Polayvi®
UkrainaSpis treści
INSTRUKCJA LEKU DO STOSOWANIA U KLINICZNEGO Polayvi® (Polivy®)
Skład:
substancja czynna: polatuzumab vedotin;
1 butelka zawiera 140 mg polatuzumabu vedotinu;
1 butelka zawiera 30 mg polatuzumabu vedotinu;
1 ml sporządzonego (odnowionego) roztworu zawiera 20 mg/ml polatuzumabu vedotinu;
substancje pomocnicze: kwas bursztynowy, wodorotlenek sodu, sacharoza, polisorbat 20.
Postać leku. Proszek do koncentratu do roztworu do infuzji.
Główne właściwości fizyko-chemiczne: masa liofilizowana od białej do szarawo-białej.
Grupa farmakoterapeutyczna. Środki przeciwnowotworowe i immunomodulujące. Środki przeciwnowotworowe. Przeciwciała monoklonalne i koniugaty przeciwciał z lekiem. Inne przeciwciała monoklonalne i koniugaty przeciwciał z lekiem.
Kod ATC L01FX14.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Polatuzumab vedotin to koniugat przeciwciało-lek, zawierający środek antymitotyczny monometylan aurystatyny E (MMAE), który jest kowalencyjnie przyłączony do monoklonalnego przeciwciała (rekombinowanego humanizowanego immunoglobuliny G1 [IgG1]), skierowanego przeciwko CD79b. Monoklonalne przeciwciała uzyskuje się metodą rekombinacji DNA w komórkach jajników chomika chińskiego. Monoklonalne przeciwciało wiąże się z wysokim powinowactwem i selektywnością z CD79b, składnikiem receptora komórek B na powierzchni komórek. Ekspresja CD79b ograniczona jest do normalnych komórek linii różnicowania komórek B (z wyjątkiem komórek plazmatycznych) oraz do złośliwych komórek B. Ekspresja CD79b występuje u > 95 % chorych z rozsianymi dużokomórkowymi limfoma B. Po związaniu z CD79b polatuzumab vedotin szybko ulega internaryzacji, a linker jest rozszczepiany przez proteazy lizosomalne, co umożliwia dostarczenie MMAE do wnętrza komórki. MMAE wiąże się z mikrorurkami i zabija dzielące się komórki poprzez hamowanie podziału komórkowego oraz indukcję apoptozy.
Wpływ farmakodynamiczny
Elektrofizjologia serca
Na podstawie danych elektrokardiograficznych (EKG) uzyskanych w dwóch otwartych badaniach u pacjentów z wcześniej leczonymi nowotworami złośliwymi komórek B, polatuzumab vedotin w zalecanej dawce nie powodował klinicznie istotnego wydłużenia średniego odstępu QTc.
Skuteczność kliniczna i bezpieczeństwo
Pierwotnie nieleczona DLBCL
Skuteczność Polayvi® oceniano w międzynarodowym, wieloośrodkowym, randomizowanym, podwójnie ślepym, placebo kontrolowanym badaniu (POLARIX, GO39942) z udziałem 879 pacjentów z pierwotnie nieleczoną DLBCL.
Do badania zakwalifikowano pacjentów w wieku 18–80 lat, u których wartość międzynarodowego indeksu rokowniczego (IPI) wynosiła 2–5, a wskaźnik ogólnego stanu zdrowia – 0–2 według skali Eastern Cooperative Oncology Group (ECOG). Typy histologiczne guza obejmowały DLBCL (nieinaczej określone [NIO], z aktywowanych komórek B [ABC], z komórek centrum germinacyjnego [GCB]), limfomę B o wysokim stopniu złośliwości (NIO, double-hit, triple-hit) oraz inne podtypy dużokomórkowych limfom B (EBV-poz., wzbogacone limfocytami T/makrofagami). Pacjenci nie mieli znanej limfomy ośrodkowego układu nerwowego ani neuropatii obwodowej > stopnia 1.
Pacjenci zostali losowo przydzieleni w stosunku 1:1 do otrzymywania leku Polayvi® w połączeniu z R-CHP lub R-CHOP w ciągu sześciu 21-dniowych cykli, po których obie grupy otrzymywały dodatkowo dwa cykle monoterapii rytyksymabem. Pacjenci zostali zrandomizowani według indeksu IPI (2 vs 3–5), obecności lub braku rozległego zaangażowania (≥ 7,5 cm) oraz regionu geograficznego.
Polayvi® podawano dożylnie w dawce 1,8 mg/kg w dniu 1 cykli 1–6. R-CHP lub R-CHOP podawano od dnia 1 cykli 1–6, po czym obie grupy kontynuowały leczenie monoterapią rytyksymabem w dniu 1 cykli 7–8. Dawkowanie w każdej grupie leczenia wyglądało następująco:
- Grupa Polayvi® + R-CHP: Polayvi® 1,8 mg/kg, rytyksymab 375 mg/m², cyklofosfamid 750 mg/m², doksorubicyna 50 mg/m² oraz prednizolon 100 mg/dobę w dniach 1–5 każdego cyklu, doustnie.
- Grupa R-CHOP: rytyksymab 375 mg/m², cyklofosfamid 750 mg/m², doksorubicyna 50 mg/m², winksystyna 1,4 mg/m² oraz prednizolon 100 mg/dobę w dniach 1–5 każdego cyklu, doustnie.
Obie grupy leczenia były ogólnie zrównoważone pod względem wyjściowych cech demograficznych i charakterystyki choroby. Średni wiek pacjentów wynosił 65 lat (zakres od 19 do 80 lat), 53,6 % stanowili pacjenci rasy europejskiej, 53,8 % to mężczyźni, 43,8 % miało rozległe zaangażowanie chorobowe, u 38 % indeks IPI wynosił 2, u 62 % wynosił 3–5, a 88,7 % miało stadium choroby III lub IV. Większość pacjentów (84,2 %) miała DLBCL (w tym NIO, ABC i GCB). U 211 pacjentów nie podano wyników określenia linii komórkowego pochodzenia (COO). W populacji analizowanej pod względem COO (n = 668), 33,1 % pacjentów miało DLBCL z fenotypem ABC, a 52,7 % – z fenotypem GCB.
Pierwotnym punktem końcowym badania była przeżycie wolne od progresji według oceny badacza. Mediana czasu obserwacji wyniosła 28,2 miesiąca.
Tabela 1
Streszczenie skuteczności u pacjentów z pierwotnie nieleczoną DLBCL w badaniu GO39942 (POLARIX)
| Polayvi® + R-CHP N = 440 |
R-CHOP N = 439 |
|
| Podstawowy punkt końcowy |
||
| Przeżycie wolne od progresji1,* |
||
| Liczba (%) pacjentów z zdarzeniami |
107 (24,3 %) |
134 (30,5 %) |
| HR (95 % CI) |
0,73 [0,57, 0,95] |
|
| wartość p3,** |
0,0177 |
|
| Wskaźnik 2-letniego PFS (%) |
76,7 |
70,2 |
| [95 % CI] |
[72,65, 80,76] |
[65,80, 74,61] |
| Kluczowe wtórne punkty końcowe |
||
| Przeżycie wolne od zdarzeń (EFSeff)1 |
||
| Liczba (%) pacjentów z zdarzeniem |
112 (25,5 %) |
138 (31,4 %) |
| HR [95% CI] |
0,75 [0,58, 0,96] |
|
| wartość p3,** |
0,0244 |
|
| Szybkość odpowiedzi obiektywnej (ORR) na końcu leczenia2 |
||
| Pacjenci odpowiadający na leczenie (%) (pełna odpowiedź, częściowa odpowiedź) |
376 (85,5%) |
368 (83,8%) |
| Różnica w częstości odpowiedzi (%) [95 % CI] |
1,63 [-3,32, 6,57] |
|
| Częstość pełnej odpowiedzi (%) (CR)2,* |
||
| Pacjenci odpowiadający na leczenie (%) |
343 (78%) |
325 (74%) |
| Różnica w częstości odpowiedzi (%) [95% CI] |
3,92 [-1,89, 9,70] |
|
| Częściowa odpowiedź (%) (PR) |
33 (7,5%) |
43 (9,8%) |
| 95 % CI metodą Cloppera – Pearsona |
[5,22, 10,37] |
[7,18, 12,97] |
DI – przedział ufności; HR – stosunek ryzyka; PFS – przeżycie wolne od progresji; EFSeff – przeżycie wolne od zdarzeń związanych z efektywnością: służy do opisania przeżycia wolnego od zdarzeń spowodowanych skutecznością leczenia, zdefiniowanego jako czas od daty randomizacji do najwcześniejszego wystąpienia dowolnego z poniższych zdarzeń: postępowanie/reaktywacja choroby, śmierć z dowolnej przyczyny, pierwotna przyczyna skuteczności ustalona przez badacza inna niż postępowanie/reaktywacja choroby, która doprowadziła do rozpoczęcia dowolnego nieokreślonego w protokole leczenia chłoniaka (NALT), jeśli biopsja została wykonana po zakończeniu leczenia i była pozytywna pod względem choroby resztkowej, niezależnie od tego, czy rozpoczęto leczenie NALT, czy nie; CMH: Cochrane – Mantel – Haenszel.
1 Ocena badacza.
2 Ocena BICR (ocena niezależna, scentralizowana, w sposób ślepy).
3 Test logarytmiczny, uwarstwiony.
* Zgodnie z kryteriami oceny odpowiedzi Lugano z 2014 roku.
** Uwarstowione według IPI (2 vs 3–5), obecność lub brak rozległego zaangażowania, region geograficzny.
W trakcie analizy pośredniej kluczowy wtórny punkt końcowy, przeżycie ogólne, był nie dojrzały i nie różnił się istotnie statystycznie [uwarstwione stosunek ryzyka 0,94 (95 % DI, 0,65, 1,37); p = 0,7524].
| Prawdopodobieństwo przeżycia bez postępu (%) |
|
|||||||
| Czas, miesiące |
||||||||
| Pacjenci z ryzykiem |
||||||||
| R-CHOP |
439 |
389 |
330 |
296 |
220 |
78 |
3 |
NE |
| Polayvi® + R-CHP |
440 |
404 |
353 |
327 |
246 |
78 |
NE |
NE |
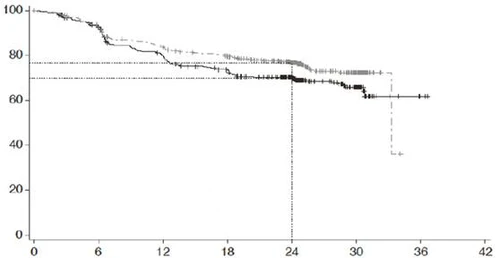
NE – nie można określić
Grupy leczenia: R-CHOP (n = 439); Polayvi® + R-CHP; dane cenzurowane
Wartość p (test log-rank) = 0,0177; HR (95% CI) = 0,73 (0,57 – 0,95)
Rys. 1. Krzywa przeżycia bez progresji (PFS) według Kaplana-Meiera według oceny badacza w badaniu GO39942 (POLARIX).
Powracająca lub oporna DLBCL
Skuteczność Polayvi® oceniano w międzynarodowym, wieloośrodkowym, otwartym badaniu (GO29365), w którym wzięło udział 80 pacjentów z wcześniej leczoną rozsianą dużokomórkową chłoniaką B (DLBCL), przydzielonych losowo do dwóch grup. Pacjenci byli losowani w stosunku 1:1 do otrzymywania Polayvi® + bendamustyna i rytymuksymab (BR) lub samego BR w ciągu sześciu 21-dniowych cykli. Pacjenci byli stertyfikowani według długości trwania odpowiedzi na ostatnie poprzednie leczenie: ≤ 12 miesięcy lub > 12 miesięcy.
Pacjenci nie byli kandydatami do autologicznej transplantacji komórek macierzystych (HSCT) i mieli chorobę nawrotową lub oporną po co najmniej jednym poprzednim trybie chemioterapii systemowej. Do badania nie włączano pacjentów po allogenicznej HSCT, z chłoniaką układu nerwowego środkowego, z transformowaną chłoniaką indolentną, chłoniakiem folikularnym stadium 3b, istotnymi chorobami serca lub płuc, aktywnymi zakażeniami, poziomem aspataminotransferazy (AST) lub alaninotransferazy (ALT) > 2,5 × górnej granicy normy (UGN) lub poziomem bilirubiny całkowitej ≥ 1,5 × UGN, poziomem kreatyniny > 1,5 × UGN (lub klirens kreatyniny < 40 ml/min), chyba że przyczyną takiego wzrostu była badana chłoniaka.
Polayvi® podawano dożylnie w dawce 1,8 mg/kg w dzień 2 cyklu 1 oraz w dzień 1 cykli 2–6. Bendamustynę podawano w dawce 90 mg/m²/dzień dożylnie w dniach 2 i 3 cyklu 1 oraz w dniach 1 i 2 cykli 2–6. Rytymuksymab podawano w dawce 375 mg/m² w dzień 1 cykli 1–6.
Spośród 80 pacjentów losowanych do otrzymywania Polayvi® + BR (n = 40) lub samego BR (n = 40), większość była rasy europejskiej (71%) i płci męskiej (66%). Średni wiek wynosił 69 lat (zakres: 30–86 lat). 64 spośród 80 pacjentów (80%) miało wskaźnik stanu ogólnego 0–1 według skali ECOG, a 14 spośród 80 pacjentów (18%) miało wskaźnik stanu ogólnego 2 według skali ECOG. Większość pacjentów (98%) miała DLBCL bez dodatkowego sprecyzowania. Ogółem 48% pacjentów miało DLBCL z aktywowanymi komórkami B, a 40% miało DLBCL z komórkami B centrum germinacyjnego. Głównymi przyczynami, dla których pacjenci nie byli kandydatami do przeprowadzenia HSCT, były wiek (40%), niewystarczająca odpowiedź na terapię ratunkową (26%) i niepowodzenie poprzedniej transplantacji (20%). Mediana liczby wcześniejszych terapii wynosiła 2 (zakres: 1–7), przy czym 29% (n = 23) otrzymało jedną poprzednią terapię, 25% (n = 20) – 2 poprzednie terapie, a 46% (n = 37) – 3 lub więcej poprzednich terapii. Wszyscy pacjenci, z wyjątkiem jednego w grupie Polayvi® + BR z badania fazy II, wcześniej nie otrzymywali bendamustyny. 80% pacjentów miało chorobę oporną. U pacjentów, którzy otrzymywali polatuzumab vedotin + BR i u których oznaczono liczbę limfocytów CD3+, bezwzględna liczba limfocytów CD3+ była > 200 komórek/μl odpowiednio u 95%, 79% i 83% osób, u których oznaczenia wykonano przed rozpoczęciem leczenia (n = 134), na końcu leczenia (n = 72) oraz 6 miesięcy po zakończeniu leczenia (n = 18).
Pierwotnym punktem końcowym tego badania była częstość całkowitej odpowiedzi (CR) na końcu leczenia (6–8 tygodni po dniu 1 cyklu 6 lub po ostatniej dawce badanej terapii) według oceny niezależnego komitetu oceny metodą PET-CT.
Tabela 2
Podsumowanie skuteczności u pacjentów z wcześniej leczoną DLBCL (badanie GO29365)
| Wskaźnik |
Polayvi® + bendamustyna + rytyksymab N = 40 |
Bendamustyna + rytyksymab N = 40 |
| Mediana czasu obserwacji 22 miesiące |
||
| Pierwotny punkt końcowy |
||
| Częstość odpowiedzi całkowitej* (według oceny niezależnego komitetu nadzorczego) na końcu leczenia** |
||
| Pacjenci odpowiadający na leczenie (%) |
16 (40,0) |
7 (17,5) |
| Różnica częstości odpowiedzi (%) [95% CI] |
22,5 [2,6, 40,2] |
|
| Wartość p (test chi-kwadrat, test Cochrana-Mantela-Haenszela***) |
0,0261 |
|
| Punkty końcowe wtórne i poszukiwane punkty końcowe |
||
| Czas trwania odpowiedzi (według oceny badacza) |
||
| Liczba pacjentów włączonych do analizy Liczba (%) pacjentów z zdarzeniem |
28 17 (60,7) |
13 11 (84,6) |
| Mediana czasu trwania odpowiedzi (95% CI), miesiące HR [95% CI] |
10,3 (5,6, OT) |
4,1 (2,6, 12,7) |
| 0,44 [0,20, 0,95] |
||
| Wartość p (test log-rank, uwarstwiony***) |
0,0321 |
|
| Częstość odpowiedzi ogólnej* (według oceny badacza) na końcu leczenia** |
||
| Pacjenci odpowiadający na leczenie (%) (PC, CZ) |
19 (47,5) |
7 (17,5) |
| Różnica częstości odpowiedzi (%) [95% CI] |
30,0 [9,5, 47,4] |
|
| Wartość p (test chi-kwadrat, test Cochrana-Mantela-Haenszela***) |
0,0036 |
|
| Odpowiedź całkowita (%) (PC) |
17 (42,5) |
6 (15,0) |
| Różnica częstości odpowiedzi (%) [95% CI] |
27,5 [7,7, 44,7] |
|
| Wartość p (test chi-kwadrat, test Cochrana-Mantela-Haenszela***) |
0,0061 |
|
| Odpowiedź częściowa (%) (CZ) 95% CI metodą Cloppera-Pearsona |
2 (5,0) [0,6, 16,9] |
1 (2,5) [0,06, 13,2] |
| Częstość najlepszej ogólnej odpowiedzi* (według oceny badacza) |
||
| Pacjenci odpowiadający na leczenie (%) (PC, CZ) |
28 (70,0) |
13 (32,5) |
| Różnica częstości odpowiedzi (%) [95% CI] |
37,5 [15,6, 54,7] |
|
| Odpowiedź całkowita (%) (PC) |
23 (57,5) |
8 (20,0) |
| 95% CI metodą Cloppera-Pearsona |
[40,9, 73,0] |
[9,1, 35,7] |
| Odpowiedź częściowa (%) (CZ) 95% CI metodą Cloppera-Pearsona |
5 (12,5) [4,2, 26,8] |
5 (12,5) [4,2, 26,8] |
DI – przedział ufności; HR – hazard ratio; OWE – oszacowanie nie dostępne; CR – kompletna odpowiedź; PR – częściowa odpowiedź.
* Według zmodyfikowanych kryteriów Lugano 2014: wymagane potwierdzenie kompletną odpowiedzi ze strony szpiku kostnego metodą PET-CT. PR potwierdzona metodą tomografii emisyjnej pozytonowej (PET-CT) musi spełniać kryteria PET-CT i tomografii komputerowej (TK).
** 6–8 tygodni po dniu 1 cyklu 6 lub po ostatniej dawce badanego leczenia.
*** Stratyfikacja według długości trwania odpowiedzi na poprzednią terapię (≤ 12 miesięcy vs > 12 miesięcy).
Całkowita przeżycie (OS) było punktem końcowym poszukiwania, który nie był kontrolowany pod kątem błędu typu I. Mediana OS w grupie Polayvi® + BR wyniosła 12,4 miesiąca (95 % DI: 9,0, OWE) w porównaniu do 4,7 miesiąca (95 % DI: 3,7, 8,3) w grupie kontrolnej. Nieskorygowane oszacowanie HR dla OS wyniosło 0,42. Po uwzględnieniu wpływu podstawowych współzmiennych HR dla OS zostało skorygowane do 0,59. Współzmienne obejmowały status pierwotnej refrakcyjności, liczbę poprzednich linii terapii, międzynarodowy indeks prognostyczny oraz poprzednią transplantację komórek macierzystych.
Przeżycie wolne od progresji (PFS) według oceny badacza było punktem końcowym poszukiwania, który nie był kontrolowany pod kątem błędu typu I. Mediana PFS w grupie Polayvi® + BR wyniosła 7,6 miesiąca (95 % DI: 6,0, 17,0) w porównaniu do 2,0 miesiąca (95 % DI: 1,5, 3,7) w grupie kontrolnej. Nieskorygowane oszacowanie HR dla PFS wyniosło 0,34.
Immunogenność
Jak wszystkie białka o działaniu terapeutycznym, polatuzumab vedotin ma potencjał do wywoływania odpowiedzi immunologicznej. W badaniach GO39442 (POLARIX) oraz GO29365 odpowiednio u 1,4 % (6/427) oraz 5,2 % (12/233) pacjentów wykryto przeciwciała przeciwko polatuzumabowi vedotinowi, z których żadne nie były przeciwciałami neutralizującymi. Ze względu na ograniczoną liczbę pacjentów, u których wykryto przeciwciała przeciwko polatuzumabowi vedotinowi, niemożliwe jest wyciągnięcie wniosku dotyczących potencjalnego wpływu immunogenności na skuteczność lub bezpieczeństwo.
Wyniki oceny immunogenności zależą istotnie od kilku czynników, w tym czułości i specyficzności metody analitycznej, metodologii analizy, sposobu postępowania z próbkami, czasu pobrania próbek, towarzyszącego leczenia oraz podstawowej choroby. Z tych powodów porównywanie częstości wykrywania przeciwciał przeciwko polatuzumabowi vedotinowi z częstością wykrywania przeciwciał przeciwko innym lekom może prowadzić do błędnej interpretacji danych.
Farmakokinetyka.
Ekspozycja MMAE skonjugowanego z przeciwciałem (acMMAE) we krwi wzrastała proporcjonalnie do dawki w zakresie dawek polatuzumabu vedotinu od 0,1 do 2,4 mg/kg. Po podaniu pierwszej dawki polatuzumabu vedotinu w dawce 1,8 mg/kg średnie stężenie maksymalne (Cmax) acMMAE wyniosło 803 (± 233) ng/ml, a pole pod krzywą „stężenie–czas” od 0 do nieskończoności (AUCinf) wyniosło 1860 (± 966) dzień•ng/ml. Na podstawie populacyjnej analizy farmakokinetycznej AUC acMMAE w cyklu 3 wzrosła o około 30 % w porównaniu z AUC w cyklu 1 i osiągnęła ponad 90 % AUC w cyklu 6. Okres półtrwania acMMAE w cyklu 6 wyniósł około 12 dni (95 % DI 8,1–19,5 dnia). Na podstawie populacyjnej analizy farmakokinetycznej przewidywane stężenie acMMAE na końcu cyklu 6 wynosi około 80 % teoretycznej wartości w stanie równowagi. Ekspozycja niekonjugowanego MMAE, cytotoksycznego składnika polatuzumabu vedotinu, wzrastała proporcjonalnie do dawki w zakresie dawek polatuzumabu vedotinu od 0,1 do 2,4 mg/kg. Dla stężeń MMAE we krwi charakterystyczna była kinetyka ograniczona szybkością jego powstawania.
Po podaniu pierwszej dawki polatuzumabu vedotinu w dawce 1,8 mg/kg Cmax wyniosła 6,82 (± 4,73) ng/ml, czas do osiągnięcia maksymalnego stężenia we krwi wyniósł około 2,5 dnia, a okres półtrwania końcowy wyniósł około 4 dni. Ekspozycja niekonjugowanego MMAE we krwi stanowi < 3 % ekspozycji acMMAE. Dane z populacyjnej analizy farmakokinetycznej wskazują na zmniejszenie ekspozycji (AUC) niekonjugowanego MMAE we krwi po powtarzanym stosowaniu co trzy tygodnie.
Na podstawie modelowania populacyjnej farmakokinetyki z wyników analizy post-hoc przewidywana ekspozycja niekonjugowanego MMAE u pacjentów z masą ciała powyżej 100 kg wzrośnie nie więcej niż o 55 %.
Wchłanianie
Polayvi® podaje się dożylnie w formie wlewu. Nie przeprowadzono badań z innymi drogami podania.
Rozkład
Szacowany objętość centralnego rozkładu acMMAE w populacji wynosił 3,15 l, co odpowiada w przybliżeniu objętości osocza krwi. In vitro MMAE umiarkowanie wiąże się (71–77 %) z białkami osocza krwi człowieka. MMAE nie przenika istotnie do erytrocytów człowieka in vitro; stosunek zawartości we krwi pełnej do osocza wynosi od 0,79 do 0,98.
Dane in vitro wskazują, że MMAE jest substratem P-gp, ale nie hamuje P-gp w klinicznie istotnych stężeniach.
Biotransformacja
Oczekuje się, że u pacjentów polatuzumab vedotin ulega katabolizmowi, w wyniku którego powstają małe peptydy, aminokwasy, niekonjugowany MMAE oraz metabolity powiązane z niekonjugowanym MMAE. Poziomy metabolitów MMAE nie były oznaczane w osoczu krwi człowieka.
Badania in vitro wskazują, że MMAE jest substratem CYP3A4/5, ale nie stymuluje głównych enzymów CYP. MMAE jest słabym, zależnym od czasu inhibitorem CYP3A4/5, ale nie hamuje konkurencyjnie CYP3A4/5 w klinicznie istotnych stężeniach.
MMAE nie hamuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ani CYP2D6.
Wydalanie
Na podstawie danych populacyjnej analizy farmakokinetycznej koniugat (acMMAE) jest wydalany głównie przez niespecyficzny, liniowy klirens na poziomie 0,9 l/dzień. Badania in vivo na szczurach, którym podano polatuzumab vedotin (oznaczony izotopem promieniotwórczym na MMAE), wykazały, że większość radioaktywności wydalana jest z kałem, a mniejsza część – z moczem.
Osobliwe grupy pacjentów
Dzieci
Badania farmakokinetyki polatuzumabu vedotinu u dzieci (w wieku < 18 lat) nie były prowadzone.
Pacjenci starsi
Na podstawie populacyjnej analizy farmakokinetycznej danych pacjentów w wieku 19–89 lat wiek nie wpływa na farmakokinetykę acMMAE i niekonjugowanego MMAE. Na podstawie populacyjnej analizy farmakokinetycznej nie zaobserwowano istotnych różnic w farmakokinetyce acMMAE i niekonjugowanego MMAE między pacjentami w wieku < 65 lat (n = 394) a pacjentami w wieku ≥ 65 lat (n = 495).
Upośledzenie funkcji nerek
U pacjentów z łagodnym (klirens kreatyniny 60–89 ml/min, n = 361) lub umiarkowanym (klirens kreatyniny 30–59 ml/min, n = 163) upośledzeniem funkcji nerek ekspozycja acMMAE i niekonjugowanego MMAE była podobna do ekspozycji u pacjentów z normalną funkcją nerek (klirens kreatyniny ≥ 90 ml/min, n = 356) na podstawie wyników populacyjnej analizy farmakokinetycznej. Brak wystarczających danych, aby ocenić wpływ ciężkiego upośledzenia funkcji nerek (klirens kreatyniny 15–29 ml/min, n = 4) na farmakokinetykę. Brak danych dotyczących pacjentów z nerek w stadium końcowym oraz/lub pacjentów poddawanych dializie.
Upośledzenie funkcji wątroby
Na podstawie populacyjnej analizy farmakokinetycznej u pacjentów z łagodnym upośledzeniem funkcji wątroby (AST lub ALT > 1,0–2,5 × GGN lub bilirubina ogólna > 1,0–1,5 × GGN, n = 133) ekspozycja acMMAE jest podobna do ekspozycji u pacjentów z normalną funkcją wątroby (n = 737), natomiast wartość AUC niekonjugowanego MMAE jest wyższa nie więcej niż o 40 %.
Brak wystarczających danych, aby ocenić wpływ umiarkowanego upośledzenia funkcji wątroby (bilirubina ogólna > 1,5–3 × GGN, n = 11) na farmakokinetykę. Dostępne są ograniczone dane dotyczące pacjentów z ciężkim upośledzeniem funkcji wątroby lub po przeszczepieniu wątroby.
Właściwości kliniczne.
Wskazania.
Preparat leczniczy Polayvi® w połączeniu z rytymusybabem, cyklofosfamidem, doksorubicyną i prednizonom (R-CHP) wskazany jest w leczeniu dorosłych pacjentów z nieleczonym wcześniej rozlanym wielokomórkowym limfoma B (DLBCL).
Preparat leczniczy Polayvi® w połączeniu z bendamustyną i rytymusybabem wskazany jest w leczeniu dorosłych pacjentów z nawrotową/refrakteryjną rozlanych wielokomórkowym limfoma B (DLBCL), którzy nie są kandydatami do przeszczepienia hematopoezyjnych komórek macierzystych.
Przeciwwskazania.
Podwyższona wrażliwość na polatuzumab wodotin lub którykolwiek ze składników pomocniczych preparatu leczniczego.
Aktywne ciężkie infekcje (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Interakcje z innymi lekami i inne rodzaje oddziaływań.
Nie przeprowadzono odpowiednich badań klinicznych dotyczących interakcji polatuzumabu wodotinu z innymi lekami u ludzi.
Interakcje przy jednoczesnym stosowaniu leków będących inhibitorami, substratami lub induktorami CYP3A4 oraz przy jednoczesnym stosowaniu leków będących inhibitorami białka P-glikoproteinowego (P-gp)
Na podstawie wyników modelu symulacji farmakokinetyki uwolnienia MMAE z polatuzumabu wodotinu opartego na fizjologicznych podstawach, silne inhibitory CYP3A4 i P-gp (np. ketokonazol) mogą zwiększać pole pod krzywą „stężenie–czas” (AUC) niekonjugowanego MMAE o 48%. Zaleca się zachowanie ostrożności przy jednoczesnym stosowaniu inhibitora CYP3A4. U pacjentów otrzymujących jednocześnie silne inhibitory CYP3A4 (np. boceprevir, klaritromycyna, kobicystat, indynawir, itrakonazol, nefazodon, nelfinawir, posakonazol, rytonawir, sakuinawir, telaprewir, telitromycyna, worykonazol) należy dokładnie obserwować objawy toksyczności.
Nie przewiduje się, że niekonjugowany MMAE zmienia AUC leków stosowanych jednocześnie, które są substratami CYP3A4 (np. midazolam).
Silne induktory CYP3A4 (np. ryfampicyna, karbamazepina, fenytoin, fenobarbital, ziele św. Jana [Hypericum perforatum]) mogą zmniejszać ekspozycję na niekonjugowany MMAE.
Interakcje przy stosowaniu leków rytymusybab, bendamustyna, cyklofosfamid i doksorubicyna w połączeniu z polatuzumabem wodotinem
Polatuzumab wodotin nie wpływa na farmakokinetykę rytymusybabu, bendamustyny, cyklofosfamidu i doksorubicyny. Jednoczesne stosowanie rytymusybabu wiąże się ze wzrostem AUC przeciwciała skoniugowanego z MMAE (acMMAE) we krwi o 24% oraz zmniejszeniem AUC niekonjugowanego MMAE we krwi o 37% na podstawie analizy farmakokinetyki populacyjnej. Wartości AUC acMMAE i niekonjugowanego MMAE we krwi dla preparatu Polayvi® w połączeniu z R-CHP są zgodne z danymi uzyskanymi w innych badaniach preparatu Polayvi®. Korekty dawki nie wymaga się.
Bendamustyna nie wpływa na AUC acMMAE i niekonjugowanego MMAE we krwi.
Szczególne zagadnienia dotyczące stosowania.
Śledzenie
W celu poprawy jakości śledzenia leków biologicznych w dokumentacji medycznej pacjenta należy jasno i czytelnie wpisać nazwę handlową oraz numer serii podawanego leku.
Myelosupresja
U pacjentów otrzymujących Polayvi® w pierwszym cyklu leczenia zgłaszano ciężką i poważną neutropenię oraz neutropenię febrilną. Należy rozważyć zastosowanie czynnika stymulującego kolonie granulocytów (G-CSF), którego stosowanie profilaktyczne było wymagane w programie klinicznego rozwoju leku. Podczas stosowania Polayvi® może również wystąpić trombocytopenia lub anemia stopnia 3 lub 4. Przed podaniem każdej dawki Polayvi® należy kontrolować morfologię krwi. U pacjentów z neutropenią i/lub trombocytopenią stopnia 3 lub 4 należy rozważyć częstsze badania laboratoryjne i/lub odroczenie lub przerwanie stosowania Polayvi® (patrz sekcja „Sposób i dawki stosowania”).
Neuropatia obwodowa (PNS)
U pacjentów otrzymujących Polayvi® w pierwszym cyklu leczenia zgłaszano PNS, a ryzyko wzrastało wraz z kolejnymi dawkami. U pacjentów z istniejącą już PNS stan ten może się nasilić. PNS zgłaszana podczas leczenia Polayvi® jest głównie sensoryczna. Jednakże zgłaszano również postacie motoryczne i sensoryczno-motoryczne PNS. Pacjentów należy obserwować pod kątem rozwoju objawów PNS, takich jak hipestezja, hiperestejzja, parestezje, dysestezje, ból neuropatyczny, uczucie pieczenia, osłabienie mięśni lub zaburzenia chodu. Pacjentom, u których wystąpiła nowa PNS lub pogorszenie istniejącej, może być konieczne odroczenie podania, zmniejszenie dawki lub przerwanie stosowania Polayvi® (patrz sekcja „Sposób i dawki stosowania”).
Zakażenia
U pacjentów otrzymujących leczenie Polayvi® zgłaszano poważne, zagrażające życiu lub śmiertelne infekcje, w tym infekcje oportunistyczne, takie jak zapalenie płuc (w tym wywołane przez Pneumocystis jirovecii oraz inne grzybicze zapalenia płuc), bakteriemię, sepsę, infekcje wirusem opryszczki oraz cytomegalowirusa (patrz sekcja „Efekty uboczne”). Zgłoszono reaktywację infekcji utajonych. Pacjentów należy dokładnie obserwować podczas leczenia pod kątem pojawienia się objawów bakteryjnych, grzybiczych lub wirusowych infekcji, a w przypadku ich wystąpienia należy skonsultować się z lekarzem. Podczas leczenia Polayvi® należy rozważyć profilaktykę infekcji. Polayvi® nie należy stosować w przypadku aktywnej ciężkiej infekcji. U pacjentów, u których rozwinęła się poważna infekcja, należy przerwać leczenie Polayvi® oraz wszelką współistniejącą chemioterapię.
Wirus HIV
Polayvi® nie był badany u pacjentów zakażonych wirusem HIV. Informacje dotyczące współistniejącego stosowania z inhibitorami CYP3A znajdują się w sekcji „Interakcje z innymi lekami i inne formy interakcji”.
Immunizacja
Nie należy stosować szczepionek żywych ani osłabionych w trakcie leczenia. Pacjenci, którzy niedawno otrzymali szczepionki żywe, nie brali udziału w badaniach klinicznych.
Postępująca wieloogniskowa leukoencefalopatia (PML)
Podczas leczenia Polayvi® zgłaszano przypadki PML (patrz sekcja „Efekty uboczne”). Pacjentów należy dokładnie obserwować pod kątem pojawienia się nowych lub nasilania się objawów neurologicznych, kognitywnych lub zachowawczych, które mogą wskazywać na PML. Leczenie Polayvi® oraz wszelką współistniejącą chemioterapię należy przerwać w przypadku podejrzenia PML i całkowicie odstawić po potwierdzeniu rozpoznania.
Zespół lizy nowotworowej (TLS)
U pacjentów z dużym obciążeniem guzem oraz z szybko proliferującymi guzami istnieje zwiększone ryzyko wystąpienia TLS. Przed rozpoczęciem leczenia Polayvi® należy podjąć odpowiednie środki zapobiegawcze zgodnie z lokalnymi zaleceniami. Podczas leczenia Polayvi® należy dokładnie monitorować stan pacjentów pod kątem rozwoju TLS.
Reakcje podczas infuzji
Polayvi® może powodować reakcje podczas infuzji, w tym ciężkie. Zdarzały się opóźnione reakcje infuzyjne nawet do 24 godzin po podaniu leku Polayvi®. Przed podaniem Polayvi® należy podać lek przeciwhistaminowy i środek przeciwgorączkowy oraz dokładnie monitorować stan pacjenta przez cały czas trwania infuzji. W przypadku wystąpienia reakcji infuzyjnych należy przerwać infuzję i rozpocząć odpowiednie leczenie farmakologiczne (patrz sekcja „Sposób i dawki stosowania”).
Toxyczność dla embrionu i płodu
Ze względu na mechanizm działania oraz wyniki badań przedklinicznych Polayvi® może powodować szkodę płodowi, gdy jest podawany kobiecie w ciąży. Kobiety w ciąży należy poinformować o ryzyku dla płodu.
Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznych metod antykoncepcji podczas leczenia Polayvi® oraz co najmniej przez 9 miesięcy po podaniu ostatniej dawki (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”). Mężczyznom, którzy mają partnerki w wieku rozrodczym, należy zalecić stosowanie skutecznych metod antykoncepcji podczas leczenia Polayvi® oraz co najmniej przez 6 miesięcy po podaniu ostatniej dawki (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”).
Plodność
W badaniach przedklinicznych stosowanie polatuzumabu wedotinu wiązało się z toksycznym działaniem na jądra, co może prowadzić do zaburzeń funkcji rozrodczej i niepłodności u mężczyzn. Mężczyznom otrzymującym terapię Polayvi® zaleca się przed rozpoczęciem leczenia zabezpieczenie i przechowanie próbek nasienia (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”).
Pacjenci w wieku podeszłym
Spośród 435 wcześniej nieleczonych pacjentów z DLBCL, którzy otrzymywali leczenie Polayvi® w połączeniu ze schematem R-CHP w badaniu GO39942, 227 (52,2%) miało wiek ≥ 65 lat. U pacjentów w wieku ≥ 65 lat częstość poważnych działań niepożądanych wyniosła 39,2%, a u pacjentów w wieku < 65 lat – 28,4%. Podobna częstość poważnych działań niepożądanych obserwowana była u pacjentów starszych w grupie leczonych schematem R-CHOP.
Spośród 151 wcześniej leczonych pacjentów z DLBCL, którzy otrzymywali leczenie Polayvi® w połączeniu z bendamustyną i rytyksymabą w badaniu GO29365, 103 (68%) miało wiek ≥ 65 lat. U pacjentów w wieku ≥ 65 lat częstość poważnych działań niepożądanych (55%) była podobna do tej u pacjentów w wieku < 65 lat (56%). Badania kliniczne leku Polayvi® nie obejmowały wystarczającej liczby pacjentów w wieku ≥ 65 lat, aby można było ustalić różnicę w odpowiedzi w porównaniu z młodszych pacjentami.
Hepatotoksyczność
U pacjentów otrzymujących leczenie Polayvi® wystąpiły poważne przypadki hepatotoksyczności, wskazujące na uszkodzenie hepatocytów, w tym podwyższenie poziomów transaminaz i/lub bilirubiny (patrz sekcja „Efekty uboczne”). Istniejące wcześniej choroby wątroby, pierwotnie podwyższone poziomy enzymów wątrobowych oraz współistniejące stosowanie innych leków mogą zwiększać ryzyko hepatotoksyczności. Należy kontrolować poziomy enzymów wątrobowych i bilirubiny (patrz sekcja „Sposób i dawki stosowania”).
Substancje pomocnicze
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, co oznacza, że jest praktycznie wolny od sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym / antykoncepcja u kobiet i mężczyzn
Kobiety
Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznych metod antykoncepcji podczas leczenia polatuzumabem wedotinem oraz przez co najmniej 9 miesięcy po podaniu ostatniej dawki.
Mężczyźni
Mężczyznom, którzy mają partnerki w wieku rozrodczym, należy zalecić stosowanie skutecznych metod antykoncepcji podczas leczenia polatuzumabem wedotinem oraz przez co najmniej 6 miesięcy po podaniu ostatniej dawki.
Ciąża
Brak danych dotyczących stosowania Polayvi® u kobiet w ciąży. Badania na zwierzętach wykazały toksyczność rozrodczą. Ze względu na mechanizm działania oraz wyniki badań przedklinicznych polatuzumab wedotin może powodować szkodę płodowi, gdy jest podawany kobiecie w ciąży. Przed leczeniem należy zbadać kobiety w wieku rozrodczym pod kątem ciąży. Polayvi® nie powinno być stosowane w czasie ciąży ani u kobiet w wieku rozrodczym nie stosujących antykoncepcji, chyba że potencjalna korzyść dla kobiety przewyższa potencjalne ryzyko dla płodu.
<Karmienie piersią>
Nie wiadomo, czy polatuzumab wedotin lub jego metabolity wydzielają się w mleku matki. Nie można wykluczyć ryzyka dla niemowląt karmionych piersią. Kobiety powinny przerwać karmienie piersią podczas leczenia Polayvi® oraz przez co najmniej 3 miesiące po podaniu ostatniej dawki.
Plodność
W badaniach przedklinicznych stosowanie polatuzumabu wedotinu wiązało się z toksycznym działaniem na jądra i mogło prowadzić do zaburzeń funkcji rozrodczej i niepłodności u samców.
Dlatego mężczyznom otrzymującym leczenie tym lekiem zaleca się przed leczeniem zabezpieczenie i przechowanie próbek nasienia. Mężczyznom otrzymującym lek Polayvi® nie zaleca się zapładniania partnerki podczas leczenia oraz przez 6 miesięcy po podaniu ostatniej dawki.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwanie maszyn.
Polayvi® ma nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn. Podczas leczenia Polayvi® mogą wystąpić reakcje infuzyjne, neuropatia obwodowa, zmęczenie i zawroty głowy (patrz sekcje „Szczególne zagadnienia dotyczące stosowania” i „Efekty uboczne”).
Sposób stosowania i dawki
Polayvi® należy podawać pod ścisłym nadzorem lekarza posiadającego doświadczenie w diagnozowaniu i leczeniu pacjentów z chorobami onkologicznymi.
Dozowanie
Chłoniak rozlany wielokomórkowy B
Pacjenci wcześniej nielеченi
Zalecana dawka Polayvi® wynosi 1,8 mg/kg w postaci wlewu dożylnego co 21 dzień w połączeniu z rytyksymabem, cyklofosfamidem, doksorubicyną i prednizolonem (R-CHP) przez 6 cykli. Polayvi®, rytyksymab, cyklofosfamid i doksorubicynę można podawać w dowolnej kolejności w dniu 1 po podaniu prednizolonu. Prednizolon podaje się w dniach 1–5 każdego cyklu. Cykle 7 i 8 obejmują stosowanie rytyksymabu jako monoterapii.
Zobacz instrukcję do stosowania medycznego leków chemioterapeutycznych stosowanych w połączeniu z Polayvi® u pacjentów z wcześniej nielеченym chłoniakiem rozlanym wielokomórkowym B.
Pacjenci z nawrotem lub chorobą oporną
Zalecana dawka Polayvi® wynosi 1,8 mg/kg w postaci wlewu dożylnego w połączeniu z bendamustyną i rytyksymabem co 21 dzień przez 6 cykli. Polayvi®, bendamustynę i rytyksymab można podawać w dowolnej kolejności w dniu 1 każdego cyklu. W przypadku stosowania w połączeniu z Polayvi® zalecana dawka bendamustyny wynosi 90 mg/m²/dzień w dniu 1 i dniu 2 każdego cyklu, zalecana dawka rytyksymabu to 375 mg/m² w dniu 1 każdego cyklu. Ze względu na ograniczone doświadczenie kliniczne nie zaleca się przekraczania dawki 240 mg/na cykl u pacjentów leczonych Polayvi® w dawce 1,8 mg/kg z dawką skumulowaną > 240 mg.
Pacjenci wcześniej nielеченi oraz pacjenci z nawrotem lub chorobą oporną
Jeśli nie przeprowadzono wcześniejszej premedykacji, przed podaniem Polayvi® należy wykonać premedykację lekami przeciwhistaminowymi i przeciwgorączkowymi.
Opóźnione lub pominięte podanie
Jeśli zaplanowana dawka Polayvi® nie została podana, należy ją podać jak najszybciej, a harmonogram podania dostosować tak, aby zachować 21-dniowy odstęp między dawkami.
Korekta dawki
Przy wystąpieniu reakcji podczas wlewania należy zmniejszyć prędkość wlewu Polayvi® lub wstrzymać wlew. Podawanie Polayvi® należy natychmiast przerwać i trwale odstawić lek w przypadku wystąpienia reakcji zagrażającej życiu.
Sposób korekty dawki Polayvi® różni się dla pacjentów z wcześniej nielеченym chłoniakiem rozlanym wielokomórkowym B i dla pacjentów z nawrotem lub chorobą oporną.
Informacje dotyczące korekty dawki w przypadku wystąpienia neuropatii obwodowej (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”) znajdują się w tabeli 3.
Tabela 3
Korekta dawki leku Polayvi® w przypadku wystąpienia neuropatii obwodowej (NO)
| Wskazania |
Stopień ciężkości NP w dniu 1 dowolnego cyklu |
Modyfikacja dawki |
| Nieleczona wcześniej DLBCL |
Stopień 2a |
Neuropatia czuciowa:
Neuropatia ruchowa:
W przypadku jednoczesnego wystąpienia neuropatii czuciowej i ruchowej należy stosować się do najbardziej restrykcyjnych zaleceń podanych powyżej. |
| Stopień 3a |
Neuropatia czuciowa:
Neuropatia ruchowa:
W przypadku jednoczesnego wystąpienia neuropatii czuciowej i ruchowej należy stosować się do najbardziej restrykcyjnych zaleceń podanych powyżej. |
|
| Stopień 4 |
Przerwać stosowanie leku Polayvi®. |
|
| DLBCL nawracająca/oporna |
Stopień 2–3 |
Przerwać stosowanie leku Polayvi® do poprawy stanu do stopnia ≤ 1. Jeśli stan wróci do stopnia ≤ 1 w dniu 14 lub wcześniej, kontynuować leczenie Polayvi® w stałej, obniżonej dawce 1,4 mg/kg. Jeśli wcześniej dawka została już zmniejszona do 1,4 mg/kg, leczenie Polayvi® należy przerwać. Jeśli stan nie wróci do stopnia ≤ 1 w dniu 14 lub wcześniej, leczenie Polayvi® należy przerwać. |
| Stopień 4 |
Przerwać stosowanie leku Polayvi®. |
a Leczenie R-CHP można kontynuować.
Informacje dotyczące modyfikacji dawki w przypadku wystąpienia mielosupresji (patrz punkt «Szczególne ostrzeżenia i środki ostrożności przed zastosowaniem») znajdują się w tabeli 4.
Tabela 4
Modyfikacja dawki Polayvi®, chemioterapii i rytuksymabu w przypadku wystąpienia mielosupresji
| Wskazania |
Wymiar mielosupresji w dniu 1 każdego cyklu |
Modyfikacja dawki |
| Nieleczona wcześniej DLBCL |
Neutropenia stopnia 3–4 |
Przestać stosować wszystkie leki do odzyskania absolutnej liczby neutrofili (ALN)* do > 1000/μl. Jeśli ALN odzyska wartość > 1000/μl do dnia 7 lub wcześniej, należy kontynuować stosowanie wszystkich leków bez zmniejszania dawki. Jeśli ALN odzyska wartość > 1000/μl po dniu 7:
|
| Trombocytopenia stopnia 3–4 |
Przestać stosować wszystkie leki do odzyskania liczby płytek krwi do > 75 000/μl. Jeśli liczba płytek krwi odzyska wartość > 75 000/μl do dnia 7 lub wcześniej, należy kontynuować stosowanie wszystkich leków bez zmniejszania dawki. Jeśli liczba płytek krwi odzyska wartość > 75 000/μl po dniu 7:
|
|
| DLBCL nawrotowa/refrakteryjna |
Neutropenia stopnia 3–41 |
Przestać stosować wszystkie leki do odzyskania ALN do > 1000/μl. Jeśli ALN odzyska wartość > 1000/μl do dnia 7 lub wcześniej, należy kontynuować stosowanie wszystkich leków bez zmniejszania dawki. Jeśli ALN odzyska wartość > 1000/μl po dniu 7:
|
| Trombocytopenia stopnia 3–41 |
Przestać stosować wszystkie leki do odzyskania liczby płytek krwi do > 75 000/μl. Jeśli liczba płytek krwi odzyska wartość > 75 000/μl do dnia 7 lub wcześniej, należy kontynuować stosowanie wszystkich leków bez zmniejszania dawki. Jeśli liczba płytek krwi odzyska wartość > 75 000/μl po dniu 7:
|
1Jeśli przyczyną główną jest chłoniak, dawka bendamustyny może nie wymagać zmniejszenia.
Informacje dotyczące modyfikacji dawki w przypadku wystąpienia reakcji infuzyjnych (patrz sekcja „Szczególne wytyczne stosowania”) znajdują się w tabeli 5.
Tabela 5
Modyfikacja dawki leku Polayvi® w przypadku wystąpienia reakcji infuzyjnych (IR)
| Wskazania |
Stopień ZI w dniu 1 dowolnego cyklu |
Modyfikacja dawki |
| Wcześniej nielеченiona i nawrotowa/refrakteryjna DLBCL |
ZI stopnia 1–3 |
Przerwać infuzję Polayvi® i podać leczenie wspierające. W przypadku pierwszego wystąpienia świstów, skurczu oskrzeli lub uogólnionej pokrzywki stopnia 3 Polayvi® należy odstawić trwale. W przypadku nawrotowych świstów lub pokrzywki stopnia 2 lub nawrotu jakichkolwiek objawów stopnia 3 Polayvi® należy odstawić trwale. W innych przypadkach po całkowitym ustąpieniu objawów infuzję można wznowić z prędkością 50% prędkości osiągniętej przed przerwaniem. W przypadku braku objawów reakcji infuzyjnej prędkość infuzji można zwiększać o 50 mg/godz. co 30 minut. W kolejnym cyklu infuzję Polayvi® należy przeprowadzać przez 90 minut. W przypadku braku reakcji infuzyjnych kolejne infuzje można podawać w ciągu 30 minut. Profilaktykę (premedykację) należy stosować we wszystkich cyklach. |
| ZI stopnia 4 |
Natychmiast przerwać infuzję leku Polayvi®. Podać leczenie wspierające. Odstawić Polayvi® trwale. |
Osoby szczególne
Pacjenci w wieku podeszłym
U pacjentów w wieku ≥ 65 lat nie jest wymagana korekta dawki leku Polayvi® (patrz punkt „Farmakokinetyka”).
Zaburzenia funkcji nerek
U pacjentów z klirem kreatyniny ≥ 30 ml/min nie jest wymagana korekta dawki leku Polayvi®. Zalecana dawka u pacjentów z klirem kreatyniny < 30 ml/min nie została określona ze względu na ograniczone dane.
Zaburzenia funkcji wątroby
Należy unikać stosowania Polayvi® u pacjentów z umiarkowanym lub ciężkim zaburzeniem funkcji wątroby (poziom bilirubiny powyżej 1,5 × górnej granicy normy [GGN]).
Nie jest wymagana korekta dawki wstępnej Polayvi® u pacjentów z łagodnym zaburzeniem funkcji wątroby (poziom bilirubiny powyżej GGN – poniżej lub równy 1,5 × GGN lub poziom AST powyżej GGN).
W badanej populacji pacjentów z łagodnym zaburzeniem funkcji wątroby (określonym jako poziom AST lub ALT > 1,0–2,5 × GGN lub poziom całkowitej bilirubiny > 1,0–1,5 × GGN) zaobserwowano zwiększenie ekspozycji na niemodyfikowanego MMAE o nie więcej niż 40%, co nie zostało uznane za klinicznie istotne.
Dzieci
Bezpieczeństwo i skuteczność stosowania leku u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Sposób podania
Lek Polayvi® przeznaczony jest do dożylnej infuzji.
Pierwszą dawkę Polayvi® należy podawać w postaci dożylnej infuzji trwającej 90 minut. Pacjentów należy obserwować pod kątem wystąpienia reakcji infuzyjnych/reakcji nadwrażliwości podczas infuzji oraz przez co najmniej 90 minut po podaniu pierwszej dawki.
Jeśli poprzednia infuzja została dobrze tolerowana, kolejną dawkę leku Polayvi® można podawać w ciągu 30 minut, a pacjentów należy obserwować podczas infuzji oraz przez co najmniej 30 minut po jej zakończeniu.
Lek Polayvi® należy rekonstytuować i rozcieńczać z zachowaniem zasad aseptyki pod nadzorem personelu medycznego. Lek należy podawać za pomocą dożylnej infuzji przez oddzielny zestaw infuzyjny wyposażony w sterylny, apirogenny wbudowany lub dodatkowy filtr o niewielkim wiązaniu białek (o wielkości por 0,2 lub 0,22 µm) i kaniulę. Leku Polayvi® nie wolno podawać dożylnie w sposób strułkowy ani bolusowy.
Środki ostrożności, które należy podjąć przed przygotowaniem lub podaniem leku
Lek Polayvi® zawiera składnik cytotoksyczny kowalencyjnie związany z przeciwciałem monoklonalnym. Należy przestrzegać odpowiednich środków ostrożności przy obchodzeniu się z lekiem i jego utylizacji.
Ogólne środki ostrożności
Lek Polayvi® zawiera składnik cytotoksyczny. Lek należy podawać pod nadzorem lekarza mającego doświadczenie w stosowaniu środków cytotoksycznych. Należy przestrzegać odpowiednich zasad obchodzenia się i utylizacji leków przeciwnowotworowych i cytotoksycznych.
Odtworzony roztwór nie zawiera środków konserwujących i przeznaczony jest wyłącznie do jednorazowego użytku. Należy przestrzegać odpowiednich zasad aseptyki podczas obchodzenia się z tym lekiem.
Lek Polayvi® należy rekonstytuować za pomocą wody do wstrzykiwań i rozcieńczyć przed podaniem w worku do infuzji dożylnej zawierającym roztwór chlorku sodu do wstrzykiwań 9 mg/ml (0,9 %) lub roztwór chlorku sodu do wstrzykiwań 4,5 mg/ml (0,45 %), albo 5 % roztwór glukozy.
Nie należy zamrażać odtworzonego roztworu ani narażać go na bezpośrednie działanie promieni słonecznych.
Instrukcje dotyczące rekonstytucji
- Polayvi®, 30 mg: za pomocą sterylnego strzykawki powoli wstrzyknąć 1,8 ml wody do wstrzykiwań do fiolki zawierającej 30 mg leku Polayvi®, aby uzyskać jednodawkowy roztwór zawierający 20 mg/ml polatuzumabu vedotinu. Strumień kierować na ściankę fiolki, a nie bezpośrednio na masę liofilizowaną.
- Polayvi®, 140 mg: za pomocą sterylnego strzykawki powoli wstrzyknąć 7,2 ml wody do wstrzykiwań do fiolki zawierającej 140 mg leku Polayvi®, aby uzyskać jednodawkowy roztwór zawierający 20 mg/ml polatuzumabu vedotinu. Strumień kierować na ściankę fiolki, a nie bezpośrednio na masę liofilizowaną.
- Delikatnie obracać fiolkę aż do całkowitego rozpuszczenia. Nie wstrząsać.
- Sprawdzić odtworzony roztwór pod kątem obecności cząstek stałych i zmiany koloru. Odtworzony roztwór powinien mieć barwę od bezbarwnej do lekko brązowatej, być przejrzysty do lekko opalescencyjnego i nie zawierać widocznych cząstek. Nie należy stosować odtworzonego roztworu o zmienionym kolorze, zmętniałego lub zawierającego widoczne cząstki.
Instrukcje dotyczące rozcieńczania
- Lek Polayvi® należy rozcieńczyć do końcowej stężenia 0,72–2,7 mg/ml w worku do infuzji dożylnej o minimalnej objętości 50 ml zawierającym roztwór chlorku sodu do wstrzykiwań 9 mg/ml lub roztwór chlorku sodu do wstrzykiwań 4,5 mg/ml, albo 5 % roztwór glukozy.
- Określić objętość odtworzonego roztworu 20 mg/ml niezbędną do uzyskania przepisanej dawki (patrz niżej):
| Ogólna dawka leku Polayvi® (ml) do dalszego rozcieńczenia |
= |
dawka leku Polayvi® (mg/kg) × masa ciała pacjenta (kg) |
| stężenie odtworzonego roztworu w fiolce (20 mg/ml) |
-
Za pomocą sterylnego strzykawki odciągnąć wymaganą objętość odtworzonego roztworu leku Polayvi® z fiolki i rozcieńczyć w worku do wlewów dożylnych. Odpadki z niezużytej części leku pozostałej w fiolce należy wyrzucić.
-
Ostrożnie mieszać zawartość worka do wlewu poprzez delikatne jego obracanie. Nie wstrząsać.
-
Sprawdzić zawartość worka do wlewu pod kątem obecności cząstek i w przypadku ich wykrycia wyrzucić zawartość.
Należy unikać transportowania przygotowanego roztworu do wlewu, ponieważ stres mechaniczny spowodowany mieszaniem może prowadzić do agregacji. Jeżeli konieczny jest transport przygotowanego roztworu, należy usunąć powietrze z worka do wlewu i ograniczyć czas transportu do 30 minut w temperaturze pokojowej (9–25 °C) lub do 24 godzin w lodówce (2–8 °C). Jeżeli usuwa się powietrze, należy zastosować zestaw do wlewu z metalową igłą do połączenia, aby zapewnić dokładne dawkowanie podczas infuzji. Całkowity czas przechowywania plus czas transportu rozcieńczonego roztworu nie powinien przekraczać okresu przechowywania wskazanego w tabeli 6.
Lek Polayvi® należy podawać za pomocą oddzielnego systemu do wlewu wyposażonego w sterylny, apirogenny wbudowany lub dodatkowy filtr o niewielkim wiązaniu białek (o wielkości por 0,2 lub 0,22 μm) oraz za pomocą kaniuli.
Polayvi® jest kompatybilny z workami do wlewów dożylnych wykonanymi z polichlorku winylu (PVC) lub poliolefinów, takich jak polietylen (PE) i polipropylen. Ponadto nie zaobserwowano niezgodności zestawów do wlewu lub materiałów z produktami zawierającymi PVC, PE, poliuretan, polibutylenodien, akrylonitrylo-butyleno-styren, poliwęglan, polietero-uretan, fluorowany etylenopropylen lub politetrafluoroetylen) oraz z membranami filtrującymi wykonanymi z polietersulfonu lub polisulfonu.
Okres ważności
Odtworzony roztwór
Z mikrobiologicznego punktu widzenia odtworzony roztwór należy użyć natychmiast. Jeżeli lek nie jest stosowany natychmiast, okres i warunki przechowywania odtworzonego roztworu są odpowiedzialnością użytkownika. Okres przechowywania w lodówce (2–8 °C) zazwyczaj nie powinien przekraczać 24 godzin, pod warunkiem że odtworzenie odbyło się w kontrolowanych i zwalidowanych warunkach bezpyłowych. Stabilność chemiczna i fizyczna odtworzonego roztworu została potwierdzona przez okres do 72 godzin podczas przechowywania w lodówce (2–8 °C) oraz do 24 godzin w temperaturze pokojowej (9–25 °C).
Rozcieńczony roztwór
Z mikrobiologicznego punktu widzenia rozcieńczony roztwór do wlewu należy użyć natychmiast. Jeżeli lek nie jest stosowany natychmiast, okres i warunki przechowywania rozcieńczonego roztworu są odpowiedzialnością użytkownika. Okres przechowywania w lodówce (2–8 °C) zazwyczaj nie powinien przekraczać 24 godzin, pod warunkiem że rozcieńczenie odbyło się w kontrolowanych i zwalidowanych warunkach bezpyłowych. Stabilność chemiczna i fizyczna rozcieńczonego roztworu do wlewu została potwierdzona przez okresy wskazane w tabeli 6. Rozcieńczony roztwór należy wyrzucić, jeżeli czas przechowywania przekracza okresy wskazane w tabeli 6.
Tabela 6
Okresy, przez które potwierdzono stabilność chemiczną i fizyczną przygotowanego roztworu do infuzji
| Roztwórnik stosowany do przygotowania roztworu do wlewu |
Warunki przechowywania roztworu do wlewu1 |
| Chlorek sodu 9 mg/ml (0,9 %) |
Do 72 godzin w lodówce (2–8 °C) lub do 4 godzin w temperaturze pokojowej (9–25 °C) |
| Chlorek sodu 4,5 mg/ml (0,45 %) |
Do 72 godzin w lodówce (2–8 °C) lub do 8 godzin w temperaturze pokojowej (9–25 °C) |
| 5 % roztwór glukozy |
Do 72 godzin w lodówce (2–8 °C) lub do 8 godzin w temperaturze pokojowej (9–25 °C) |
1W celu zapewnienia stabilności leku nie należy przekraczać wskazanych okresów przechowywania.
Preparat Polayvi® przeznaczony jest wyłącznie do jednorazowego użytku.
Wszelkie niewykorzystane leki lub odpady należy zutylizować zgodnie z lokalnymi przepisami.
Dzieci
Bezpieczeństwo i skuteczność stosowania preparatu u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Przedawkowanie.
Nie ma doświadczeń z przedawkowaniem u ludzi w badaniach klinicznych. Najwyższą zbadaną dawką do tej pory jest dawka 2,4 mg/kg podana dożylnie; dawka ta była związana z większą częstością i ciężkością przypadków neuropatii obwodowej. W przypadku przedawkowania należy natychmiast przerwać infuzję i dokładnie obserwować stan pacjenta.
Efekty uboczne.
Podsumowanie profilu bezpieczeństwa
Bezpieczeństwo leku Polayvi® oceniano u 435 pacjentów w badaniu GO39942 (POLARIX). Zaobserwowano następujące efekty uboczne opisane w tej sekcji:
- podczas leczenia i w okresie dalszej obserwacji u wcześniej nieleczonych pacjentów z DLBCL uczestniczących w głównym badaniu klinicznym GO39942 (POLARIX), którzy otrzymywali Polayvi® w połączeniu z R-CHP (n = 435) lub R-CHOP (n = 438). W grupie leczenia Polayvi® + R-CHP 91,7% pacjentów otrzymało 6 cykli leku Polayvi® w porównaniu do 88,5% pacjentów, którzy otrzymali 6 cykli winkrystyny w grupie R-CHOP.
U wcześniej nieleczonych pacjentów z DLBCL, którzy otrzymywali leczenie Polayvi® w połączeniu z R-CHP:
- Najczęściej występującymi efektami ubocznymi (≥ 30%) u pacjentów leczonych Polayvi® w połączeniu z R-CHP z powodu wcześniej nieleczonego DLBCL były neuropatia obwodowa (52,9%), nudności (41,6%), neutropenia (38,4%) i biegunka (30,8%).
- O poważne efekty uboczne zgłaszano u 24,1% pacjentów leczonych Polayvi® w połączeniu z R-CHP.
- Najczęstsze poważne efekty uboczne występujące u ≥ 5% pacjentów to febrilna neutropenia (10,6%) i zapalenie płuc (5,3%).
- Efektem ubocznym prowadzącym do przedwczesnego zakończenia leczenia u > 1% pacjentów leczonych Polayvi® w połączeniu z R-CHP było zapalenie płuc (1,1%).
Bezpieczeństwo stosowania leku Polayvi® oceniano u 151 pacjentów w badaniu GO29365. Zaobserwowano następujące efekty uboczne opisane w tej sekcji:
- podczas leczenia i okresu obserwacji u wcześniej leczonych pacjentów z DLBCL (n = 151) w głównym badaniu klinicznym GO29365. Obejmuje to pacjentów z fazy wprowadzającej (n = 6), pacjentów zrandomizowanych (n = 39) oraz pacjentów z kohorty rozszerzonej (n = 106), którzy otrzymywali Polayvi® + BR, w porównaniu z pacjentami zrandomizowanymi (n = 39), którzy otrzymywali tylko BR. Pacjenci w grupach leczenia otrzymali średnio 5 cykli leczenia, podczas gdy pacjenci zrandomizowani do grupy porównawczej otrzymali średnio 3 cykle leczenia.
U wcześniej leczonych pacjentów z DLBCL, którzy otrzymywali leczenie Polayvi® w połączeniu z bendamustyną i rytyksymabą:
- Najczęściej występującymi efektami ubocznymi (≥ 30%) (wszystkich stopni) u pacjentów leczonych Polayvi® w połączeniu z bendamustyną i rytyksymabą z powodu wcześniej leczonego DLBCL były neutropenia (45,7%), biegunka (35,8%), nudności (33,1%), trombocytopenia (32,5%), anemia (31,8%) i neuropatia obwodowa (30,5%).
- O poważne efekty uboczne zgłaszano u 41,7% pacjentów leczonych Polayvi® w połączeniu z bendamustyną i rytyksymabą.
- Najczęstsze poważne efekty uboczne występujące u ≥ 5% pacjentów to: febrilna neutropenia (10,6%), sepsa (9,9%), zapalenie płuc (8,6%) i podwyższona temperatura ciała (7,9%).
- Efektem ubocznym prowadzącym do przedwczesnego zakończenia leczenia u > 5% pacjentów leczonych Polayvi® w połączeniu z bendamustyną i rytyksymabą była trombocytopenia (7,9%).
Poniżej przedstawiono efekty uboczne występujące u 586 pacjentów, którzy otrzymywali leczenie lekiem Polayvi® w badaniach klinicznych.
Efekty uboczne są wymienione poniżej według klas układów organizmu MedDRA i kategorii częstości występowania. Odpowiednia kategoria częstości dla każdego efektu ubocznego oparta jest na: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), rzadziej (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000). W każdej grupie według częstości efekty uboczne są wymienione w kolejności malejącej według nasilenia.
Zakażenia i inwazje: bardzo często – zapalenie płuca, zakażenie dróg oddechowych górnych; często – sepsaa, zakażenie wirusem Herpesa, zakażenie cytomegalowirusem, zakażenie dróg moczowychc.
Zaburzenia układu krwi i chłonnego: bardzo często – febrilna neutropenia, neutropenia, trombocytopenia, anemia, leukopenia; często – limfopenia, pancytopenia.
Zaburzenia przemiany materii i odżywiania: bardzo często – hipokaliemia, obniżenie apetytu; często – hipokalcemia, hiperalbuminemia.
Zaburzenia układu nerwowego: bardzo często – neuropatia obwodowa; często – zawroty głowy.
Zaburzenia oka: rzadziej – zamazanie widzeniab.
Zaburzenia układu oddechowego, klatki piersiowej i jamy opłucnowej: bardzo często – kaszel; często – zapalenie płuc, dusznośćc.
Zaburzenia układu pokarmowego: bardzo często – biegunka, nudności, zaparcia, wymioty, mukoryza, ból brzucha.
Zaburzenia skóry i tkanki podskórnej: bardzo często – alopecjaa; często – świąd, zakażenia skóry, wysypka, suchość skóry.
Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej: często – artralgia, mialgia.
Zaburzenia ogólne i stan w miejscu podania: bardzo często – osłabienie, podwyższenie temperatury ciała, osłabienie fizyczne; często – obrzęk obwodowy, dreszcze.
Odchylenia wyników badań laboratoryjnych: bardzo często – utrata masy ciała; często – podwyższenie poziomu transaminaz, podwyższenie poziomu lipazyb, hipofosfatemia.
Urazy, zatrucia i komplikacje procedur: bardzo często – reakcja infuzyjna.
a Efekty uboczne związane z zakończeniem śmiertelnym.
b Efekty uboczne obserwowane wyłącznie u pacjentów z nawrotową lub oporną DLBCL.
c Efekty uboczne obserwowane wyłącznie u wcześniej nieleczonych pacjentów z DLBCL.
Wymienione efekty uboczne obserwowano zarówno u wcześniej nieleczonych pacjentów z DLBCL, jak i u pacjentów z nawrotową lub oporną DLBCL, z wyjątkiem tych wskazanych w przypisach.
Efekty uboczne występujące rzadko i bardzo rzadko: brak.
Opis poszczególnych efektów ubocznych obserwowanych w badaniach klinicznych
Mielosupresja
W badaniu placebo-kontrolowanym GO39942 (POLARIX) 0,5% pacjentów w grupie leczenia Polayvi® + R-CHP przedwcześnie zakończyło badane leczenie z powodu neutropenii. Żaden pacjent nie zakończył leczenia w grupie R-CHOP z powodu neutropenii. Trombocytopenia spowodowała przedwczesne zakończenie leczenia u 0,2% pacjentów w grupie Polayvi® + R-CHP w porównaniu z brakiem takich przypadków w grupie R-CHOP. Żaden pacjent nie zakończył przedwcześnie leczenia z powodu anemii ani w grupie Polayvi® + R-CHP, ani w grupie R-CHOP.
W otwartym badaniu GO29365 lek Polayvi® został przedwcześnie odstawiony z powodu neutropenii u 4% pacjentów w grupach leczenia Polayvi® + bendamustyna + rytyksymab w porównaniu do 2,6% pacjentów w grupie bendamustyna + rytyksymab, u których leczenie zostało zakończone z powodu neutropenii. Trombocytopenia spowodowała zakończenie leczenia u 7,9% pacjentów w grupach Polayvi® + BR i u 5,1% pacjentów w grupie BR. Żaden pacjent w żadnej z grup (grupy leczenia Polayvi® + BR lub tylko grupa BR) nie zakończył leczenia z powodu anemii. W grupach leczenia Polayvi® + bendamustyna + rytyksymab o neutropenii, trombocytopenii i anemii stopnia 3 lub wyższego zgłoszono odpowiednio u 40,4%, 25,8% i 12,6% pacjentów.
Neuropatia obwodowa (PNO)
W badaniu placebo-kontrolowanym GO39942 (POLARIX) w grupie leczenia Polayvi® + R-CHP o PNO stopnia 1, 2 i 3 zgłoszono odpowiednio u 39,1%, 12,2% i 1,6% pacjentów. W grupie R-CHOP o PNO stopnia 1, 2 i 3 zgłoszono odpowiednio u 37,2%, 15,5% i 1,1% pacjentów. O przypadkach PNO stopnia 4–5 nie zgłaszano ani w grupie Polayvi® + R-CHP, ani w grupie R-CHOP. W grupie Polayvi® + R-CHP 0,7% pacjentów przedwcześnie zakończyło leczenie z powodu PNO w porównaniu do 2,3% w grupie R-CHOP. W grupie Polayvi® + R-CHP dawkę leczenia zmniejszono u 4,6% pacjentów z powodu PNO w porównaniu do 8,2% w grupie R-CHOP. W grupie Polayvi® + R-CHP mediana czasu do pierwszego objawu PNO wynosiła 2,27 miesiąca w porównaniu do 1,87 miesiąca w grupie R-CHOP. Na dzień zakończenia zbierania danych klinicznych objawy PNO ustąpiły u 57,8% pacjentów w grupie Polayvi® + R-CHP w porównaniu do 66,9% w grupie R-CHOP. Mediana czasu ustąpienia neuropatii obwodowej wynosiła 4,04 miesiąca w grupie Polayvi® + R-CHP w porównaniu do 4,6 miesiąca w grupie R-CHOP.
W otwartym badaniu GO29365 w grupach leczenia Polayvi® + bendamustyna + rytyksymab o PNO stopnia 1 i 2 zgłoszono odpowiednio u 15,9% i 12,6% pacjentów. W grupie bendamustyna + rytyksymab przypadki PNO stopnia 1 i 2 odnotowano u 2,6% i 5,1% pacjentów odpowiednio. O jednym przypadku PNO stopnia 3 zgłoszono w grupach Polayvi® + BR i nie zgłoszono żadnego przypadku w grupie BR. O przypadkach PNO stopnia 4–5 nie zgłoszono w żadnej grupie (grupy leczenia Polayvi® + BR lub tylko grupa BR). Leczenie lekiem Polayvi® przedwcześnie zakończono u 2,6% pacjentów z powodu PNO i dawkę Polayvi® zmniejszono u 2% pacjentów z powodu PNO. Żaden pacjent w grupie BR nie zakończył przedwcześnie leczenia ani nie miał zmniejszonej dawki z powodu PNO. W grupach Polayvi® + BR mediana czasu do pierwszego zdarzenia PNO wynosiła 1,6 miesiąca, a u 39,1% pacjentów z PNO zgłoszono ustąpienie objawów.
Zakażenia
W badaniu placebo-kontrolowanym GO39942 (POLARIX) o zakażeniach, w tym zapaleniu płuc i innych typach zakażeń, zgłoszono u 49,7% pacjentów w grupie Polayvi® + R-CHP i u 42,7% pacjentów w grupie R-CHOP. Zakażenia stopnia 3–4 wystąpiły u 14% pacjentów w grupie Polayvi® + R-CHP i u 11,2% pacjentów w grupie R-CHOP. W grupie leczenia Polayvi® + R-CHP o poważne zakażenia zgłoszono u 14% pacjentów, a zakażenia zakończone śmiercią odnotowano u 1,1% pacjentów. W grupie R-CHOP o poważne zakażenia zgłoszono u 10,3% pacjentów, a zakażenia zakończone śmiercią odnotowano u 1,4% pacjentów. Leczenie z powodu zakażenia przerwano u 7 pacjentów (1,6%) w grupie Polayvi® + R-CHP w porównaniu do 10 pacjentów (2,3%) w grupie R-CHOP.
W otwartym badaniu GO29365 o zakażeniach, w tym zapaleniu płuc i innych typach zakażeń, zgłoszono u 48,3% pacjentów w grupach Polayvi® + BR i u 51,3% pacjentów w grupie BR. W grupach Polayvi® + BR o poważne zakażenia zgłoszono u 27,2% pacjentów, a o zakażenia zakończone śmiercią – u 6,6% pacjentów. W grupie BR o poważne zakażenia zgłoszono u 30,8% pacjentów, a o zakażenia zakończone śmiercią – u 10,3% pacjentów. Leczenie z powodu zakażenia przedwcześnie zakończono u 4 pacjentów (2,6%) w grupach Polayvi® + BR w porównaniu do 2 pacjentów (5,1%) w grupie BR.
Postępująca wielofocalna leukoencefalopatia (PML)
W badaniu placebo-kontrolowanym GO39942 (POLARIX) nie zgłoszono przypadków postępującej wielofocalnej leukoencefalopatii.
W otwartym badaniu GO29365 jeden przypadek PML zakończony śmiercią zaobserwowano u jednego pacjenta, który otrzymywał leczenie Polayvi® + bendamustyna + obinutuzumab. Pacjent ten wcześniej otrzymał trzy linie terapii, w tym przeciwciała przeciwko CD20.
Hepatotoksyczność
W badaniu placebo-kontrolowanym GO39942 (POLARIX) o hepatotoksyczności zgłoszono u 10,6% pacjentów w grupie leczenia Polayvi® + R-CHP i u 7,3% pacjentów w grupie R-CHOP. W grupie Polayvi® + R-CHP większość zdarzeń miała stopień 1–2 (8,7%); o zdarzeniach stopnia 3 zgłoszono u 1,8% pacjentów. Nie zaobserwowano zdarzeń stopnia 4 lub 5. U jednego pacjenta (0,2%) odnotowano poważną hepatotoksyczność, która była odwracalna.
W innym badaniu zgłoszono dwa przypadki poważnej hepatotoksyczności (uszkodzenie hepatocytarne i stłuszczenie wątroby), które były odwracalne.
Toxiczność przewodu pokarmowego
W badaniu placebo-kontrolowanym GO39942 (POLARIX) zdarzenia toksyczności przewodu pokarmowego odnotowano u 76,1% pacjentów w grupie Polayvi® + R-CHP w porównaniu do 71,9% pacjentów w grupie R-CHOP. Większość zdarzeń miała stopień 1–2, a zdarzenia ≥ 3 stopnia odnotowano u 9,7% pacjentów w grupie Polayvi® + R-CHP w porównaniu do 8,2% pacjentów w grupie R-CHOP. Najczęstsze zdarzenia toksyczności przewodu pokarmowego to nudności i biegunka.
W otwartym badaniu GO29365 o zdarzeniach toksyczności przewodu pokarmowego zgłoszono u 72,8% pacjentów w grupie Polayvi® + BR w porównaniu do 66,7% pacjentów w grupie BR. Większość zdarzeń miała stopień 1–2. O zdarzeniach stopnia 3–4 zgłoszono u 16,5% pacjentów w grupie Polayvi® + BR w porównaniu do 12,9% pacjentów w grupie BR. Najczęstsze zdarzenia toksyczności przewodu pokarmowego to biegunka i nudności.
Okres ważności.
30 miesięcy.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8 ºC w oryginalnym opakowaniu w celu ochrony przed światłem. Nie zamrażać. Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Nie należy mieszać ani rozcieńczać tego leku innymi lekami, z wyjątkiem tych wskazanych w sekcji „Środki ostrożności przed przygotowaniem lub podaniem leku”.
Opakowanie.
Fiolka o pojemności 6 ml z bezbarwnego szkła (szkło borokrzemowe, klasa I), zamknięta butylorubberową zawleczką (laminowaną folią fluororubberową) i uszczelniona aluminiową pokrywką z plastikowym dyskiem typu „flip-off”. Po 30 mg w fiolce. Po 1 fiolce w tekturowym pudełku.
Fiolka o pojemności 20 ml z bezbarwnego szkła (szkło borokrzemowe, klasa I), zamknięta butylorubberową zawleczką (laminowaną folią fluororubberową) i uszczelniona aluminiową pokrywką z plastikowym dyskiem typu „flip-off”. Po 140 mg w fiolce. Po 1 fiolce w tekturowym pudełku.
Kategoria wydawania.
Na receptę.
Producent.
F. Hoffmann-La Roche Ltd
Miejsce produkcji i adres działalności.
Wurmisweg, 4303 Kaiseraugst, Szwajcaria