Nuviq / Nuwiq®
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU Nuviq / Nuwiq®
Skład:
Substancja czynna: simoctokog alfa (rekombinowany czynnik krzepnięcia krwi VIII);
1 fiolka proszku do sporządzenia roztworu do wstrzykiwań zawiera simoctokog alfa (rekombinowany czynnik krzepnięcia krwi VIII) 1500 J, lub 2500 J, lub 3000 J, lub 4000 J;
Substancje pomocnicze: sodu chlorek; sacharoza; L-argininy chlorowodorek; wapnia chlorek, dwuwodny; poloksymer 188; sodu cytrynian, dwuwodny.
Roztwórnik: woda do wstrzykiwań.
Postać farmaceutyczna. Proszek i roztwórnik do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne:
Proszek: biała grudka. Możliwa niewielka ilość białego proszku.
Roztwórnik: przejrzysta, bezbarwna ciecz, bez cząstek.
Grupa farmakoterapeutyczna. Środki przeciwkrwotoczne. Czynnik krzepnięcia krwi VIII.
Kod ATC B02B D02.
Właściwości farmakologiczne.
Farmakodynamika.
Czynnik krzepnięcia krwi VIII wiąże się z czynnikiem von Willebranda w krążeniu pacjenta. Aktywny czynnik VIII działa jako kofaktor dla aktywnego czynnika IX, skracając czas przekształcania czynnika X w aktywny czynnik X. Aktywny czynnik X przekształca protrombinę w trombinę. Następnie trombina przekształca fibrynogen w fibrynę, w wyniku czego powstaje skrzep.
Hemofilia A to dziedziczna choroba krwiotoczna związana z płcią, spowodowana wrodzonym niedoborem czynnika krzepnięcia VIII:C, prowadzącym do krwawień do stawów, mięśni, narządów wewnętrznych, które pojawiają się spontanicznie lub w wyniku urazu przypadkowego lub chirurgicznego. W leczeniu zastępczym poziomy czynnika VIII w osoczu wzrastają, co prowadzi do tymczasowej korekty niedoboru czynnika krzepnięcia krwi VIII i skłonności do krwawień.
Populacja dorosłych i pacjentów w wieku 12–65 lat.
Profilaktyka: W badaniu klinicznym u 32 dorosłych pacjentów z ciężką hemofilią A średnia dawka profilaktyczna leku Nuviq wynosiła 468,7 J/ kg/miesiąc. Leczenie krwawień: Średnia dawka stosowana w leczeniu krwawień u pacjentów otrzymujących profilaktykę wynosiła 33,0 J/kg. W innym badaniu klinicznym 22 dorosłych pacjentów otrzymywało leczenie na żądanie. W 986 przypadkach krwawień zastosowano średnią dawkę 30,9 J/kg. Ogólnie niewielkie krwawienia wymagały mniejszych dawek, natomiast silne krwawienia wymagały średnich dawek trzykrotnie wyższych.
Indywidualizowana profilaktyka: Oceny indywidualizowanej profilaktyki opartej na FVIII poddano 66 dorosłych pacjentów z ciężką hemofilią A, którzy wcześniej otrzymywali leczenie (PTPs). Po 1–3 miesięcznym standardowym okresie profilaktyki (dawka co drugi dzień lub 3 razy w tygodniu), 44 (67%) pacjentów przełożono na schemat dawkowania oparty na ocenie ich FVIII, a 40 pacjentów ukończyło 6-miesięczną profilaktykę zgodnie z przepisanym trybem i dawkowaniem. Spośród tych pacjentów, 34 (85%) otrzymywało leczenie dwa razy w tygodniu lub rzadziej. 33 (82,5%) pacjentów nie miało żadnych krwawień, a 36 (90,0%) pacjentów nie miało krwawień spontanicznych. Średnia + SD roczna częstość krwawień (obliczona rocznie) wynosiła 1,2 + 3,9, a średnia + SD dawka wynosiła 52,2 + 12,2 J/kg na zastrzyk oraz 99,7 + 25,6 J/kg na tydzień.
Należy zauważyć, że roczna częstość krwawień (ABR) nie jest porównywalna między różnymi koncentratami czynnika ani między różnymi badaniami klinicznymi.
Populacja pediatryczna
Dane uzyskano u 29 wcześniej leczonych dzieci w wieku od 2 do 5 lat, 31 dzieci w wieku od 6 do 12 lat oraz jednego nastolatka w wieku 14 lat. Średnia profilaktyczna dawka dożylna wynosiła 37,8 J/kg. Dwadzieścia pacjentów otrzymało średnie dawki powyżej 45 J/kg. Średnia profilaktyczna dawka leku Nuviq miesięcznie wynosiła 521,9 J/kg. W leczeniu krwawień dzieciom podawano wyższą średnią dawkę leku Nuviq (43,9 J/kg) niż dorosłym (33,0 J/kg), a także wyższą średnią dawkę w leczeniu zarówno mniejszych, jak i większych krwawień (78,2 J/kg vs 41,7 J/kg). Dzieciom w młodszym wieku ogólnie wymagane były wyższe średnie dawki (6–12 lat – 43,9 J/kg; 2–5 lat – 52,6 J/kg). Te dane potwierdzono w długoterminowej obserwacji medycznej 49 takich dzieci leczonych przez dodatkowy średni okres około 30 miesięcy (zakres od 9,5 do 52 miesięcy); w tym okresie 45% dzieci nie miało krwawień spontanicznych.
Dane od 108 pacjentów z ciężką hemofilią A (˂ 1% FVIII:C), którzy wcześniej nie otrzymywali leczenia, uzyskano w prospektywnym otwartym badaniu klinicznym. U większości pacjentów leczenie profilaktyczne rozpoczęto po wystąpieniu pierwszego krwawienia wymagającego leczenia.
Farmakokinetyka.
Populacja dorosła
Tabela 1.
Parametry farmakokinetyczne leku Nuviq (dawka 50 J/kg) u dorosłych w wieku od 18 do 65 lat z ciężką hemofilią A, którzy wcześniej otrzymywali leczenie (n=20).
| Parametry farmakokinetyczne |
Analiza chromogeniczna |
|
| Średnia ± SD |
Mediana (zakres) |
|
| AUC (godz*MO/ml) |
22,6 ± 8,0 |
22,3 (8,4 – 38,1) |
| T 1/2 (godz) |
14,7 ± 10,4 |
12,5 (5,4 – 55,6) |
| IVR (%/MO/kg) |
2,5 ± 0,4 |
2,5 (1,7 – 3,2) |
| CL (ml/godz/kg) |
3,0 ± 1,2 |
2,7 (1,5 – 6,4) |
AUC – pole pod krzywą (FVIII:C), T½ – okres półwydalenia
IVR – odzysk in vivo, CL – klirens, SD – odchylenie standardowe
Tabela 2.
Parametry farmakokinetyczne leku Nuviq (dawka 50 J/ kg) u dzieci w wieku od 6 do 12 lat z ciężką hemofilią A, które wcześniej były leczone (n=12).
| Farmakokinetyczne parametry |
Analiza chromogeniczna |
|
| Średnia ± SD |
Mediana (zakres) |
|
| AUC (godz*MO/ml) |
13,2 ± 3,4 |
12,8 (7,8 – 19,1) |
| T 1/2 (godz) |
10,0 ± 1,9 |
9,9 (7,6 – 14,1) |
| IVR (%/MO/kg) |
1,9 ± 0,4 |
1,9 (1,2 – 2,6) |
| CL (ml/godz/kg) |
4,3 ± 1,2 |
4,2 (2,8 – 6,9) |
AUC – pole pod krzywą (FVIII:C), T½ – okres półwydalania
IVR – odzysk in vivo, CL – klirens, SD – odchylenie standardowe
Tabela 3.
Parametry farmakokinetyczne leku Nuviq (dawka 50 JM/kg) u dzieci w wieku od 2 do 5 lat z ciężką hemofilią typu A, które wcześniej były leczone (n = 13).
| Parametry farmakokinetyczne |
Analiza chromogeniczna |
|
| Średnia ± SD |
Mediana (zakres) |
|
| AUC (godz*MO/ml) |
11,7 ± 5,3 |
10,5 (4,9 – 23,8) |
| T 1/2 (godz) |
9,5 ± 3,3 |
8,2 (4,3 – 17,3) |
| IVR (%/MO/kg) |
1,9 ± 0,3 |
1,8 (1,5 – 2,4) |
| CL (ml/godz/kg) |
5,4 ± 2,4 |
5,1 (2,3 – 10,9) |
AUC – pole pod krzywą (FVIII:C), T½ – okres półtrwania
IVR – odzysk in vivo, CL – klirens, SD – odchylenie standardowe
Populacja pediatryczna
Zgodnie z opublikowanymi danymi, wskaźniki odzysku i okres półtrwania były niższe u dzieci w młodszym wieku w porównaniu z dorosłymi, natomiast wskaźnik klirensu był wyższy, co może częściowo wynikać z większej objętości osocza na kilogram masy ciała u młodszych pacjentów.
Podgrupy według masy ciała
Tabela 4.
Parametry farmakokinetyczne leku Nuviq (dawka 50 J/ kg) u dorosłych w wieku od 18 do 65 lat z ciężką hemofilią typu A, którzy wcześniej byli leczeni (n=20), według masy ciała pacjenta
| Parametry farmakokinetyczne |
Wszyscy pacjenci (n =20) |
O prawidłowej masie ciała (n =14) |
Przed otyłością (n =4) |
Otyłość (n =2) |
| Analiza chromogenna, średnia ± SD |
||||
| AUC (godz*MO/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
| T 1/2 (godz) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
| IVR (%/MO/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
| CL (ml/godz/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
| Analiza chromogenna, mediana (zakres) |
||||
| AUC (godz*MO/ml) |
22,3 (8,4 – 38,1) |
21,2 (8,4– 32,6) |
23,3 (17,4 – 35,5) |
33,5 (28,9 – 38,1) |
| T 1/2 (godz) |
12,5 (5,4 – 55,6) |
12,3 (5,4– 55,6) |
11,2 (9,3 – 22,0) |
17,2 (13,8 – 20,6) |
| IVR (%/MO/kg) |
2,5 (1,7 – 3,2) |
2,4 (1,7 – 3,1) |
2,8 (2,3 – 3,2) |
2,8 (2,6 – 3,0) |
| CL (ml/godz/kg) |
2,7 (1,5 – 6,4) |
2,8 (1,7 – 6,4) |
2,5 (1,6 – 3,7) |
1,8 (1,5 – 2,0) |
Prawidłowa masa ciała BMI – 18,5–25 kg/m², nadmiar masy ciała: BMI 25–30 kg/m², otyłość: BMI > 30 kg/m², SD – odchylenie standardowe
Dane kliniczne.
Wskazania.
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (wrodzony niedobór czynnika VIII).
Preparat Nuviq może być stosowany u pacjentów wszystkich grup wiekowych.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek ze składników pomocniczych preparatu leczniczego.
Interakcje z innymi lekami i inne rodzaje oddziaływań.
Nie przeprowadzono żadnych badań dotyczących interakcji leku Nuviq z innymi lekami.
Szczególne środki ostrożności.
Śledzenie produktu
W celu poprawy śledzenia produktów biologicznych należy dokładnie rejestrować nazwę handlową oraz numer serii podanego leku.
Specyficzna aktywność leku Nuviq wynosi około 9500 j.m./mg białka.
Simoctokog alfa (czynnik krzepnięcia krwi VIII (rDNA)) to czysty białek zawierający 1440 aminokwasów. Sekwencja aminokwasów jest podobna do formy czynnika VIII osocza ludzkiego o masie 90+80 kDa (tj. z usuniętym domeną B). Nuviq jest wytwarzany metodą rekombinowanego DNA w genetycznie zmodyfikowanych komórkach nerek embrionalnych ludzkich (HEK) 293F. W trakcie procesu wytwarzania oraz w końcowym produkcie nie stosuje się materiałów pochodzenia zwierzęcego ani ludzkiego.
Zwiększona wrażliwość
Tak jak w przypadku stosowania każdego leku białkowego podawanego dożylnie, istnieje ryzyko wystąpienia reakcji alergicznych i nadwrażliwości. Nuviq zawiera śladowe ilości białek ludzkich komórek, które różnią się od czynnika VIII. W przypadku wystąpienia objawów nadwrażliwości należy natychmiast przerwać stosowanie leku i skonsultować się z lekarzem. Pacjentów należy poinformować o wczesnych objawach reakcji nadwrażliwości, takich jak pokrzywka, uogólniona pokrzywka, uczucie ucisku w klatce piersiowej, trudności w oddychaniu, świsty, hipotensja tętnicza i anafilaksja.
W przypadku wystąpienia szoku należy podjąć standardowe leczenie stanu wstrząsu.
Inhibitory
Tworzenie się przeciwciał neutralizujących (inhibitorów) czynnika VIII jest znanym powikłaniem występującym podczas leczenia osób z hemofilią A. Inhibitory te są zazwyczaj immunoglobulinami klasy IgG, których działanie skierowane jest przeciwko aktywności prokoagulacyjnej czynnika VIII i które ilościowo oznacza się w jednostkach Bethesda (BU) na 1 ml osocza, stosując test modyfikowany. Ryzyko powstawania inhibitorów koreluje z ciężkością choroby oraz zależy od ekspozycji na czynnik VIII, przy czym jest najwyższe w pierwszych 50 dniach ekspozycji, ale utrzymuje się przez całe życie, choć jest rzadkie.
Obserwowano przypadki ponownego powstawania inhibitorów (niski tytuł) u pacjentów zmieniających jeden preparat rekombinowanego czynnika VIII na inny, którzy wcześniej leczono preparatem przez ponad 100 dni i u których w wywiadzie występowało tworzenie się inhibitorów. Z tego powodu zaleca się dokładne monitorowanie wszystkich pacjentów pod kątem powstawania inhibitorów po każdej zmianie leku.
Kliniczne znaczenie powstawania inhibitorów zależy od ich tytułu: inhibitory o niskim tytule, które występują tymczasowo lub stale utrzymują niski tytuł, stanowią mniejsze ryzyko niewystarczającej odpowiedzi klinicznej niż inhibitory o wysokim tytule.
Wszyscy pacjenci leczeni rekombinowanymi czynnikami krzepnięcia krwi VIII powinni być dokładnie monitorowani pod kątem powstawania inhibitorów poprzez odpowiednią obserwację kliniczną i badania laboratoryjne. Jeśli oczekiwane poziomy aktywności czynnika VIII we krwi nie są osiągane lub krwawienie nie ustępuje mimo podania odpowiedniej dawki leku, należy przeprowadzić badania w celu wykrycia obecności inhibitorów czynnika VIII. U pacjentów z wysokimi tytorami inhibitorów terapia czynnikiem VIII może być nieskuteczna, dlatego należy rozważyć inne opcje terapeutyczne, takie jak indukcja tolerancji immunologicznej (ITI). Leczenie takich pacjentów powinno być prowadzone przez lekarzy doświadczonych w leczeniu hemofilii i przypadkach powstawania inhibitorów czynnika VIII.
Powikłania sercowo-naczyniowe
U pacjentów z istniejącymi czynnikami ryzyka chorób sercowo-naczyniowych terapia zastępcza FVIII może zwiększyć ryzyko wystąpienia chorób sercowo-naczyniowych.
Powikłania związane z zastosowaniem kaniuli
W przypadku konieczności stosowania urządzenia do dostępu do żyły centralnej (CVAD) należy rozważyć ryzyko wystąpienia powikłań związanych z jego użyciem, w tym infekcji miejscowych, bakteriemii i zakrzepicy w miejscu umiejscowienia kaniuli.
Zdecydowanie zaleca się, aby przy każdym podaniu leku Nuviq rejestrować nazwę handlową oraz numer partii (serii) leku w celu ustalenia związku między stanem pacjenta a podaną partią leku.
Wszystkie ostrzeżenia dotyczą zarówno dorosłych, jak i dzieci.
Informacje dotyczące substancji pomocniczych (zawartość sodu)
1 ml rozcieńczonego roztworu zawiera 7,35 mg (18,4 mg sodu na fiolkę), co oznacza, że „praktycznie nie zawiera sodu”.
Jednakże, w zależności od masy ciała i dawki, pacjent może otrzymać więcej niż jedną fiolkę. Należy wziąć to pod uwagę u pacjentów przestrzegających diety z kontrolowaną zawartością sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Nie przeprowadzono badań wpływu Nuviq na funkcję rozrodczą u zwierząt. Ponieważ hemofilia A rzadko występuje u kobiet, brak doświadczeń z zastosowaniem Nuviq w czasie ciąży i karmienia piersią. Dlatego Nuviq może być przepisywany w czasie ciąży i karmienia piersią w przypadku nagłej potrzeby. Brak danych dotyczących wpływu na płodność.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
Nie stwierdzono żadnego wpływu na zdolność prowadzenia pojazdów lub pracy z innymi urządzeniami mechanicznymi.
Sposób stosowania i dawki.
Leczenie powinno być prowadzone pod nadzorem lekarza posiadającego doświadczenie w leczeniu hemofilii.
Monitorowanie leczenia
W trakcie leczenia zaleca się odpowiednie oznaczanie poziomów czynnika VIII w celu kontrolowania wymaganej dawki i częstotliwości powtórnego wlewu. Indywidualni pacjenci mogą wykazywać różną odpowiedź na stosowanie czynnika VIII, co objawia się różnym okresem półtrwania i czasem regeneracji (odzyskiwania). Dawka dostosowana do masy ciała może wymagać korekty w przypadku niedostatecznej lub nadmierniej masy ciała. W przypadku rozległych zabiegów chirurgicznych konieczny jest dokładny monitoring terapii zastępczej poprzez analizę krzepnięcia krwi (aktywność czynnika VIII w osoczu).
Stosowanie jednoetapowego testu aktywności krzepnięcia krwi opartego na czasie tromboplastynowym częściowym (aPTT) in vitro do oznaczania aktywności czynnika VIII w próbkach krwi pacjentów może prowadzić do istotnego wpływu na wyniki aktywności czynnika VIII w osoczu krwi zarówno rodzaju używanego odczynnika aPTT, jak i standardowego wzorca stosowanego w teście. Mogą również występować znaczące rozbieżności między wynikami uzyskanymi w jednoetapowym teście aktywności krzepnięcia krwi opartym na aPTT a testem chromogennym zgodnie z Europejską Farmakopeą. Jest to szczególnie ważne, szczególnie podczas zmiany laboratorium i/lub odczynników stosowanych w teście.
Sposób stosowania
Dawkowanie i trwanie terapii zastępczej zależą od ciężkości niedoboru czynnika VIII, lokalizacji i stopnia krwawienia, jak również od stanu klinicznego pacjenta.
Ilość wprowadzanych jednostek czynnika VIII określa się w jednostkach międzynarodowych (j.m.), które odnoszą się do obowiązującego standardu koncentratu WHO dla leków zawierających czynnik VIII. Aktywność czynnika VIII w osoczu określa się albo w procentach (w stosunku do osocza normalnego człowieka), albo głównie w jednostkach międzynarodowych (zgodnie z międzynarodowym standardem dla czynnika VIII w osoczu).
1 jednostka międzynarodowa (j.m.) aktywności czynnika VIII odpowiada ilości czynnika VIII zawartemu w 1 ml osocza normalnego człowieka.
Leczenie na żądanie
Obliczenia wymaganej dawki czynnika VIII opierają się na danych empirycznych, że 1 jednostka międzynarodowa (j.m.) czynnika VIII na 1 kg masy ciała zwiększa aktywność czynnika VIII w osoczu krwi o około 2% w stosunku do aktywności normalnej lub o 2 j.m/dl. Wymaganą dawkę określa się za pomocą następującego wzoru:
I.
Wymagane jednostki = masa ciała (kg) × pożądany wzrost czynnika VIII (%) (j.m./dl) × 0.5 (j.m./kg na j.m./dl)
II.
| Oczekiwany wzrost czynnika VIII (% normy) = |
2 × podane j.m./ |
| masa ciała (kg) |
II.
Dawka do podania oraz częstotliwość podawania zawsze powinny być dostosowane do skuteczności klinicznej w każdym indywidualnym przypadku.
W przypadku poniżej wymienionych sytuacji krwotocznych aktywność czynnika VIII nie powinna spaść poniżej określonego poziomu aktywności osocza (w % od wartości normalnej lub j.m./dL) w odpowiednim okresie. Tabelę 5 można wykorzystać do ustalenia dawkowania w okresach krwotoków i zabiegów chirurgicznych.
Tabela 5
| Stopień krwawienia/ typ procedury chirurgicznej |
Wymagany poziom czynnika VIII (%) (JE/dl) |
Częstotliwość dawkowania (godziny)/ trwanie terapii (dni) |
| Krwawienie Wczesny hemartroza, krwawienie mięśniowe lub krwawienie jamy ustnej |
20–40 |
Powtarzaj co 12–24 godziny przez co najmniej 1 dzień, aż krwawienie towarzyszące bólowi ustąpi lub do pełnego wyleczenia |
| Bardziej nasilony hemartroza, krwawienie mięśniowe lub hematoma |
30–60 |
Powtarzaj co 12–24 godziny przez 3–4 dni lub dłużej, aż ustąpi ból i ostre zaburzenia |
| Krwawienie zagrażające życiu |
60–100 |
Powtarzaj wstrzyknięcia co 8–24 godziny, aż minie niebezpieczeństwo |
| Operacja Niewielka operacja, w tym usunięcie zęba Wielka operacja |
30–60 |
Co 24 godziny przez co najmniej 1 dzień do pełnego wygojenia |
| 80–100 (przed i po zabiegu chirurgicznym) |
Powtarzaj wstrzyknięcia co 8–24 godziny aż do istotnego zagojenia rany, następnie kontynuuj terapię przez co najmniej 7 dni, aby utrzymać aktywność czynnika VIII na poziomie 30–60% (JE/dl) |
Profilaktyka
Długotrwałe zapobieganie krwawieniom u pacjentów z ciężką hemofilią A zwykle wymaga dawkowania od 20 do 40 J mW FVIII na 1 kg masy ciała co 2–3 dni. W niektórych przypadkach, szczególnie u młodszych pacjentów, może być konieczne zwiększenie dawki lub częstotliwości podawania leku.
Zaleca się monitorowanie poziomów czynnika VIII w trakcie leczenia w celu dostosowania dawki i częstotliwości powtórnego wlewu. W przypadku dużych zabiegów chirurgicznych konieczna jest dokładna kontrola terapii zastępczej poprzez oznaczanie aktywności czynnika VIII w osoczu. W zależności od czynnika VIII, poszczególni pacjenci mogą wykazywać różne okresy półtrwania i odnowienia. Dawkowanie można dostosowywać na podstawie odpowiedzi pacjenta.
Sposób stosowania jest taki sam zarówno dla dorosłych, jak i dla dzieci oraz młodzieży, jednak u dzieci i młodzieży mogą być wymagane krótsze odstępy między dawkami lub większe dawki. Brak danych dotyczących stosowania u dzieci poniżej 2. roku życia.
Sposób podania
Lek Nuviq / Nuwiq® przeznaczony jest do wstrzykiwania dożylnego.
Zaleca się podawanie nie więcej niż 4 ml na minutę.
Instrukcje dotyczące przygotowania leku przed podaniem.
Proszek należy rozpuszczać wyłącznie za pomocą dołączonego rozpuszczalnika (2,5 ml wody do wstrzykiwań), przy użyciu zestawu do przygotowania roztworu do wstrzykiwań. Fiolkę należy delikatnie wymieszać aż do całkowitego rozpuszczenia proszku. Po rozpuszczeniu roztwór należy za pomocą strzykawki, w której znajdował się rozpuszczalnik, wciągnąć do strzykawki.
Przygotowany roztwór należy sprawdzić wizualnie pod kątem obecności cząstek stałych i zmiany barwy przed podaniem. Rozcieńczony roztwór powinien być przezroczysty i bezbarwny, bez obcych cząstek oraz mieć wartość pH od 6,5 do 7,5. Nie wolno stosować roztworów nieprzezroczystych ani roztworów zawierających osad.
Instrukcje dotyczące przygotowania i podania leku.
|
Rys. 1 |
|
Rys. 2 |
|
Rys. 3 |
|
Rys. 4 |
|
Rys. 5 |
|
Rys. 6 |
|
Rys. 7 |
|
Rys. 8 |
|
|
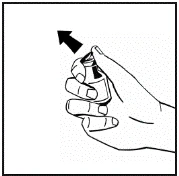
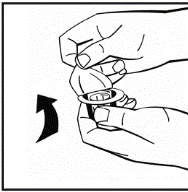
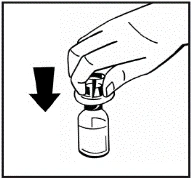
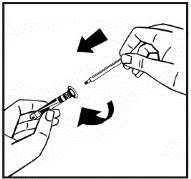
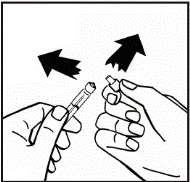
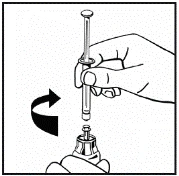
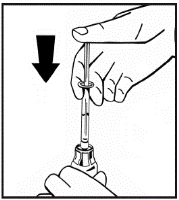
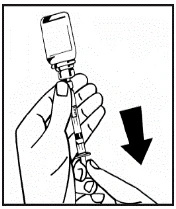
Jeśli do jednej procedury stosuje się więcej niż jeden fiolki z proszkiem, można ponownie użyć tej samej igły do wstrzykiwań. Nakładka fiolki i strzykawka przeznaczone są wyłącznie do jednorazowego użytku.
Niezużyty lek oraz odpady należy zutylizować zgodnie z lokalnymi przepisami.
Dzieci.
Nuviq można stosować dzieciom w każdym wieku, jednak brakuje danych dotyczących stosowania u dzieci poniżej 2. roku życia.
Przedawkowanie.
Nie zgłaszano przypadków przedawkowania.
Działania niepożądane
Podsumowanie profilu bezpieczeństwa
Reakcje nadwrażliwościowe i alergiczne (mogą obejmować obrzęk naczynioruchowy, uczucie pieczenia i mrowienia w miejscu infuzji, dreszcze, zaczerwienienie, ogólne pokrzywki, ból głowy, pokrzywkę, hipotensję tętniczą, senność, nudności, wysypkę, pobudzenie, tachykardię, duszność, mrowienie, pokrzywkę, ogólne pokrzywki, wymioty, chrypkę/szmer oddychania) rzadko występują po podaniu leków zawierających czynnik FVIII, ale w niektórych przypadkach mogą postępować do ciężkiej anafilaksji (w tym wstrząsu).
U pacjentów z hemofilią A leczonych lekami zawierającymi czynnik VIII, w tym lekiem Nuviq / Nuwiq®, mogą rozwijać się przeciwciała neutralizujące (inhibitory) przeciwko czynnikowi VIII. W przypadku pojawienia się inhibitorów za przyczynę niewystarczającej odpowiedzi klinicznej uważa się trwające lub częste krwawienia mimo odpowiedniej profilaktyki. W takich przypadkach zaleca się kontakt z wyspecjalizowanym ośrodkiem hemofilii.
Lista działań niepożądanych w formie tabeli
Poniższa tabela 6 została ułożona według klasyfikacji układów narządów MedDRA (Medical Dictionary for Regulatory Activities). Częstość została określona na podstawie danych z badań klinicznych przeprowadzonych u 355 pacjentów z ciężką hemofilią A, w tym u 247 pacjentów wcześniej leczonych (PTP) i 108 pacjentów wcześniej nieleczonych (PUP).
Częstość została oszacowana według następujących kryteriów: bardzo często (≥1/10); często (≥1/100 do <1/10); rzadko (≥1/1000 do <1/100); rzadko (≥1/10000 do <1/1000); bardzo rzadko (<1/10000); nieznana (nie można oszacować na podstawie dostępnych danych).
W każdej grupie częstości działania niepożądane są wymienione w kolejności zmniejszającego się nasilenia.
Tabela 6.
| Standardowa klasa układów i narządów MedDRA |
Reakcje niepożądane |
Częstotliwość |
| Zaburzenia układu krwiotwórczego i limfatycznego |
Anemia Inhibicja czynnika VIII Anemia hemoragiczna |
rzadko* rzadko (PTPs)# bardzo często (PUPs)# rzadko* |
| Zaburzenia układu odpornościowego |
Zwiększona wrażliwość |
często* |
| Zaburzenia układu nerwowego |
Zawroty głowy Bóle głowy Paraesthesia (zaburzenia czucia) |
rzadko* rzadko* rzadko* |
| Zaburzenia narządu słuchu i równowagi |
Zawroty głowy |
rzadko* |
| Zaburzenia układu oddechowego, narządów klatki piersiowej i jamy śródpiersia |
Dyspnea (trudności w oddychaniu) |
rzadko* |
| Zaburzenia układu pokarmowego |
Suchość w ustach |
rzadko* |
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
Ból pleców |
rzadko* |
| Zaburzenia ogólne oraz w miejscu podania |
Pirogę (gorączka) Ból w klatce piersiowej Zapalenie w miejscu iniekcji Ból w miejscu iniekcji Niedowolność |
często* rzadko* rzadko* rzadko* rzadko* |
| Badania |
Pozytywne przeciwciała nie mające aktywności neutralizującej (u PTPs) |
rzadko* |
*Obliczono jako liczbę pacjentów z reakcjami niepożądanymi na ogólną liczbę 355 badanych pacjentów, z których 247 pacjentów wcześniej otrzymywało leczenie (PTPs), a 108 pacjentów nie otrzymywało wcześniej leczenia (PUPs).
Częstość oparta na badaniach ze wszystkimi lekami czynnika VIII, które obejmowały pacjentów z ciężką hemofilią A.
PTPs = pacjenci wcześniej leczeni, PUPs = pacjenci nieleczeni wcześniej.
Opis pojedynczych reakcji niepożądanych
Antyciała przeciwko antygenom czynnika VIII bez aktywności neutralizującej wykryto u jednego dorosłego pacjenta (patrz tabela 6). Wynik był pozytywny tylko w roztworze czynnika 1, a miana przeciwciał były bardzo niskie. Aktywność inhibitorów, mierzona zmodyfikowanym testem Bethesda, nie została wykryta u tego pacjenta. Skuteczność kliniczna oraz in vivo odzyskanie leku Nuviq nie zostały u tego pacjenta zaburzone.
Dzieci
Częstość, rodzaj i ciężkość reakcji niepożądanych u dzieci są przewidywane jako takie same jak u dorosłych.
Zgłaszanie potencjalnych reakcji niepożądanych
Zgłaszanie potencjalnych reakcji niepożądanych po rejestracji produktu leczniczego jest ważne. Pozwala to na kontynuację monitorowania bilansu korzyści i ryzyka leku. Specjaliści opieki zdrowotnej powinni zgłaszać potencjalne reakcje niepożądane.
Okres ważności.
Proszek: 2 lata.
Roztwórnik (woda do wstrzykiwań): 5 lat.
Przez cały okres ważności możliwe jest przechowywanie leku nie dłużej niż 1 miesiąc w temperaturze pokojowej (do 25 °C). Jeśli lek został już wyjęty z lodówki, nie można go ponownie do niej umieścić.
Na opakowaniu leku należy zaznaczyć datę rozpoczęcia okresu przechowywania w temperaturze pokojowej.
Po przygotowaniu roztworu do wstrzykiwań, stabilność chemiczna i fizyczna była potwierdzona przez 24 godziny przy przechowywaniu w temperaturze pokojowej.
Z mikrobiologicznego punktu widzenia lek należy zastosować natychmiast po przygotowaniu. Jeśli nie zostanie użyty natychmiast, użytkownik leku ponosi osobistą odpowiedzialność za okres ważności oraz warunki stosowania leku.
Warunki przechowywania.
Proszek
Przechowywać w temperaturze od 2 do 8 °C. Nie zamrażać.
Przechowywać w tekturowym pudełku w celu ochrony przed światłem.
Roztwórnik (woda do wstrzykiwań)
Przechowywać w temperaturze od 2 do 8 °C.
Przygotowany roztwór przechowywać w temperaturze pokojowej.
Nie zamrażać przygotowanego roztworu.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Ze względu na brak badań zgodności, tego leku nie należy mieszać z innymi lekami.
Należy stosować wyłącznie dostarczone zestawy do wstrzykiwań, ponieważ leczenie może zakończyć się niepowodzeniem w wyniku adsorpcji czynnika krzepnięcia krwi VIII na wewnętrznych powierzchniach niektórych urządzeń do wstrzykiwań.
Opakowanie. 1500 J, 2500 J, 3000 J lub 4000 J proszku w fiolce; 1 fiolka z proszkiem, 1 wstępnie wypełniony strzykacz z 2,5 ml roztwornika (woda do wstrzykiwań) oraz zestaw do rozpuszczenia i podania dożylnego (1 adapter do otwierania fiolki, 1 igła motylkowa, 2 przesączane alkoholem tampony) w tekturowym pudełku.
Kategoria wydawania.
Na receptę.
Producent.
Octapharma AB.
Miejsce produkcji i adres działalności.
Lars Forssells gata 23, Sztokholm, 11275, Szwecja.