó la inflamación. Además, estudios en anim游戏副本
| Indicador |
Wegovy FlexTouch |
Placebo |
| Número de pacientes (N) |
1306 |
655 |
| Peso corporal (kg) |
||
| Nivel basal (kg) |
105,4 |
105,2 |
| Cambio (%) respecto al nivel basal1,2 |
-14,9 |
-2,4 |
| Diferencia (%) respecto al placebo1 [IC del 95 %] |
-12,4 [-13,4; -11,5]* |
- |
| Cambio (kg) respecto al nivel basal |
-15,3 |
-2,6 |
| Diferencia (kg) respecto al placebo1 [IC del 95 %] |
-12,7 [-13,7; -11,7] |
- |
| Pacientes (%) que alcanzaron una pérdida de peso ≥ 5 %3 |
83,5* |
31,1 |
| Pacientes (%) que alcanzaron una pérdida de peso ≥ 10 %3 |
66,1* |
12,0 |
| Pacientes (%) que alcanzaron una pérdida de peso ≥ 15 %3 |
47,9* |
4,8 |
| Circunferencia de la cintura (cm) |
||
| Nivel basal |
114,6 |
114,8 |
| Cambio respecto al nivel basal1 |
-13,5 |
-4,1 |
| Diferencia respecto al placebo1 [IC del 95 %] |
-9,4 [-10,3; -8,5]* |
- |
| Presión arterial sistólica (mm Hg) |
||
| Nivel basal |
126 |
127 |
| Cambio respecto al nivel basal1 |
-6,2 |
-1,1 |
| Diferencia respecto al placebo1 [IC del 95 %] |
-5,1 [-6,3; -3,9]* |
- |
* p < 0,0001 (no corregido bilateral) para ventaja.
1 Evaluado utilizando un modelo ANCOVA con imputación múltiple basado en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros fármacos contra la obesidad o cirugía bariátrica.
2 Durante el estudio, el tratamiento fue definitivamente interrumpido en el 17,1 % y el 22,4 % de los pacientes aleatorizados a semaglutida 2,4 mg y placebo, respectivamente. Suponiendo que todos los pacientes aleatorizados continuaron el tratamiento y no recibieron tratamiento adicional contra la obesidad, los cambios estimados desde el inicio hasta la semana 68 en el peso corporal, basados en un modelo mixto para medidas repetidas, incluyendo todas las observaciones hasta la primera interrupción, fueron de -16,9 % y -2,4 % para semaglutida 2,4 mg y placebo, respectivamente.
3 Evaluado mediante un modelo de regresión binaria basado en el mismo procedimiento de imputación que en el análisis primario.
HTMLIMG0END
Valores observados en pacientes que completaron cada visita programada y estimaciones con imputación múltiple (IM) de datos procedentes de pacientes que abandonaron el estudio.
Fig. 1. STEP 1: Cambio medio del peso corporal (%) desde el valor basal hasta la semana 68
Tras el período de estudio de 68 semanas, se realizó un período de 52 semanas sin tratamiento que incluyó a 327 pacientes que finalizaron el período principal del estudio en la dosis de mantenimiento de semaglutida o placebo. Durante el período sin tratamiento, desde la semana 68 hasta la semana 120, el peso corporal medio aumentó en ambos grupos de tratamiento. Sin embargo, en los pacientes que recibieron semaglutida durante el período principal del estudio, el peso corporal permaneció un 5,6 % por debajo del valor basal en comparación con un 0,1 % en el grupo placebo.
Estudio STEP 2. Control del peso corporal en pacientes con diabetes mellitus tipo 2
En un ensayo clínico doble ciego de 68 semanas de duración, 1210 pacientes con sobrepeso u obesidad (IMC ≥ 27 kg/m²) y con diabetes mellitus tipo 2 fueron aleatorizados para recibir semaglutida una vez por semana a dosis de 2,4 mg, semaglutida 1 mg o placebo. Los pacientes incluidos en el estudio tenían diabetes mellitus tipo 2 mal controlada (HbA1c entre 7 % y 10 %) y recibían tratamiento con dieta y ejercicio físico únicamente, o con 1 a 3 fármacos antidiabéticos orales. Todos los pacientes siguieron una dieta hipocalórica y aumentaron su actividad física durante todo el estudio.
El tratamiento con semaglutida durante 68 semanas produjo una reducción significativa y clínicamente relevante del peso corporal y de la HbA1c en comparación con el placebo (véase la tabla 2 y la figura 2).
Tabla 2. Estudio STEP 2: resultados en la semana 68
| Indicador |
Vegovy FlexTouch |
Placebo |
| Número de pacientes (N) |
404 |
403 |
| Peso corporal (kg) |
||
| Nivel basal (kg) |
99,9 |
100,5 |
| Cambio (%) respecto al nivel basal1,2 |
-9,6 |
-3,4 |
| Diferencia (%) respecto al placebo1 [IC 95 %] |
-6,2 [-7,3; -5,2]* |
- |
| Cambio (kg) respecto al nivel basal |
-9,7 |
-3,5 |
| Diferencia (kg) respecto al placebo1 [IC 95 %] |
-6,1 [-7,2; -5,0] |
- |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 5 %3 |
67,4* |
30,2 |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 10 %3 |
44,5* |
10,2 |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 15 %3 |
25,0* |
4,3 |
| Circunferencia de la cintura (cm) |
||
| Nivel basal |
114,5 |
115,5 |
| Cambio respecto al nivel basal1 |
-9,4 |
-4,5 |
| Diferencia respecto al placebo1 [IC 95 %] |
-4,9 [-6,0; -3,8]* |
- |
| Presión arterial sistólica (mm Hg) |
||
| Nivel basal |
130 |
130 |
| Cambio respecto al nivel basal1 |
-3,9 |
-0,5 |
| Diferencia respecto al placebo1 [IC 95 %] |
-3,4 [-5,6; -1,3]** |
- |
| HbA1c (mmol/mol (%)) |
||
| Nivel basal |
65,3 (8,1) |
65,3 (8,1) |
| Cambio respecto al nivel basal1 |
-17,5 (-1,6) |
-4,1 (-0,4) |
| Diferencia respecto al placebo1 [IC 95 %] |
-13,5 [-15,5; -11,4] (-1,2 [-1,4; -1,1])* |
- - |
* p < 0,0001 (no corregido bilateral) a favor; ** p < 0,05 (no corregido bilateral) a favor.
1 Evaluado utilizando un modelo ANCOVA con imputación múltiple basado en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros fármacos contra la obesidad o cirugía bariátrica.
2 Durante el estudio, el tratamiento se interrumpió definitivamente en el 11,6 % y el 13,9 % de los pacientes aleatorizados a semaglutida 2,4 mg y placebo, respectivamente. Suponiendo que todos los pacientes aleatorizados continuaron con el tratamiento y no recibieron tratamiento adicional para la obesidad, los cambios estimados desde el inicio aleatorizado hasta la semana 68 en el peso corporal, basados en un modelo mixto para medidas repetidas que incluye todas las observaciones hasta la primera interrupción, fueron del -10,6 % y -3,1 % para semaglutida 2,4 mg y placebo, respectivamente.
3 Evaluado mediante un modelo de regresión binaria basado en el mismo procedimiento de imputación que en el análisis primario.
**
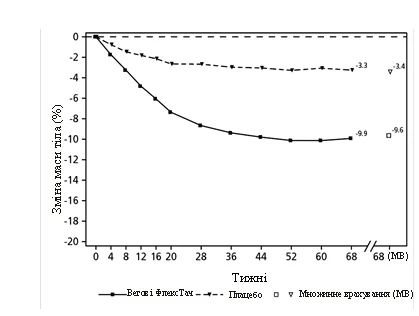
**
Valores observados en pacientes que completaron cada visita programada y estimaciones con imputación múltiple (IM) de datos de pacientes que abandonaron el estudio.
Fig. 2. STEP 2: Cambio medio del peso corporal (%) desde el valor basal hasta la semana 68
Estudio STEP 3. Control del peso corporal con terapia conductual intensiva
En un ensayo clínico doble ciego de 68 semanas de duración, 611 pacientes con obesidad (IMC ≥ 30 kg/m²) o con sobrepeso (IMC ≥ 27 kg/m² y < 30 kg/m²) y al menos una comorbilidad relacionada con el peso corporal fueron aleatorizados a recibir semaglutida o placebo. Todos los pacientes recibieron terapia conductual intensiva (TCI), que consistió en una dieta muy restrictiva, aumento de la actividad física y asesoramiento conductual durante toda la duración del estudio.
El tratamiento con semaglutida y TCI durante 68 semanas condujo a una reducción significativa y clínicamente relevante del peso corporal en comparación con placebo (véase la tabla 3).
Tabla 3. Estudio STEP 3: resultados en la semana 68
| Indicador |
Wegovy FlexTouch |
Placebo |
| Número de pacientes (N) |
407 |
204 |
| Peso corporal (kg) |
||
| Nivel basal (kg) |
106,9 |
103,7 |
| Cambio (%) respecto al nivel basal1,2 |
-16,0 |
-5,7 |
| Diferencia (%) frente al placebo1 [95 % IC] |
-10,3 [-12,0; -8,6]* |
- |
| Cambio (kg) respecto al nivel basal |
-16,8 |
-6,2 |
| Diferencia (kg) frente al placebo1 [95 % IC] |
-10,6 [-12,5; -8,8] |
- |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 5 %3 |
84,8* |
47,8 |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 10 %3 |
73,0* |
27,1 |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 15 %3 |
53,5* |
13,2 |
| Circunferencia de la cintura (cm) |
||
| Nivel basal |
113,6 |
111,8 |
| Cambio respecto al nivel basal1 |
-14,6 |
-6,3 |
| Diferencia frente al placebo1 [95 % IC] |
-8,3 [-10,1; -6,6]* |
- |
| Presión arterial sistólica (mmHg) |
||
| Nivel basal |
124 |
124 |
| Cambio respecto al nivel basal1 |
-5,6 |
-1,6 |
| Diferencia frente al placebo1 [95 % IC] |
-3,9 [-6,4; -1,5]* |
- |
*p < 0,0001 (bilateral, sin ajustar) a favor del semaglutido.
1 Evaluado utilizando un modelo ANCOVA con imputación múltiple basada en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros medicamentos contra la obesidad o cirugía bariátrica.
2 Durante el estudio, el tratamiento fue definitivamente interrumpido en el 16,7 % y el 18,6 % de los pacientes aleatorizados a semaglutida 2,4 mg y placebo, respectivamente. Suponiendo que todos los pacientes aleatorizados continuaron el tratamiento y no recibieron tratamiento adicional contra la obesidad, los cambios estimados desde la aleatorización hasta la semana 68 en el peso corporal, basados en un modelo mixto para medidas repetidas que incluye todas las observaciones hasta la primera interrupción, fueron del -17,6 % y -5,0 % para semaglutida 2,4 mg y placebo, respectivamente.
3 Evaluado mediante un modelo de regresión binaria basado en el mismo procedimiento de imputación que en el análisis primario.
Estudio STEP 4. Mantenimiento sostenido de la pérdida de peso
En un ensayo clínico doble ciego de 68 semanas de duración, se incluyeron 902 pacientes con obesidad (IMC ≥ 30 kg/m²) o con sobrepeso (IMC ≥ 27 kg/m² y < 30 kg/m²) y al menos una comorbilidad relacionada con el peso. Todos los pacientes siguieron una dieta hipocalórica y aumentaron su actividad física durante todo el estudio. De la semana 0 a la semana 20 (fase de inducción), todos los pacientes recibieron semaglutida. En la semana 20 (línea basal), los pacientes que alcanzaron la dosis de mantenimiento de 2,4 mg fueron aleatorizados para continuar con semaglutida o cambiar a placebo. En la semana 0 (inicio de la fase de inducción), los pacientes tenían un peso corporal medio de 107,2 kg y un IMC medio de 38,4 kg/m².
Los pacientes que alcanzaron la dosis de mantenimiento de 2,4 mg en la semana 20 (línea basal) y continuaron con semaglutida durante 48 semanas (semanas 20 a 68) siguieron perdiendo peso y mostraron una reducción de peso más pronunciada y clínicamente significativa en comparación con aquellos que cambiaron a placebo (véase la tabla 4 y la figura 3). El peso corporal aumentó de forma estable desde la semana 20 hasta la semana 68 en los pacientes que cambiaron a placebo en la semana 20 (línea basal). Sin embargo, el peso corporal medio observado en la semana 68 fue inferior al del inicio de la fase de inducción (semana 0) (véase la figura 3). Los pacientes que recibieron semaglutida desde la semana 0 (fase de inducción) hasta la semana 68 (final del tratamiento) alcanzaron una reducción media del peso del -17,4 %; además, el 87,8 % de estos pacientes lograron una pérdida de peso ≥ 5 %, el 78,0 % ≥ 10 %, el 62,2 % ≥ 15 % y el 38,6 % ≥ 20 %.
Tabla 4. Estudio STEP 4: resultados desde la semana 20 hasta la semana 68
| Indicador |
Wegovy FlexTouch |
Placebo |
| Número de pacientes (N) |
535 |
268 |
| Peso corporal (kg) |
||
| Nivel basal1 (kg) |
96,5 |
95,4 |
| Cambio (%) desde el nivel basal1,2,3 |
-7,9 |
6,9 |
| Diferencia (%) respecto al placebo2 [IC del 95 %] |
-14,8 [-16,0; -13,5]* |
- |
| Cambio (kg) desde el nivel basal |
-7,1 |
6,1 |
| Diferencia (kg) respecto al placebo2 [IC del 95 %] |
-13,2 [-14,3; -12,0] |
- |
| Circunferencia de la cintura (cm) |
||
| Nivel basal |
105,5 |
104,7 |
| Cambio desde el nivel basal1 |
-6,4 |
3,3 |
| Diferencia respecto al placebo2 [IC del 95 %] |
-9,7 [-10,9; -8,5]* |
- |
| Presión arterial sistólica (mm Hg) |
||
| Nivel basal |
121 |
121 |
| Cambio desde el nivel basal1,2 |
0,5 |
4,4 |
| Diferencia respecto al placebo2 [IC del 95 %] |
-3,9 [-5,8; -2,0]* |
- |
* p < 0,0001 (no corregido bilateral) a favor.
1 Nivel basal = semana 20.
2 Evaluado utilizando un modelo ANCOVA con imputación múltiple basado en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros fármacos contra la obesidad o cirugía bariátrica.
3 Durante el estudio, el tratamiento fue definitivamente interrumpido en el 5,8 % y el 11,6 % de los pacientes aleatorizados a semaglutida 2,4 mg y placebo, respectivamente. Suponiendo que todos los pacientes aleatorizados continuaron con el tratamiento y no recibieron tratamiento adicional contra la obesidad, los cambios estimados desde la aleatorización hasta la semana 68 en la masa corporal, basados en un modelo mixto para mediciones repetidas que incluye todas las observaciones hasta la primera interrupción, fueron de -8,1 % y -6,5 % para semaglutida 2,4 mg y placebo, respectivamente.
**
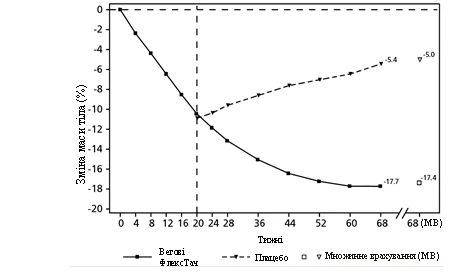
**
Valores observados en pacientes que completaron cada visita planificada y estimaciones con imputación múltiple (IM) de datos de pacientes que abandonaron el estudio.
Fig. 3. STEP 4: Cambio medio en la masa corporal (%) desde la semana 0 hasta la semana 68
Estudio STEP 5. Datos a 2 años
En un ensayo clínico doble ciego de 104 semanas de duración, 304 pacientes con obesidad (IMC ≥ 30 kg/m²) o con sobrepeso (IMC ≥ 27 kg/m² y < 30 kg/m²) y al menos una comorbilidad relacionada con el peso corporal fueron aleatorizados para recibir semaglutida o placebo. Todos los pacientes siguieron una dieta hipocalórica y aumentaron su actividad física durante todo el estudio. Al inicio, los pacientes tenían un IMC medio de 38,5 kg/m² y una masa corporal media de 106,0 kg.
El tratamiento con semaglutida durante 104 semanas produjo una reducción significativa y clínicamente relevante de la masa corporal en comparación con placebo. La masa corporal media disminuyó desde el nivel basal hasta la semana 68 con semaglutida, tras lo cual se alcanzó una meseta. Con placebo, la masa corporal media disminuyó menos y se alcanzó una meseta aproximadamente a las 20 semanas de tratamiento (ver tabla 5 y figura 4). Los pacientes que recibieron semaglutida lograron una reducción media de la masa corporal del -15,2 %; en el 74,7 % de estos pacientes la pérdida de masa corporal fue ≥ 5 %, en el 59,2 % fue ≥ 10 % y en el 49,7 % fue ≥ 15 %. El 80 % y el 37 % de los pacientes con prediabetes al inicio del tratamiento alcanzaron un estado glucémico normal al final del tratamiento con semaglutida y placebo, respectivamente.
Tabla 5. Estudio STEP 5: resultados en la semana 104
| Indicador |
Wegovy FlexTouch |
Placebo |
| Número de pacientes (N) |
152 |
152 |
| Peso corporal (kg) |
||
| Nivel basal (kg) |
105,6 |
106,5 |
| Cambio (%) respecto al nivel basal1,2 |
-15,2 |
-2,6 |
| Diferencia (%) respecto al placebo1 [95 % IC] |
-12,6 [-15,3; -9,8]* |
- |
| Cambio (kg) respecto al nivel basal |
-16,1 |
-3,2 |
| Diferencia (kg) respecto al placebo1 [95 % IC] |
-12,9 [-16,1; -9,8] |
- |
| Pacientes (%) que lograron una pérdida de peso ≥ 5 %3 |
74,7* |
37,3 |
| Pacientes (%) que lograron una pérdida de peso ≥ 10 %3 |
59,2* |
16,8 |
| Pacientes (%) que lograron una pérdida de peso ≥ 15 %3 |
49,7* |
9,2 |
| Circunferencia de la cintura (cm) |
||
| Nivel basal |
115,8 |
115,7 |
| Cambio respecto al nivel basal1 |
-14,4 |
-5,2 |
| Diferencia respecto al placebo1 [95 % IC] |
-9,2 [-12,2; -6,2]* |
- |
| Presión arterial sistólica (mm Hg) |
||
| Nivel basal |
126 |
125 |
| Cambio respecto al nivel basal1 |
-5,7 |
-1,6 |
| Diferencia respecto al placebo1 [95 % IC] |
-4,2 [-7,3; -1,0]* |
- |
* p < 0,0001 (bilateral no ajustado) a favor.
1 Evaluado utilizando un modelo ANCOVA con ajuste múltiple basado en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros fármacos contra la obesidad o cirugía bariátrica.
2 Durante el estudio, el tratamiento fue definitivamente interrumpido en el 13,2 % y el 27,0 % de los pacientes aleatorizados a semaglutida y placebo, respectivamente. Suponiendo que todos los pacientes aleatorizados continuaron el tratamiento y no recibieron tratamiento adicional contra la obesidad, los cambios estimados desde la aleatorización hasta la semana 68 en el peso corporal, basados en un modelo mixto para medidas repetidas, incluyendo todas las observaciones hasta la primera interrupción, fueron del -16,7 % y -0,6 % para semaglutida y placebo, respectivamente.
3 Evaluado mediante un modelo de regresión binaria basado en el mismo procedimiento de ajuste que en el análisis primario.
HTMLIMG3END
Valores observados en pacientes que completaron cada visita programada y estimaciones con ajuste múltiple (AM) de datos obtenidos de pacientes que abandonaron el estudio.
Fig. 4. STEP 5: Cambio medio en el peso corporal (%) desde la semana 0 hasta la semana 104
Estudio STEP 8. Comparación de semaglutida con liraglutida
En un estudio aleatorizado, abierto, controlado con placebo y de comparación directa, de 68 semanas de duración, 338 pacientes con obesidad (IMC ≥ 30 kg/m²) o con sobrepeso (IMC ≥ 27 kg/m² y < 30 kg/m²) y con al menos una comorbilidad relacionada con el peso corporal, fueron aleatorizados para recibir semaglutida una vez por semana, liraglutida 3 mg una vez al día o placebo. La administración de semaglutida una vez por semana y de liraglutida 3 mg fue abierta, pero cada grupo de tratamiento activo fue doblemente ciego respecto al placebo, que se administraba con la misma frecuencia de dosificación. En el momento basal, los pacientes tenían un IMC medio de 37,5 kg/m² y un peso corporal medio de 104,5 kg.
El tratamiento con semaglutida una vez por semana durante 68 semanas produjo una reducción significativa y clínicamente relevante del peso corporal en comparación con liraglutida. El peso corporal medio disminuyó desde el valor basal hasta la semana 68 con semaglutida. Con liraglutida, el peso corporal medio disminuyó con menor intensidad (ver Tabla 6). El 37,4 % de los pacientes que recibieron semaglutida perdieron ≥ 20 % del peso corporal, en comparación con el 7,0 % de los pacientes que recibieron liraglutida. En la Tabla 6 se muestran los resultados de los puntos finales confirmatorios de pérdida de peso corporal ≥ 10 %, ≥ 15 % y ≥ 20 %.
Tabla 6. Estudio STEP 8: Resultados en la semana 68 en la comparación de semaglutida y liraglutida
| Indicador |
Wegovy FlexTouch |
Liraglutida 3 mg |
| Número de pacientes (N) |
126 |
127 |
| Peso corporal (kg) |
||
| Nivel inicial (kg) |
102,5 |
103,7 |
| Cambio (%) respecto al nivel inicial1,2 |
-15,8 |
-6,4 |
| Diferencia (%) respecto a liraglutida1 [95 % IC] |
‑9,4 [‑12,0;‑6,8]* |
- |
| Cambio (kg) respecto al nivel inicial |
‑15,3 |
‑6,8 |
| Diferencia (kg) respecto a liraglutida1 [95 % IC] |
‑8,5 [‑11,2;‑5,7] |
- |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 10 %3 |
69,4* |
27,2 |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 15 %3 |
54,0* |
13,4 |
| Pacientes (%) que alcanzaron una pérdida de peso corporal ≥ 20 %3 |
37,4* |
7,0 |
* p < 0,0001 (no corregido bilateral) a favor.
1 Evaluado utilizando un modelo ANCOVA con ajuste múltiple basado en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros medicamentos contra la obesidad o cirugía bariátrica.
2 Durante el estudio, el tratamiento se interrumpió definitivamente en el 13,5 % y el 27,6 % de los pacientes aleatorizados a semaglutida y liraglutida, respectivamente. Suponiendo que todos los pacientes aleatorizados hubieran continuado el tratamiento y no hubieran recibido tratamiento adicional contra la obesidad, los cambios estimados desde la aleatorización hasta la semana 68 en el peso corporal, basados en un modelo mixto para medidas repetidas que incluye todas las observaciones hasta la primera interrupción, fueron del -16,7 % y -6,7 % para semaglutida y liraglutida, respectivamente.
3 Evaluado mediante un modelo de regresión binaria basado en el mismo procedimiento de ajuste que en el análisis primario.
Efecto sobre la composición corporal
En un subestudio de STEP 1 (N = 140), la composición corporal se midió mediante absorciometría de rayos X de doble energía (DEXA). Los resultados del análisis DEXA mostraron que el tratamiento con semaglutida se asoció con una mayor reducción de la masa grasa en comparación con la masa muscular, lo que condujo a una mejora en la composición corporal en comparación con placebo a las 68 semanas. Además, la reducción de la grasa corporal total se acompañó de una disminución de la grasa visceral. Estos resultados indican que la mayor parte de la pérdida total de peso estuvo relacionada con una reducción del tejido adiposo, incluida la grasa visceral.
Mejora del funcionamiento físico
La semaglutida mostró una ligera mejora en los índices de funcionamiento físico. Las funciones físicas se evaluaron mediante un cuestionario general de calidad de vida relacionada con la salud, el Short Form-36v2 Health Survey, Acute Version (SF-36), así como mediante el cuestionario Impact of Weight on Quality of Life Clinical Trials Version (IWQOL-Lite)-CT.
Evaluación cardiovascular
En el estudio SUSTAIN 6, 3297 pacientes con diabetes mellitus tipo 2 mal controlada y alto riesgo cardiovascular fueron aleatorizados para recibir semaglutida subcutánea a una dosis de 0,5 mg o 1 mg una vez por semana o placebo, además del tratamiento estándar. La duración del tratamiento fue de 104 semanas. La edad media de los pacientes fue de 65 años y el IMC medio fue de 33 kg/m².
El punto final primario fue el tiempo desde la aleatorización hasta la primera ocurrencia de un evento cardiovascular adverso mayor (MACE): muerte por causas cardiovasculares, infarto de miocardio no mortal o accidente cerebrovascular no mortal. La tasa total de MACE fue de 254, incluyendo 108 (6,6 %) con semaglutida y 146 (8,9 %) con placebo.
La seguridad cardiovascular del tratamiento con semaglutida 0,5 mg o 1 mg fue confirmada, ya que la razón de riesgos (RR) para semaglutida en comparación con placebo fue de 0,74 [0,58; 0,95] [IC del 95 %], lo que se debió a una reducción en la frecuencia de infarto de miocardio no mortal e ictus no mortal, sin diferencias en la frecuencia de muertes por causas cardiovasculares (ver figura 5).
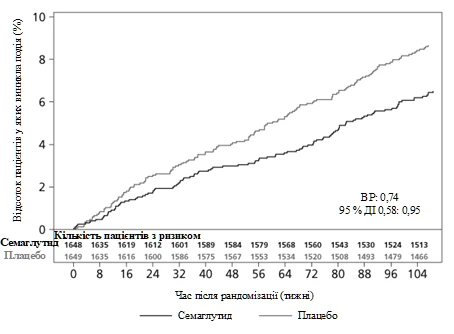
Fig. 5. Curva de Kaplan-Meier del tiempo hasta el primer evento compuesto: muerte por causas cardiovasculares, infarto de miocardio no mortal y accidente cerebrovascular no mortal (ensayo clínico SUSTAIN 6)
Pacientes pediátricos
La Agencia Europea de Medicamentos ha pospuesto la obligación de presentar los resultados de los estudios con el medicamento Wegovy FlexTouch en una o varias subgrupos de la población pediátrica para el control del peso corporal (ver la sección «Posología y forma de administración» para información sobre el uso en niños).
Estudio STEPTEENS: Control del peso corporal en pacientes de 12 a 18 años de edad
En un estudio doble ciego de 68 semanas, 201 adolescentes en pubertad de 12 a 18 años con obesidad o sobrepeso y al menos una comorbilidad relacionada con el peso corporal fueron aleatorizados en una proporción 2:1 para recibir semaglutida o placebo. Todos los pacientes siguieron una dieta hipocalórica y aumentaron su actividad física durante todo el estudio.
Al final del tratamiento (semana 68), la mejora del IMC con semaglutida fue mayor y clínicamente significativa en comparación con placebo (ver tabla 6 y figura 6). Además, una proporción mayor de pacientes alcanzó una pérdida de peso corporal ≥ 5 %, ≥ 10 % y ≥ 15 % con semaglutida en comparación con placebo (ver tabla 6).
Tabla 6. STEPTEENS: Resultados a la semana 68
| Indicador |
Wegovy FlexTouch |
Placebo |
| Número de pacientes (N) |
134 |
67 |
| IMC |
||
| Nivel basal (IMC) |
37,7 |
35,7 |
| Cambio (%) respecto al nivel basal1,2 |
-16,1 |
0,6 |
| Diferencia (%) respecto al placebo1 [95 % IC] |
-16,7 [-20,3; -13,2] |
- |
| Nivel basal (IMC EEE (índice de desviación estándar)) |
3,4 |
3,1 |
| Cambio respecto al nivel basal IMC EEE1 |
-1,1 |
-0,1 |
| Diferencia respecto al placebo1 [95 % IC] |
-1,0 [-1,3; -0,8] |
- |
| Masa corporal |
||
| Nivel basal (kg) |
109,9 |
102,6 |
| Cambio (%) respecto al nivel basal1 |
-14,7 |
2,8 |
| Diferencia (%) respecto al placebo1 [95 % IC] |
-17,4 [-21,1; -13,8] |
- |
| Cambio (kg) respecto al nivel basal1 |
-15,3 |
2,4 |
| Diferencia (kg) respecto al placebo1 [95 % IC] |
-17,7 [-21,8; -13,7] |
- |
| Pacientes (%) que alcanzaron una pérdida de masa corporal ≥ 5 %3 |
72,5* |
17,7 |
| Número de pacientes (N) |
134 |
67 |
| Pacientes (%) que alcanzaron una pérdida de masa corporal ≥ 10 %3 |
61,8 |
8,1 |
| Pacientes (%) que alcanzaron una pérdida de masa corporal ≥ 15 %3 |
53,4 |
4,8 |
| Circunferencia de la cintura (cm) |
||
| Nivel basal |
111,9 |
107,3 |
| Cambio respecto al nivel basal1 |
-12,7 |
-0,6 |
| Diferencia respecto al placebo1 [95 % IC] |
-12,1 [-15,6; -8,7]* |
- |
| Presión arterial sistólica (mm Hg) |
||
| Nivel basal |
120 |
120 |
| Cambio respecto al nivel basal1 |
-2,7 |
-0,8 |
| Diferencia respecto al placebo1 [95 % IC] |
-1,9 [-5,0; -1,1]* |
- |
*p < 0,0001 (bilateral sin ajustar) para la ventaja.
1 Evaluado utilizando un modelo ANCOVA con imputación múltiple basado en todos los datos, independientemente de la interrupción del tratamiento aleatorizado o del inicio de otros medicamentos contra la obesidad o cirugía bariátrica.
2 Durante el estudio, el tratamiento fue definitivamente interrumpido en el 10,4 % y el 10,4 % de los pacientes aleatorizados a semaglutida 2,4 mg y placebo, respectivamente. Suponiendo que todos los pacientes aleatorizados continuaron el tratamiento y no recibieron tratamiento adicional contra la obesidad, los cambios estimados desde la aleatorización hasta la semana 68 en el peso corporal, basados en un modelo mixto para medidas repetidas, incluyendo todas las observaciones hasta la primera interrupción, fueron de -17,9 % y -0,6 % para semaglutida 2,4 mg y placebo, respectivamente.
3 Evaluado mediante un modelo de regresión binaria basado en el mismo procedimiento de imputación que en el análisis primario.
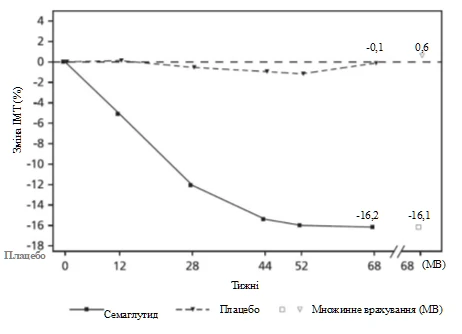
Valores observados para pacientes que completaron cada visita planificada y estimaciones con imputación múltiple (IM) de datos obtenidos de pacientes que abandonaron el estudio.
Fig. 6. STEP TEENS: Cambio medio del IMC (%) desde el valor basal hasta la semana 68
Farmacocinética.
Comparado con el GLP-1 nativo, la semaglutida presenta un periodo de semidesintegración prolongado de aproximadamente 1 semana, lo que permite su administración subcutánea una vez por semana. El mecanismo principal de esta acción prolongada es la unión a la albúmina, lo que reduce el aclaramiento renal y protege contra la degradación metabólica. Además, la semaglutida está estabilizada frente a la degradación por la enzima DPP-4.
Absorción
La concentración media en estado estacionario de semaglutida tras la administración subcutánea de la dosis de mantenimiento fue de aproximadamente 75 nmol/l en pacientes con sobrepeso (IMC ≥ 27 kg/m² hasta < 30 kg/m²) u obesidad (IMC ≥ 30 kg/m²), basado en datos de estudios de fase 3, donde el 90 % de los pacientes tuvieron concentraciones medias entre 51 nmol/l y 110 nmol/l. Con dosis de 0,25 mg a 2,4 mg administradas una vez por semana, la exposición en estado estacionario de semaglutida aumentó de forma dependiente de la dosis. La exposición en estado estacionario fue estable en el tiempo, como se determinó hasta la semana 68. Se alcanzó una exposición similar tras la administración subcutánea de semaglutida en el abdomen, muslo o hombro. La biodisponibilidad absoluta de semaglutida tras su administración subcutánea fue del 89 %.
Distribución
El volumen medio de distribución de semaglutida tras administración subcutánea en pacientes con sobrepeso u obesidad fue de aproximadamente 12,4 l. La semaglutida se une ampliamente a la albúmina plasmática (> 99 %).
Metabolismo/biotransformación
Antes de la excreción, la semaglutida se metaboliza activamente mediante escisión proteolítica del esqueleto peptídico de la proteína y posterior beta-oxidación de la cadena lateral de ácidos grasos. La enzima endopeptidasa neutra (NEP) se ha identificado como uno de los enzimas metabólicos activos.
Eliminación
La sustancia relacionada con semaglutida se elimina principalmente por orina y heces. Aproximadamente el 3 % de la dosis administrada se excreta por orina como semaglutida sin cambios. El aclaramiento de semaglutida en pacientes con sobrepeso (IMC ≥ 27 kg/m² hasta < 30 kg/m²) u obesidad (IMC ≥ 30 kg/m²) fue de aproximadamente 0,05 l/h. Con un periodo de semidesintegración de aproximadamente 1 semana, la semaglutida permanecerá en circulación durante aproximadamente 7 semanas tras la administración de la última dosis de 2,4 mg.
Grupos especiales de pacientes
Pacientes de edad avanzada
La edad del paciente no influye en la farmacocinética de semaglutida según datos de estudios de fase 3 que incluyeron pacientes de 18 a 86 años.
Sexo, raza y origen étnico
El sexo, la raza (caucásica, negra o asiática) y el origen étnico (hispano o latinoamericano, no hispano o no latinoamericano) del paciente no influyeron en la farmacocinética de semaglutida según datos de estudios de fase 3a.
Peso corporal
El peso corporal del paciente influye en la exposición a semaglutida. Un peso corporal más alto conduce a una exposición más baja del fármaco; una diferencia del 20 % en peso corporal entre pacientes dará lugar a casi un 18 % de diferencia en la exposición. La dosis de semaglutida de 2,4 mg una vez por semana proporciona una exposición sistémica adecuada en un rango de peso corporal de 54,4 a 245,6 kg, evaluado para la respuesta a la exposición durante los estudios clínicos.
Alteración de la función renal
La alteración de la función renal no tuvo un impacto clínicamente relevante en la farmacocinética de semaglutida. Esto se demostró tras la administración de una dosis única de 0,5 mg de semaglutida en pacientes con alteración renal de diversa gravedad (leve, moderada, grave y pacientes en diálisis) en comparación con pacientes con función renal normal. Esto también fue confirmado por datos de estudios clínicos de fase 3a en pacientes con sobrepeso (IMC ≥ 27 kg/m² hasta < 30 kg/m²) u obesidad (IMC ≥ 30 kg/m²) y con alteración renal de leve a moderada.
Alteración de la función hepática
La alteración de la función hepática no influyó en la exposición a semaglutida. La farmacocinética de semaglutida fue evaluada en pacientes con diversos grados de alteración hepática (leve, moderada, grave) en comparación con pacientes con función hepática normal en un estudio con dosis única de 0,5 mg de semaglutida.
Prediabetes y diabetes mellitus
La prediabetes y la diabetes mellitus no tuvieron ningún impacto clínicamente relevante en la exposición a semaglutida según datos de estudios de fase 3.
Inmunogenicidad
La formación de anticuerpos contra semaglutida durante el tratamiento con semaglutida fue infrecuente (ver sección «Reacciones adversas»), y la respuesta aparentemente no influyó en la farmacocinética de semaglutida.
Pacientes pediátricos
Las propiedades farmacocinéticas de semaglutida fueron evaluadas en un estudio clínico con pacientes adolescentes con obesidad o sobrepeso y al menos una comorbilidad relacionada con el peso corporal, de edades entre 12 y < 18 años (124 pacientes, peso corporal 61,6–211,9 kg). La exposición a semaglutida en niños fue similar a la observada en adultos con obesidad o sobrepeso.
La seguridad y eficacia de semaglutida en niños menores de 12 años no han sido estudiadas.
Datos preclínicos de seguridad
Los datos preclínicos, basados en estudios de seguridad farmacológica, toxicidad de dosis repetidas y genotoxicidad, no revelaron riesgo alguno para el ser humano.
Las neoplasias no letales derivadas de las células C de la glándula tiroides observadas en roedores pertenecen a efectos característicos de la clase de agonistas del receptor de GLP-1. En un estudio de carcinogenicidad de 2 años de duración en ratas y ratones, semaglutida indujo la aparición de tumores en las células C de la tiroides a niveles de exposición clínicamente relevantes. No se observaron otros tumores cuya aparición pudiera relacionarse con el tratamiento. Los tumores en células C en roedores están mediados por un mecanismo específico no genotóxico, dependiente del receptor de GLP-1, al que los roedores son parcialmente sensibles. La relevancia de este mecanismo en humanos es muy baja, aunque no puede excluirse completamente.
En estudios de fertilidad en ratas, semaglutida no afectó la eficacia del apareamiento ni la fertilidad en machos. En hembras de rata se observó un alargamiento del ciclo estral y una ligera reducción en el número de cuerpos amarillos (ovulación) a dosis que se asociaron con pérdida de peso corporal en hembras.
En estudios de desarrollo embrionario y fetal en ratas, semaglutida indujo efectos embriotóxicos a exposiciones inferiores a los niveles clínicamente relevantes. Semaglutida provocó una reducción notable del peso corporal en hembras y una disminución en la supervivencia y crecimiento de embriones. En fetos se observaron malformaciones esqueléticas y viscerales importantes, incluyendo alteraciones en huesos largos, costillas, columna vertebral, huesos de la cola, vasos sanguíneos y ventrículos cerebrales. Una evaluación mecanicista indicó que el efecto embriotóxico incluye una alteración mediada por receptores de GLP-1 en el suministro de nutrientes al embrión a través del saco vitelino en ratas. Debido a diferencias anatómicas y funcionales del saco vitelino entre especies y a la ausencia de expresión de receptores de GLP-1 en el saco vitelino de primates no humanos, este mecanismo se considera poco probable en humanos. Sin embargo, no puede descartarse la posibilidad de un efecto directo de semaglutida sobre el feto.
En estudios sobre toxicidad del desarrollo en conejos y macacos de Java, a niveles de exposición clínicamente relevantes, se observó un aumento en la frecuencia de pérdidas gestacionales y un ligero incremento en anomalías fetales. Estos resultados coincidieron con una pérdida notable de peso corporal en hembras, que alcanzó hasta un 16 %. No se sabe si estos efectos están relacionados con la reducción en la ingesta de alimento por parte de las hembras debido al efecto directo de GLP-1.
El crecimiento y desarrollo postnatal fueron evaluados en macacos de Java. Las crías nacieron ligeramente más pequeñas, pero su peso corporal se normalizó durante el período de lactancia.
En ratas jóvenes, semaglutida provocó un retraso en la maduración sexual tanto en machos como en hembras. Este retraso no afectó ni la fertilidad ni la capacidad reproductiva de ambos sexos, ni la capacidad de las hembras para mantener el embarazo.
Características clínicas.
Indicaciones
Adultos
El medicamento Vegovi FlexTouch está indicado para el control del peso corporal como complemento de una dieta hipocalórica y de un aumento de la actividad física en pacientes adultos con un índice de masa corporal (IMC) inicial
- superior a 30 kg/m² (obesidad) o
- entre 27 y 30 kg/m² (sobrepeso) con al menos una comorbilidad relacionada con el peso corporal, como disglucemia (prediabetes o diabetes tipo 2), hipertensión, dislipidemia, apnea obstructiva del sueño o enfermedad cardiovascular.
Niños ≥ 12 años
El medicamento Vegovi FlexTouch está indicado para el control del peso corporal como complemento de una dieta hipocalórica y de un aumento de la actividad física en niños a partir de 12 años con
- obesidad* y
- un peso corporal superior a 60 kg.
Debe suspenderse y reevaluarse el uso de Vegovi FlexTouch si en los niños el IMC no se reduce al menos en un 5 % tras 12 semanas de tratamiento con 2,4 mg o con la dosis máxima tolerada.
* Obesidad (IMC ≥ percentil 95), según se define en las tablas de crecimiento del IMC por sexo y edad (CDC.gov) (ver tabla 7).
Tabla 7. Valores umbral del IMC para obesidad (≥ percentil 95) por sexo y edad en niños a partir de 12 años (criterios CDC)
| Edad (años) |
IMC (kg/m2) en el percentil 95 |
|
| Sexo masculino |
Sexo femenino |
|
| 12 |
24,2 |
25,2 |
| 12,5 |
24,7 |
25,7 |
| 13 |
25,1 |
26,3 |
| 13,5 |
25,6 |
26,8 |
| 14 |
26,0 |
27,2 |
| 14,5 |
26,4 |
27,7 |
| 15 |
26,8 |
28,1 |
| 15,5 |
27,2 |
28,5 |
| 16 |
27,5 |
28,9 |
| 16,5 |
27,9 |
29,3 |
| 17 |
28,2 |
29,6 |
| 17,5 |
28,6 |
30,0 |
Contraindicaciones.
Hipersensibilidad a la sustancia activa o a alguno de los excipientes del medicamento (véase la sección «Composición»).
Interacción con otros medicamentos y otras formas de interacción.
El semaglutido retrasa el vaciamiento gástrico y potencialmente puede afectar la velocidad de absorción de medicamentos orales administrados simultáneamente. No se ha observado un efecto clínicamente relevante sobre la velocidad de vaciamiento gástrico tras la administración de semaglutido en una dosis de 2,4 mg, probablemente debido a un efecto de tolerancia. El semaglutido debe administrarse con precaución a pacientes que toman medicamentos orales que requieren una rápida absorción en el tracto gastrointestinal.
Paracetamol
Según los resultados del estudio de farmacocinética del paracetamol durante una prueba estandarizada con alimento, el semaglutido retrasa la velocidad de vaciamiento gástrico. Tras la administración concomitante de paracetamol y semaglutido en una dosis de 1 mg, los valores de AUC0-60 min y Cmax del paracetamol disminuyeron un 27 % y un 23 %, respectivamente. La exposición total al paracetamol (AUC0-5 h) no cambió. No se observó un efecto clínicamente relevante del semaglutido sobre el paracetamol. No se requiere ajuste de dosis cuando se administra paracetamol junto con semaglutido.
Anticonceptivos orales
No se espera que el semaglutido reduzca la eficacia de los anticonceptivos orales. Así, cuando se administró conjuntamente con un medicamento anticonceptivo oral combinado (etinilestradiol 0,03 mg/levonorgestrel 0,15 mg), el semaglutido no afectó clínicamente de forma significativa a la exposición total al etinilestradiol y al levonorgestrel. La exposición al etinilestradiol no se alteró; la exposición al levonorgestrel aumentó un 20 % en estado de equilibrio. La Cmax de ninguna de estas sustancias cambió.
Atorvastatina
El semaglutido no alteró la exposición total a la atorvastatina tras la administración de una dosis única de atorvastatina (40 mg). La Cmax de atorvastatina disminuyó un 38 %. Esto fue considerado clínicamente no significativo.
Digoxina
El semaglutido no alteró la exposición total ni la Cmax de digoxina tras la administración de una dosis única de digoxina (0,5 mg).
Metformina
El semaglutido no alteró la exposición total ni la Cmax de metformina tras la administración de metformina a una dosis de 500 mg dos veces al día durante 3,5 días.
Warfarina
El semaglutido no alteró la exposición total ni la Cmax de la warfarina R- y S-tras la administración de una dosis única de warfarina (25 mg), y los efectos farmacodinámicos de la warfarina, medidos mediante la razón normalizada internacional (INR), no cambiaron de forma clínicamente significativa. Sin embargo, al iniciar el tratamiento con semaglutido en pacientes que toman warfarina u otros derivados cumarínicos, se recomienda realizar un monitoreo frecuente del INR.
Pacientes pediátricos
Los estudios de interacción se han realizado únicamente con participación de pacientes adultos.
Características de uso.
Seguimiento
Con el fin de mejorar el seguimiento de los medicamentos biológicos, el nombre y número de lote del medicamento administrado deben registrarse claramente en el envase.
Deshidratación
La administración de agonistas del receptor GLP-1 puede asociarse con reacciones adversas gastrointestinales que podrían provocar deshidratación, lo cual en casos raros podría conducir a un deterioro de la función renal. Los pacientes deben ser advertidos sobre el riesgo potencial de deshidratación debido a reacciones adversas gastrointestinales y deben tomarse medidas para prevenir la deshidratación.
Pancreatitis aguda
Se han observado casos de pancreatitis aguda durante el tratamiento con agonistas del receptor GLP-1 (ver sección «Reacciones adversas»). Los pacientes deben informarse sobre los síntomas característicos de pancreatitis aguda. Si se sospecha pancreatitis, el tratamiento con semaglutida debe suspenderse; si se confirma pancreatitis, no se debe reanudar el tratamiento con semaglutida.
La semaglutida debe usarse con precaución en pacientes con antecedentes de pancreatitis.
En ausencia de otros signos y síntomas de pancreatitis aguda, el mero aumento de los niveles de enzimas pancreáticas no es un indicador predictivo de pancreatitis aguda.
Diabetes mellitus tipo 2
La semaglutida no debe usarse como sustituto de la insulina en pacientes con diabetes mellitus tipo 2.
La semaglutida no debe usarse en combinación con otros agonistas del receptor GLP-1. Esta combinación no ha sido evaluada, por lo que se considera probable un aumento del riesgo de reacciones adversas relacionadas con sobredosis.
Hipoglucemia en pacientes con diabetes mellitus tipo 2
Se sabe que la insulina y las sulfonilureas provocan hipoglucemia. En pacientes tratados con semaglutida en combinación con sulfonilurea o insulina, puede aumentar el riesgo de hipoglucemia. El riesgo de hipoglucemia puede reducirse disminuyendo la dosis de sulfonilurea o insulina al iniciar el tratamiento con un agonista del receptor GLP-1. No se ha evaluado la adición del medicamento Wegovy FlexTouch al tratamiento de pacientes que reciben insulina.
Retinopatía diabética en pacientes con diabetes mellitus tipo 2
En pacientes con retinopatía diabética que recibieron semaglutida, se observó un riesgo aumentado de complicaciones de la retinopatía diabética (ver sección «Reacciones adversas»). La rápida mejora en el control de la glucosa se ha asociado con un empeoramiento temporal de la retinopatía diabética, aunque no puede descartarse la posibilidad de otros mecanismos de acción. Los pacientes con retinopatía diabética deben ser cuidadosamente monitoreados y tratados de acuerdo con las recomendaciones clínicas. No existe experiencia en el uso del medicamento Wegovy FlexTouch en pacientes con diabetes mellitus tipo 2 que presenten retinopatía diabética descontrolada o potencialmente inestable. No se recomienda el uso de Wegovy FlexTouch en estos pacientes.
Poblaciones no estudiadas
No se han estudiado la seguridad y eficacia del medicamento Wegovy FlexTouch en pacientes:
- que utilizan otros medicamentos para el control del peso corporal,
– con diabetes mellitus tipo 1,
- con insuficiencia renal grave (ver sección «Instrucciones de uso y dosis»),
- con insuficiencia hepática grave (ver sección «Instrucciones de uso y dosis»),
- con insuficiencia cardíaca congestiva clase IV según la clasificación de la New York Heart Association (NYHA).
No se recomienda su uso en estos pacientes.
Existe una experiencia limitada con el medicamento Wegovy FlexTouch en pacientes:
- de 75 años o más (ver sección «Instrucciones de uso y dosis»),
- con disfunción hepática leve o moderada (ver sección «Instrucciones de uso y dosis»),
– con enfermedades inflamatorias intestinales,
- con gastroparesia diabética.
En estos pacientes, el medicamento Wegovy FlexTouch debe usarse con precaución.
Contenido de sodio
Este medicamento contiene menos de 1 mmol de sodio (23 mg)/dosis, por lo que puede considerarse prácticamente exento de sodio.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil
Se recomienda a las mujeres en edad fértil el uso de métodos anticonceptivos durante el tratamiento con semaglutida (ver sección «Interacción con otros medicamentos e interacciones de otros tipos»).
Embarazo
Los estudios en animales han demostrado toxicidad reproductiva (ver sección «Datos preclínicos de seguridad»). Los datos sobre el uso de semaglutida en mujeres embarazadas son limitados. Por tanto, no debe usarse semaglutida durante el embarazo. Si una paciente planea quedar embarazada o ya está embarazada, el tratamiento con semaglutida debe suspenderse. Debido al largo periodo de semivida de la semaglutida, su uso debe suspenderse al menos 2 meses antes de una concepción planeada (ver sección «Farmacocinética»).
Lactancia
Durante la lactancia en ratas, la semaglutida se excreta en la leche. No puede descartarse el riesgo para el lactante. No debe usarse semaglutida durante el periodo de lactancia.
Fertilidad
El efecto de la semaglutida sobre la fertilidad en humanos es desconocido. El uso de semaglutida en ratas macho no afectó la fertilidad. En ratas hembra se observó alargamiento del ciclo estral y reducción del número de ovulaciones a dosis asociadas con pérdida de peso corporal.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
La semaglutida no afecta o afecta mínimamente la capacidad para conducir vehículos o manejar maquinaria. Sin embargo, puede presentarse mareo, principalmente durante el periodo de aumento de dosis. Si aparece mareo, se debe tener precaución al conducir vehículos o manejar maquinaria.
Pacientes con diabetes mellitus tipo 2
Al usar semaglutida en combinación con sulfonilurea o insulina, se debe recomendar a los pacientes adoptar medidas de seguridad para prevenir la hipoglucemia durante la conducción de vehículos o el manejo de maquinaria (ver sección «Características de uso»).
Vía de administración y dosis.
Dosificación
Adultos
La dosis de mantenimiento de semaglutida de 2,4 mg una vez por semana se alcanza comenzando con una dosis de 0,25 mg. Para reducir la probabilidad de reacciones adversas gastrointestinales, la dosis debe aumentarse progresivamente durante un período de 16 semanas hasta alcanzar la dosis de mantenimiento de 2,4 mg una vez por semana (ver tabla 8). Si aparecen reacciones gastrointestinales significativas, se deberá considerar posponer el aumento de la dosis o reducirla a la dosis previa hasta que mejore el estado del paciente. No se recomiendan dosis superiores a 2,4 mg por semana.
Tabla 8. Esquema de aumento de la dosis
| Aumento de la dosis |
Dosis semanal |
| Semana 1–4 |
0,25 mg |
| Semana 5–8 |
0,5 mg |
| Semana 9–12 |
1 mg |
| Semana 13–16 |
1,7 mg |
| Dosis de mantenimiento |
2,4 mg |
Niños
En niños a partir de 12 años de edad, debe aplicarse el mismo esquema de aumento de dosis que en adultos (véase la tabla 8). La dosis debe aumentarse hasta 2,4 mg (dosis de mantenimiento) o hasta alcanzar la dosis máxima tolerada. No se recomiendan dosis semanales superiores a 2,4 mg.
Pacientes con diabetes mellitus tipo 2
Al iniciar el tratamiento con semaglutida en pacientes con diabetes mellitus tipo 2, debe considerarse la posibilidad de reducir la dosis de insulina administrada simultáneamente o de los estimuladores de la secreción de insulina (como las sulfonilureas), con el fin de reducir el riesgo de hipoglucemia; véase la sección «Precauciones de uso».
Dosis olvidada
Si se olvida una dosis, debe administrarse tan pronto como sea posible dentro de los 5 días siguientes al olvido. Si ya han pasado más de 5 días, se debe omitir la dosis no administrada y administrar la siguiente dosis en el día programado. De esta manera, en cada caso, los pacientes pueden mantener su esquema de administración regular del medicamento una vez por semana. Si se han olvidado varias dosis, debe considerarse la posibilidad de reducir la dosis inicial al reiniciar el tratamiento.
Grupos de pacientes especiales
Pacientes de edad avanzada (más de 65 años)
No se requiere ajuste de dosis según la edad del paciente. La experiencia en el tratamiento de pacientes con ≥ 75 años de edad es limitada, y no puede descartarse una mayor sensibilidad en algunas personas de edad avanzada.
Alteración de la función renal
No se requiere ajuste de dosis en pacientes con alteración renal leve o moderada. La experiencia con semaglutida en pacientes con alteración renal grave es limitada. No se recomienda el uso de semaglutida en pacientes con alteración renal grave (TFG < 30 ml/min/1,73 m²), incluidos pacientes con enfermedad renal en fase terminal (véanse las secciones «Precauciones de uso», «Reacciones adversas» y «Farmacocinética»).
Alteración de la función hepática
No se requiere ajuste de dosis en pacientes con alteración hepática leve o moderada. La experiencia con semaglutida en pacientes con alteración hepática grave es limitada. No se recomienda el uso de semaglutida en pacientes con alteración hepática grave; en pacientes con alteración hepática leve o moderada debe administrarse con precaución (véanse las secciones «Precauciones de uso» y «Farmacocinética»).
Vía de administración
Administración subcutánea
Wegovy FlexTouch se administra una vez por semana, en cualquier momento del día, independientemente de la ingestión de alimentos.
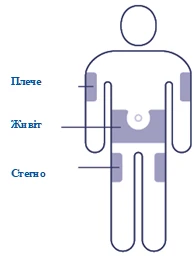
El medicamento se administra por vía subcutánea en la región del abdomen anterior, el muslo o el hombro. El sitio de inyección puede alternarse. No debe administrarse el medicamento por vía intravenosa ni intramuscular.
Si es necesario, puede cambiarse el día semanal de administración del medicamento, siempre que el intervalo entre dosis sea de al menos 3 días (> 72 horas). Tras elegir un nuevo día de administración, debe continuarse el tratamiento según el esquema de una vez por semana.
Al administrar una dosis única con el dispositivo inyectable precargado Wegovy FlexTouch, debe presionar firmemente el dispositivo contra la piel hasta que la franja amarilla deje de moverse. La inyección dura aproximadamente entre 5 y 10 segundos.
Antes de usar el medicamento, debe aconsejarse al paciente que lea cuidadosamente las instrucciones de uso del dispositivo inyectable Wegovy FlexTouch, incluidas en el prospecto.
Para obtener información adicional antes de la administración, véase la sección «Instrucciones de uso del dispositivo inyectable».
| Instrucciones para el uso del bolígrafo inyector Wegovy FlexTouch |
|||
| Antes de comenzar a usar el bolígrafo inyector Wegovy FlexTouch una vez por semana, lea cuidadosamente estas instrucciones siempre y hable con su médico, enfermera o farmacéutico sobre cómo administrar correctamente Wegovy FlexTouch. Wegovy FlexTouch es un bolígrafo inyector precargado que contiene cuatro dosis prescritas, lo que equivale a cuatro usos semanales. Por favor, utilice la tabla situada en el interior de la tapa de la caja de cartón para llevar la cuenta del número de dosis que ha utilizado y del número de dosis que le quedan en su bolígrafo inyector. Wegovy FlexTouch se suministra en cinco tipos diferentes de bolígrafos inyectores, cada uno conteniendo una de las siguientes dosis de semaglutida:
Compruebe siempre la etiqueta de su bolígrafo inyector para asegurarse de que contiene la dosis prescrita de Wegovy FlexTouch El bolígrafo está diseñado para usarse con agujas desechables de 30G, 31G y 32G, con una longitud máxima de 8 mm. El envase contiene:
|
|||
|
|
|||
|
|||
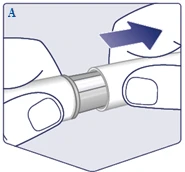
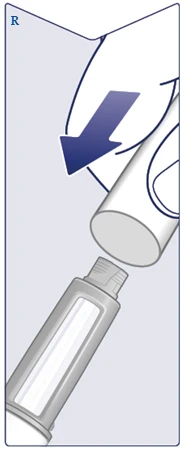
| Compruebe el nombre y la dosis de su bolígrafo inyector para asegurarse de que contiene la dosis prescrita del medicamento Wegovy FlexTouch. Retire la tapa del bolígrafo inyector. (Véase la figura A). |
|
||
| Asegúrese de que la solución en el bolígrafo inyector es transparente e incolora. Mire a través de la ventana del bolígrafo inyector. Si la solución está turbia o coloreada, no debe usar el bolígrafo inyector. (Véase la figura V). |
|
||
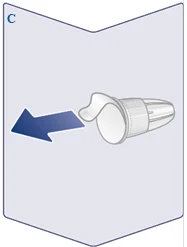
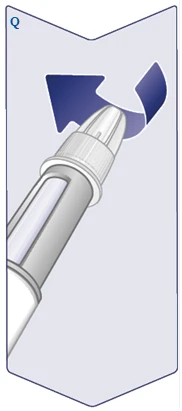
| Utilice siempre una aguja nueva para cada inyección. Coja una aguja cuando esté listo para administrar la inyección. Compruebe que la membrana de papel y la tapa externa de la aguja no estén dañadas, ya que esto podría afectar a la esterilidad. Si hay algún daño, utilice una nueva aguja. Retire la membrana de papel. (Véase la figura C). |
|
||
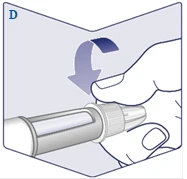
| Enrosque la aguja en el bolígrafo inyector. Gire la aguja hasta que quede firmemente sujeta al bolígrafo. (Véase la figura D). |
|
||
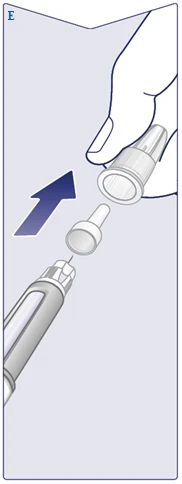
| La aguja está cubierta por dos tapas. Debe retirar ambas tapas. Si olvida retirar ambas tapas, no podrá administrar Wegovy FlexTouch. Retire la tapa externa de la aguja y guárdela. Le será necesaria después de completar la inyección para retirar la aguja del bolígrafo inyector de forma segura. Retire la tapa interna de la aguja y deséchela. Puede aparecer una gota de solución en la punta de la aguja. Sin embargo, es necesario comprobar el flujo al usar un nuevo bolígrafo inyector por primera vez. Véase el apartado «Comprobación del funcionamiento de cada nuevo bolígrafo inyector». Nunca use una aguja doblada o dañada. Para obtener más información sobre el manejo de las agujas, consulte el apartado «Sobre sus agujas» al final de estas instrucciones. (Véase la figura E). |
|
||
| Comprobación del funcionamiento de cada nuevo bolígrafo inyector |
|||
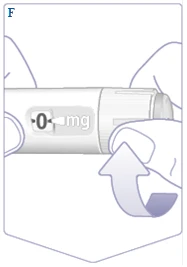
| Si ya ha usado anteriormente el bolígrafo inyector Wegovy FlexTouch, pase al apartado 2. «Ajuste de la dosis». Solo antes del primer uso de cada nuevo bolígrafo inyector Wegovy FlexTouch, compruebe el flujo. Gire el selector de dosis hasta que la ventana de dosificación muestre el símbolo de comprobación de flujo (). (Véase la figura F). |
|
||
| Asegúrese de que el símbolo de comprobación de flujo coincide con el indicador de dosis. (Véase la figura G). |
|
||
| Comprobación del flujo |
|||
| Sujete el bolígrafo inyector con la aguja hacia arriba. Presione y mantenga pulsado el botón de liberación hasta que la ventana de dosificación regrese al símbolo . Asegúrese de que el símbolo coincida con el indicador de dosis. Debe aparecer una gota de solución en la punta de la aguja. Esta gota indica que su bolígrafo inyector está listo para su uso. Si no aparece la gota, repita la comprobación del flujo. Esto solo debe hacerse dos veces como máximo. Si aún no aparece la gota, reemplace la aguja y repita la comprobación del flujo. No utilice el bolígrafo inyector si no aparece la gota de solución. (Véase la figura H). |
|
||
|
|||
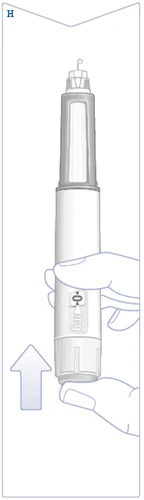
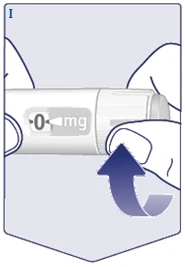
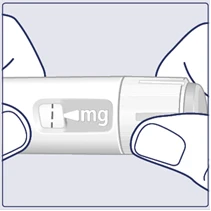
| Gire el selector de dosis hasta que la ventana de dosificación se detenga y muestre la dosis prescrita. (Véase la figura I). |
|
||
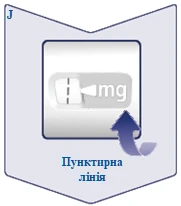
| La línea discontinua () en la ventana de dosificación ayuda a identificar la dosis. El selector de dosis hace clic al girarlo hacia adelante, hacia atrás o después de la dosis. Escuchará un clic cada vez que gire el selector. No intente ajustar la dosis contando los clics. (Véase la figura J). |
|
||
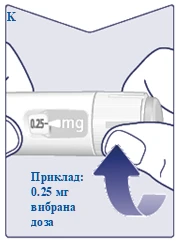
| Cuando la dosis prescrita coincida con el indicador de dosis, habrá seleccionado su dosis. En esta ilustración, la dosis se muestra como ejemplo. Si la ventana de dosificación se detiene antes de alcanzar la dosis prescrita, consulte el apartado «¿Tiene suficiente Wegovy FlexTouch?» al final de estas instrucciones. (Véase la figura K). |
|
||
| Seleccione el lugar de inyección Seleccione un lugar en el hombro, muslo o abdomen (manteniéndose al menos a 5 cm del ombligo). Puede administrar la inyección en la misma zona del cuerpo cada semana, pero asegúrese de que no sea exactamente en el mismo punto que la vez anterior. |
|
||
|
|||
| Introduzca la aguja bajo la piel. Asegúrese de que puede ver la ventana de dosificación. No tape la ventana con los dedos, ya que podría interrumpir la inyección. (Véase la figura L). |
|
||
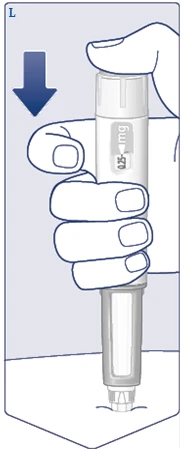
| Presione y mantenga pulsado el botón de liberación hasta que la ventana de dosificación muestre . (Véase la figura M). Mantenga la aguja bajo la piel después de que la ventana de dosificación regrese a 0 y cuenta lentamente hasta 6. debe coincidir con el indicador de dosis. Luego puede escuchar o sentir un clic cuando la ventana de dosificación regrese a . (Véase la figura N). |
|
||
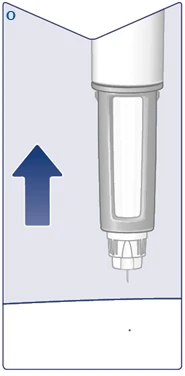
| Retire la aguja de la piel. Si retira la aguja demasiado pronto, podría ver que la solución gotea desde la punta de la aguja y no se administrará la dosis completa. Si aparece una gota de sangre en el lugar de inyección, presione suavemente sobre el área para detener el sangrado. Puede ver una gota de solución en la punta de la aguja después de la inyección. Esto es normal y no afecta al volumen de la dosis administrada. (Véase la figura O). |
|
||
|
|||
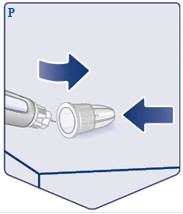
| Coloque la tapa externa de la aguja sobre una superficie plana y coloque la tapa externa sobre la aguja, sin tocar la aguja ni la tapa externa. Una vez que la aguja esté cubierta, presione firmemente sobre la tapa externa de la aguja. (Véase la figura P). |
|
||
| Desenrosque la aguja y desecharla adecuadamente según las indicaciones de su médico, enfermera, farmacéutico o autoridades locales. Nunca intente volver a colocar la tapa interna sobre la aguja. Podría pincharse con la aguja. Retire siempre la aguja del bolígrafo inmediatamente después de cada inyección para evitar obstrucciones, contaminación, infección y errores en la dosificación. Nunca guarde el bolígrafo con la aguja colocada. (Véase la figura Q). |
|
||
| Cubra el bolígrafo inyector con su tapa después de cada uso para proteger la solución de la luz. (Véase la figura R). |
|
||
| Cuando el bolígrafo inyector esté vacío, desecharlo sin la aguja según las indicaciones de su médico, enfermera, farmacéutico o autoridades locales. La tapa del bolígrafo y la caja vacía pueden desecharse junto con los residuos domésticos. |
|||
| Sobre sus agujas |
|||
| Cómo identificar una aguja obstruida o dañada
Qué hacer si la aguja está obstruida
|
|||
| Cuidado de su bolígrafo inyector |
|||
| Trate su bolígrafo inyector con cuidado. Un manejo inadecuado o un uso incorrecto podría provocar la administración de una dosis incorrecta. Si esto ocurre, podría no obtener el efecto deseado del medicamento.
|
|||
| ¿Tiene suficiente Wegovy FlexTouch? |
|||
| Si la ventana de dosificación se detiene antes de alcanzar la dosis prescrita, no habrá suficiente solución para administrar la dosis completa. Deseche este bolígrafo inyector y use un nuevo bolígrafo inyector Wegovy FlexTouch. |
|
||
| Información importante |
|||
|
|||








Niños
No se requiere ajuste de dosis para niños a partir de los 12 años de edad. La seguridad y eficacia del uso de semaglutida en niños menores de 12 años no han sido establecidas.
Sobredosis
La sobredosis de semaglutida puede estar asociada con trastornos gastrointestinales, los cuales pueden provocar deshidratación. En caso de sobredosis, se debe observar al paciente en busca de signos clínicos y comenzar el tratamiento de soporte adecuado.
Reacciones adversas.
Resumen del perfil de seguridad
En cuatro estudios clínicos de fase 3, 2650 pacientes recibieron el medicamento Vegov FlexTach. La duración de los estudios fue de 68 semanas. Las reacciones adversas más frecuentemente notificadas fueron las relacionadas con el tracto gastrointestinal, incluyendo náuseas, diarrea, estreñimiento y vómitos.
Reacciones adversas
A continuación se indican las reacciones adversas detectadas en estudios clínicos de fase 3a. La frecuencia de aparición de las reacciones adversas se determinó según los datos de los estudios clínicos de fase 3a.
Las reacciones adversas se clasifican por sistemas orgánicos y frecuencia de aparición. La evaluación de la frecuencia de las reacciones adversas se realizó según la siguiente escala: muy frecuente (≥ 1/10), frecuente (de ≥ 1/100 a < 1/10), poco frecuente (de ≥ 1/1000 a < 1/100), rara (de ≥ 1/10000 a < 1/1000), muy rara (< 1/10000).
Trastornos del sistema inmunitario: rara – reacción anafiláctica.
Trastornos del metabolismo y de la nutrición: frecuente – hipoglucemia en pacientes con diabetes mellitus tipo 2a.
Trastornos del sistema nervioso: muy frecuente – cefaleab; frecuente – mareob.
Trastornos visuales: frecuente – retinopatía diabética en pacientes con diabetes mellitus tipo 2a.
Trastornos cardíacos: poco frecuente – hipotensión, hipotensión ortostática, frecuencia cardíaca aumentadaa,c.
Trastornos gastrointestinales: muy frecuente – vómitosa,b, diarreaa,b, estreñimientoa,b, náuseasa,b, dolor abdominalb,c; frecuente – gastritisb,c, enfermedad por reflujo gastroesofágicob, dispepsiab, eructosb, flatulenciab, meteorismob, distensión abdominalb; poco frecuente – pancreatitis agudaa, retardo del vaciamiento gástrico.
Trastornos del hígado y de las vías biliares: frecuente – litiasis biliara.
Trastornos de la piel y del tejido subcutáneo: frecuente – caída del cabelloa; rara – angioedema.
Trastornos generales y alteraciones en el lugar de administración: muy frecuente – fatigab,c; frecuente – reacciones en el lugar de inyecciónc.
Análisis de laboratorio: poco frecuente – aumento del nivel de amilasa, aumento del nivel de lipasac.
a Descripción de reacciones adversas individuales indicada más abajo.
b Principalmente observado durante el período de aumento de la dosis.
c Términos agrupados de prioridad.
Descripción de reacciones adversas individuales
Reacciones adversas gastrointestinales
Durante estudios de 68 semanas de duración, las náuseas ocurrieron en el 43,9 % de los pacientes tratados con semaglutida (en el 16,1 % de los que recibieron placebo), diarrea en el 29,7 % (en el 15,9 % de los que recibieron placebo) y vómitos en el 24,5 % (en el 6,3 % de los que recibieron placebo). La mayoría de los casos fueron de intensidad leve a moderada y de corta duración. El estreñimiento ocurrió en el 24,2 % de los pacientes que recibieron semaglutida (en el 11,1 % en el grupo placebo) y fue de intensidad leve o moderada, pero más prolongado. En los pacientes que recibieron semaglutida, la duración media de las náuseas fue de 8 días, de los vómitos de 2 días, de la diarrea de 3 días y del estreñimiento de 47 días.
Los pacientes con disfunción renal moderada (TFG ≥ 30 ml/min/1,73 m²) pueden experimentar efectos gastrointestinales más intensos durante el tratamiento con semaglutida.
Las reacciones gastrointestinales llevaron a la interrupción definitiva del tratamiento en el 4,3 % de los pacientes.
Pancreatitis aguda
La frecuencia de casos de pancreatitis aguda confirmada por expertos durante los estudios clínicos de fase 3a fue del 0,2 % con semaglutida y < 0,1 % con placebo.
Enfermedad biliar aguda/litiasis biliar
La litiasis biliar se registró en el 1,6 % de los pacientes y condujo a colecistitis en el 0,6 % de los pacientes que recibieron semaglutida. La litiasis biliar y la colecistitis se observaron en el 1,1 % y 0,3 % respectivamente de los pacientes que recibieron placebo.
Caída del cabello
Se informó de caída del cabello en el 2,5 % de los pacientes que recibieron semaglutida y en el 1,0 % de los que recibieron placebo. La reacción fue principalmente de intensidad leve, y la mayoría de los pacientes se recuperaron durante la continuación del tratamiento. La caída del cabello fue más frecuente en pacientes con mayor pérdida de peso corporal (≥ 20 %).
Aumento de la frecuencia cardíaca
En estudios clínicos de fase 3a, en pacientes que recibieron semaglutida se observó un aumento medio de la frecuencia cardíaca de 3 latidos por minuto (lpm) respecto al valor medio basal de 72 lpm. La proporción de pacientes con un aumento de la frecuencia cardíaca de ≥ 10 lpm respecto al nivel basal en cualquier momento durante el período de tratamiento fue del 67,0 % en el grupo de semaglutida frente al 50,1 % en el grupo placebo.
Inmunogenicidad
Debido a las propiedades inmunogénicas potenciales de los medicamentos que contienen proteínas o péptidos, pueden formarse anticuerpos en pacientes tratados con semaglutida. El porcentaje de pacientes con resultados positivos en el análisis de anticuerpos contra semaglutida en cualquier momento tras el inicio del tratamiento fue bajo (2,9 %), y ningún paciente presentó anticuerpos neutralizantes contra semaglutida ni anticuerpos con efecto neutralizante sobre el GLP-1 al finalizar los estudios clínicos. Durante el tratamiento, altas concentraciones de semaglutida podrían haber reducido la sensibilidad de los análisis, por lo que no puede descartarse el riesgo de resultados falsos negativos. Sin embargo, en pacientes con resultados positivos en la prueba de anticuerpos durante y tras el tratamiento, la presencia de anticuerpos fue temporal y sin impacto aparente sobre la eficacia y seguridad.
Hipoglucemia en pacientes con diabetes mellitus tipo 2
En el estudio STEP 2, la hipoglucemia clínicamente significativa se observó en el 6,2 % (0,1 casos por paciente-año) de los pacientes que recibieron semaglutida, en comparación con el 2,5 % (0,03 casos por paciente-año) de los que recibieron placebo. La hipoglucemia con semaglutida se observó tanto con como sin tratamiento concomitante con sulfonilureas. Un episodio (0,2 % de los pacientes, 0,002 casos por paciente-año) fue registrado como grave en un paciente que no recibía sulfonilurea simultáneamente. El riesgo de hipoglucemia aumentó con el uso de semaglutida junto con sulfonilureas.
Complicaciones de la retinopatía diabética
En un estudio clínico de 2 años de duración sobre semaglutida 0,5 mg y 1 mg frente a placebo, participaron 3297 pacientes con diabetes mellitus tipo 2, alto riesgo cardiovascular, larga evolución de la diabetes y control insuficiente de la glucemia. En este estudio, las reacciones de empeoramiento de la retinopatía diabética ocurrieron en un mayor número de pacientes que recibieron semaglutida (3,0 %) que en aquellos que recibieron placebo (1,8 %). Esto se observó en pacientes con retinopatía diabética confirmada que recibían insulina. La diferencia entre los regímenes de tratamiento apareció temprano y se mantuvo durante todo el estudio. En el estudio STEP 2, las enfermedades de la retina se notificaron en el 6,9 % de los pacientes que recibieron Wegovy FlexTouch, en el 6,2 % de los que recibieron semaglutida 1 mg y en el 4,2 % de los que recibieron placebo. La mayoría de los casos se registraron como retinopatía diabética (4,0 %, 2,7 % y 2,7 % respectivamente) y retinopatía no proliferativa (0,7 %, 0 % y 0 % respectivamente).
Población pediátrica
En un estudio clínico realizado con niños de 12 a 18 años con obesidad o sobrepeso y al menos una comorbilidad relacionada con el peso corporal, 133 pacientes recibieron Wegovy FlexTouch. La duración del ensayo fue de 68 semanas.
En general, la frecuencia, tipo y gravedad de las reacciones adversas en niños fueron similares a las de los adultos. La litiasis biliar se registró en el 3,8 % de los pacientes que recibieron Wegovy FlexTouch y en el 0 % de los que recibieron placebo.
Tras 68 semanas de tratamiento, no se observó impacto sobre el crecimiento ni sobre el desarrollo puberal.
Notificación de reacciones adversas
La notificación de reacciones adversas tras la comercialización del medicamento es importante. Permite realizar el seguimiento continuo de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar de todos los casos sospechosos de reacciones adversas y de falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en el enlace: https://aisf.dec.gov.ua.
Período de validez. 3 años.
Después de la primera utilización: 6 semanas. Conservar a una temperatura inferior a 30 °C o en nevera (a 2 °C – 8 °C).
Condiciones de conservación.
Conservar en nevera (2 °C – 8 °C). Mantener alejado de elementos congelantes. No congelar.
Conservar la pluma precargada con la tapa puesta para protegerla de la luz.
Mantener fuera del alcance y de la vista de los niños.
Incompatibilidades.
Este medicamento no debe mezclarse con otros medicamentos, ya que no se han realizado estudios de compatibilidad.
Presentación.
0,25, 0,5 mg
Cartucho de vidrio (vidrio tipo I) de 1,5 ml de capacidad, cerrado por un lado con un émbolo de goma (clorobutilo) y por el otro con una tapa de aluminio con un revestimiento de goma laminada (bromobutilo/políisopreno). El cartucho está insertado en una pluma precargada desechable fabricada con polipropileno, polioximetileno, policarbonato y acrilonitrilo-butadieno-estireno.
1 pluma precargada y 4 agujas desechables NovoFine® Plus en una caja de cartón.
1 mg, 1,7 mg
Cartucho de vidrio (vidrio tipo I) de 3 ml de capacidad, cerrado por un lado con un émbolo de goma (clorobutilo) y por el otro con una tapa de aluminio con un revestimiento de goma laminada (bromobutilo/políisopreno). El cartucho está insertado en una pluma precargada desechable fabricada con polipropileno, polioximetileno, policarbonato y acrilonitrilo-butadieno-estireno.
1 pluma precargada y 4 agujas desechables NovoFine® Plus en una caja de cartón.
2,4 mg
Cartucho de vidrio (vidrio tipo I) de 3 ml de capacidad, cerrado por un lado con un émbolo de goma (clorobutilo) y por el otro con una tapa de aluminio con un revestimiento de goma laminada (bromobutilo/políisopreno). El cartucho está insertado en una pluma precargada desechable fabricada con polipropileno, polioximetileno, policarbonato y acrilonitrilo-butadieno-estireno.
1 o 3 plumas precargadas y 4 o 12 agujas desechables NovoFine® Plus en una caja de cartón.
Categoría de dispensación. Bajo receta médica.
Titular del registro/Producto.
A/S Novo Nordisk.
Domicilio del titular/productor y dirección del lugar de actividad.
Novo Allé
2880, Bagsværd
Dinamarca.