Mirzera®
Ukraina
Spis treści
INSTRUKCJA dot. stosowania leczniczego leku Mirzera® (Mircera®)
Skład:
substancja czynna: methoxy polietylenoglikolu-epoetyna beta;
1 strzykawka wstępnie napełniona zawierająca 0,3 ml roztworu do wstrzykiwań zawiera 50 µg lub 75 µg metoksy polietylenoglikolu-epoetyny beta;
substancje pomocnicze: L-metionina; siarczan sodu bezwodny; dwusiarczan sodu diwodorotlenowy, monohydrat; sorbitol (E 421); poloksymer 188; kwas chlorowodorowy rozcieńniony lub roztwór wodorotlenku sodu (q.s. do pH 6,2); woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: przejrzysta ciecz o barwie od bezbarwnej do lekko żółtawej.
Grupa farmakoterapeutyczna.
Inne środki przeciwanemiczne. Kod ATC B03X A03.
Właściwości farmakodynamiczne.
Farmakodynamika.
Metoksy polietylenoglikol-epoetyna beta stymuluje erytropoetynę poprzez oddziaływanie na receptory erytropoetyny znajdujące się na komórkach prekursorowych szpiku kostnego. Metoksy polietylenoglikol-epoetyna beta, substancja czynna leku Mirzera®, jest długotrwałym agonistą receptorów erytropoetyny, który w przeciwieństwie do erytropoetyny wykazuje odmienną aktywność na poziomie receptorowym, charakteryzującą się wolniejszym wiązaniem i szybszym odłączaniem się od receptora, obniżoną specyficzną aktywnością in vitro oraz zwiększoną aktywnością in vivo, a także dłuższym okresem półtrwania. Średnia masa cząsteczkowa metoksy polietylenoglikol-epoetyny beta wynosi około 60 kDa, w tym około 30 kDa masy białka i części węglowodanowej.
Dawkowanie leku odnosi się do ilości białka w cząsteczce metoksy polietylenoglikol-epoetyny beta bez uwzględnienia glikozylacji. Białko jest wytwarzane metodą rekombinowanego DNA w komórkach jajnika chomika chińskiego i kowalencyjnie sprzężone z liniowym polietylenoglikolem (PEG).
Naturalny hormon erytropoetyna, główny czynnik wzrostu dla rozwoju erytropoetycznego, jest wytwarzany przez nerki i wydzielany do krwiobiegu w odpowiedzi na hipoksję. W odpowiedzi na hipoksję erytropoetyna oddziałuje z komórkami prekursorowymi erytropoetycznymi, co prowadzi do zwiększenia produkcji erytrocytów.
Kliniczna skuteczność
Dorośli
Wyniki badań korekcji anemii u pacjentów leczonych metoksy polietylenoglikol-epoetyną beta z częstotliwością podawania raz na 2 tygodnie lub raz na 4 tygodnie wykazały, że częstość odpowiedzi hemoglobiny w grupie stosującej metoksy polietylenoglikol-epoetynę beta na końcu okresu korekcji była wysoka i porównywalna z grupą stosującą leki porównawcze. Średni czas odpowiedzi wynosił 43 dni w grupie stosującej metoksy polietylenoglikol-epoetynę beta i 29 dni w grupie leku porównawczego, przy czym wzrost hemoglobiny w ciągu pierwszych 6 tygodni wynosił odpowiednio 0,2 g/dl/tydzień i 0,3 g/dl/tydzień.
Dotychczas przeprowadzono 4 randomizowane, kontrolowane badania u pacjentów poddawanych dializie i leczonych darbepoetyną alfa lub epoetyną w momencie włączenia do badania. W momencie włączenia do badania pacjenci byli randomizowani do kontynuacji terapii epoetynami, które otrzymywali wcześniej, lub do przejścia na leczenie metoksy polietylenoglikol-epoetyną beta w celu osiągnięcia stabilnego poziomu hemoglobiny. W okresie oceny (29–36 tydzień) średni poziom i mediana poziomu hemoglobiny u pacjentów leczonych lekiem Mirzera® były praktycznie identyczne z początkowym poziomem hemoglobiny.
Metoksy polietylenoglikol-epoetyna beta nie jest dopuszczona do leczenia pacjentów z anemią indukowaną chemoterapią.
Dzieci
Przeprowadzono dwa badania z udziałem dzieci. Jedno badanie z podaniem dożylnym (i.v.) i jedno badanie z podaniem podskórnym (s.c.) metoksy polietylenoglikol-epoetyny beta.
Badanie podania i.v.: otwarte, niekontrolowane, wieloośrodkowe badanie fazy II z poszukiwaniem dawki z zastosowaniem wielokrotnych dawek (NH19707) z udziałem 64 dzieci w wieku od 5 do 17 lat z przewlekłą chorobą nerek poddawanych hemodializie, mające na celu ocenę dwóch współczynników przeliczenia (grupa 1 i grupa 2) przejścia z leczenia podtrzymującego i.v. epoetyną alfa/beta lub darbepoetyną alfa na stosowanie metoksy polietylenoglikol-epoetyny beta i.v. co 4 tygodnie przez 20 tygodni. Skuteczność oceniano na podstawie zmiany stężenia hemoglobiny (g/dl) od poziomu wyjściowego do okresu oceny. Skorygowana średnia zmiana stężenia hemoglobiny od poziomu wyjściowego do okresu oceny w grupie 1 wyniosła -0,74 g/dl [95 % CI: od -1,32 do -0,16], a w grupie 2 wyniosła -0,09 g/dl [95 % CI: od -0,45 do 0,26]. U 58 % i 75 % pacjentów utrzymywano poziom hemoglobiny w granicach ±1 g/dl od poziomu wyjściowego, a u 75 % i 81 % – w granicach 10–12 g/dl odpowiednio w grupie 1 i grupie 2. Analiza podgrup wiekowych (5–11 lat i 12–17 lat) była zgodna z obserwacjami w populacji ogólnej. Pacjenci, którzy ukończyli 20-tygodniowy okres leczenia podstawowego i u których odpowiednio utrzymywano poziom hemoglobiny, byli kwalifikowani do włączenia do opcjonalnego, 52-tygodniowego przedłużonego okresu oceny bezpieczeństwa z tą samą częstotliwością podawania leku.
Badanie podania s.c.: otwarte, niekontrolowane, wieloośrodkowe badanie fazy II z poszukiwaniem dawki (NH19708) z udziałem 40 dzieci (w wieku od 3 miesięcy do 17 lat) z przewlekłą chorobą nerek, poddawanych hemodializie lub jeszcze nie poddawanych dializie, mające na celu ocenę współczynnika przeliczenia użytego w grupie 2 badania podania i.v. dla przejścia z leczenia podtrzymującego epoetyną alfa/beta lub darbepoetyną alfa s.c. na stosowanie metoksy polietylenoglikol-epoetyny beta s.c. co 4 tygodnie przez 20 tygodni.
Pierwotnym punktem końcowym skuteczności była zmiana stężenia hemoglobiny (g/dl) od poziomu wyjściowego do okresu oceny. Średnia zmiana stężenia hemoglobiny w okresie oceny wyniosła 0,48 g/dl [95 % CI: od 0,15 do 0,82], co mieściło się w granicach równoważności od -1 do +1 g/dl. Wyniki średniej zmiany stężenia hemoglobiny według grup wiekowych (< 5 lat, 5–11 lat, ≥ 12 lat) były zgodne z wynikami pierwotnego punktu końcowego w okresie oceny. Pacjenci, którzy ukończyli 20-tygodniowy okres leczenia podstawowego i u których odpowiednio utrzymywano poziom hemoglobiny, byli kwalifikowani do włączenia do opcjonalnego, 24-tygodniowego przedłużonego okresu oceny bezpieczeństwa z tą samą częstotliwością podawania leku.
W obu badaniach średnie wartości hemoglobiny u większości pacjentów utrzymywały się w granicach 10–12 g/dl przez cały okres oceny i przedłużony okres oceny bezpieczeństwa. Profil bezpieczeństwa obserwowany u dzieci w obu badaniach odpowiadał profilowi bezpieczeństwa u dorosłych (patrz sekcja „Działania niepożądane”).
Farmakokinetyka.
Dorośli
Farmakokinetykę metoksy polietylenoglikol-epoetyny beta badano u zdrowych ochotników oraz u pacjentów z przewlekłą chorobą nerek i anemią, w tym u chorych poddawanych i nie poddawanych dializie.
Po podaniu podskórnym pacjentom z przewlekłą chorobą nerek nie poddawanym dializie maksymalne stężenia metoksy polietylenoglikol-epoetyny beta w surowicy krwi obserwowano po 95 godzinach (średnia wartość) po podaniu. Bezpośrednia biodostępność metoksy polietylenoglikol-epoetyny beta po podaniu podskórnym wyniosła 54 %. Okres półtrwania końcowego wyniósł 142 godziny u pacjentów z przewlekłą chorobą nerek nie poddawanych dializie.
Po podaniu podskórnym pacjentom z przewlekłą chorobą nerek poddawanym dializie maksymalne stężenia metoksy polietylenoglikol-epoetyny beta w surowicy krwi obserwowano po 72 godzinach (średnia wartość) po podaniu. Bezpośrednia biodostępność metoksy polietylenoglikol-epoetyny beta po podaniu podskórnym wyniosła 62 %. Okres półtrwania końcowego wyniósł 139 godzin u pacjentów z przewlekłą chorobą nerek poddawanych dializie.
Po podaniu dożylnym pacjentom z przewlekłą chorobą nerek poddawanym dializie całkowity klirens systemowy wyniósł 0,494 ml/godz/kg. Po podaniu dożylnym okres półtrwania metoksy polietylenoglikol-epoetyny beta wyniósł 134 godziny.
Porównanie stężenia metoksy polietylenoglikol-epoetyny beta w surowicy krwi, oznaczonego przed i po przeprowadzeniu hemodializy u 41 pacjentów z przewlekłą chorobą nerek, wykazało, że hemodializa nie wpływa na farmakokinetykę leku Mirzera®. Analiza danych z 126 pacjentów z przewlekłą niewydolnością nerek wykazała brak różnic w parametrach farmakokinetycznych u pacjentów poddawanych i nie poddawanych dializie.
W badaniu jednej dawki po podaniu dożylnym farmakokinetyka metoksy polietylenoglikol-epoetyny beta jest porównywalna u pacjentów z ciężką niewydolnością wątroby i u zdrowych ochotników (patrz sekcja „Sposób dawkowania i dawka”).
Dzieci
Analizę farmakokinetyki populacyjnej przeprowadzono na podstawie danych 103 dzieci w wieku od 6 miesięcy do 17 lat o masie ciała od 7 do 90 kg oraz 524 dorosłych pacjentów. Dzieci otrzymywały metoksy polietylenoglikol-epoetynę beta i.v. (wszystkie na hemodializie) lub s.c. (na dializie peritonealnej, hemodializie lub jeszcze nie na dializie). Stwierdzono, że klirens i objętość rozkładu rosną wraz ze wzrostem masy ciała, a objętość rozkładu wzrasta z wiekiem. Obserwowane maksymalne i minimalne stężenia metoksy polietylenoglikol-epoetyny beta w surowicy krwi u dzieci po ustabilizowaniu poziomu hemoglobiny były porównywalne z wartościami u dorosłych, dla obu dróg podania (i.v. i s.c.).
Właściwości kliniczne.
Wskazania.
Leczenie anemii objawowej związanej z przewlekłą chorobą nerek u dorosłych pacjentów.
Leczenie anemii objawowej związanej z przewlekłą chorobą nerek u dzieci w wieku od 3 miesięcy do 18 lat, które przechodzą z innego leku stymulującego erytropoetynę, po ustabilizowaniu poziomu hemoglobiny przy użyciu tego leku.
Przeciwwskazania.
Podwyższona wrażliwość na metoksy polietylenoglikol-epoetynę beta lub dowolny składnik pomocniczy leku (patrz dział „Składniki pomocnicze”). Niekontrolowana nadciśnienie tętnicze.
Interakcje z innymi lekami i inne rodzaje interakcji.
Badania interakcji metoksy polietylenoglikol-epoetyny beta nie były prowadzone. Nie ma danych sugerujących, że Mirzera® wpływa na metabolizm innych leków.
Szczególne wytyczne dotyczące stosowania.
Nie ustalono bezpieczeństwa i skuteczności terapii metoksy polietylenoglikolem-epoetynem beta w innych wskazaniach, w tym w przypadku anemii u pacjentów z nowotworami złośliwymi.
Należy zachować ostrożność przy zwiększaniu dawek metoksy polietylenoglikolu-epoetyny beta u pacjentów z przewlekłą niewydolnością nerek, ponieważ wysokie dawki kumulacyjne epoetyn mogą wiązać się z zwiększonym ryzykiem zgonów, poważnych powikłań sercowo-naczyniowych i mózgowo-naczyniowych. W przypadku słabej odpowiedzi pacjenta na leczenie epoetynami w zakresie stężenia hemoglobiny należy rozważyć inne możliwe przyczyny (patrz sekcja „Sposób stosowania i dawki”).
Dzieci. Dzieci, w szczególności pacjentów w wieku < 1 roku, należy dokładnie przebadać przed przejściem z innego środka stymulującego erytropoezę. Należy również ustabilizować poziom hemoglobiny przed przejściem. Po przejściu z innego środka stymulującego erytropoezę zaleca się monitorowanie poziomu hemoglobiny co 4 tygodnie.
Jeśli obecna dawka środka stymulującego erytropoezę wynosi < 9 μg/tydzień dapreepoetyny alfa lub < 2000 MI/tydzień epoetyny, pacjenta nie powinno się przekształcać na metoksy polietylenoglikol-epoetynę beta, ponieważ najmniejsza dostępna dawka w prezentowanym strzykawce wynosi 30 μg. Nie zaleca się podawania niepełnych dawek z prezentowanej strzykawki.
Dodatkowa terapia żelazem jest zalecana wszystkim pacjentom ze stężeniem ferrytyny w surowicy krwi poniżej 100 μg/l lub nasyceniem transferyny żelazem poniżej 20 %. Aby zapewnić skuteczną erytropoezę, poziom żelaza należy oznaczać u wszystkich pacjentów przed rozpoczęciem i w trakcie leczenia.
W przypadku braku odpowiedzi na leczenie należy rozpocząć poszukiwanie czynników przyczynowych. Niedobór żelaza, kwasu foliowego i witaminy B12 zmniejsza skuteczność terapii środkami stymulującymi erytropoezę, dlatego należy korygować niedobór tych substancji. Na zmniejszenie skuteczności terapii środkami stymulującymi erytropoezę mogą również wpływać współistniejące infekcje, procesy zapalne, urazy, utajona utrata krwi, hemoliza, ciężka toksyczność aluminium, współistniejące choroby krwi oraz włóknienie szpiku. Podczas badania pacjentów należy również oznaczyć liczbę retikulocytów. W przypadku wykluczenia wymienionych stanów i nagłego spadku poziomu hemoglobiny, związanego z retikulocytopenią i wykryciem przeciwciał przeciwko erytropoetynie, należy przeprowadzić badanie szpiku kostnego w celu wykluczenia rozpoznania prawdziwej aplazji czerwonych komórek krwiotwórczych. W przypadku potwierdzenia rozpoznania prawdziwej aplazji czerwonych komórek krwiotwórczych leczenie należy przerwać, a pacjentów nie należy przekształcać na leczenie innymi środkami stymulującymi erytropoezę.
Na żądanie lekarza firma Roche zaproponuje testowanie lub ponowne testowanie próbek surowicy krwi w laboratorium referencyjnym. Usługa ta jest bezpłatna w przypadku podejrzenia lub potwierdzenia prawdziwej aplazji czerwonych komórek krwiotwórczych spowodowanej przeciwciałami przeciwko erytropoetynie lub utraty efektu leczenia (np. klinicznie obserwowanej jako ciężka anemia z niską liczbą retikulocytów).
O prawdziwej aplazji czerwonych komórek krwiotwórczych spowodowanej przeciwciałami przeciwko erytropoetynie donoszono w trakcie stosowania wszystkich środków stymulujących erytropoezę, w tym metoksy polietylenoglikolu-epoetyny beta. Wykazano, że przeciwciała przeciwko erytropoetynie wykazują reakcję krzyżową ze wszystkimi środkami stymulującymi erytropoezę. Pacjentów, u których podejrzewa się lub u których wykryto przeciwciała przeciwko erytropoetynie, nie należy przekształcać na leczenie metoksy polietylenoglikolem-epoetyną beta (patrz sekcja „Działania niepożądane”).
Prawdziwa aplazja czerwonych komórek krwiotwórczych u pacjentów z wirusowym zapaleniem wątroby typu C: w przypadku paradoksalnego spadku poziomu hemoglobiny i rozwoju ciężkiej anemii związanej z niską liczbą retikulocytów należy przerwać leczenie epoetyną i przebadać pacjenta pod kątem obecności przeciwciał przeciwko erytropoetynie. Zanotowano przypadki prawdziwej aplazji czerwonych komórek krwiotwórczych u pacjentów zakażonych wirusem zapalenia wątroby typu C, którzy otrzymywali leczenie interferonem i rybawiryną równocześnie z epoetynami. Epoetyny nie są zarejestrowane do stosowania w leczeniu anemii związanej z wirusowym zapaleniem wątroby typu C.
Monitorowanie ciśnienia tętniczego: jak i w przypadku stosowania innych środków stymulujących erytropoezę, możliwe jest wzrost ciśnienia tętniczego podczas leczenia lekiem Mirzera®. Poziom ciśnienia tętniczego należy odpowiednio kontrolować u wszystkich pacjentów przed rozpoczęciem i w trakcie leczenia metoksy polietylenoglikolem-epoetyną beta. Jeśli ciśnienie tętnicze nie może być kontrolowane lekami lub dietą, należy zmniejszyć dawkę metoksy polietylenoglikolu-epoetyny beta lub przerwać leczenie (patrz sekcja „Sposób stosowania i dawki”).
Zgłaszano ciężkie reakcje skórne, w tym zespół Stevensa-Johnsona (SJS) i toksyczny epidermalny nekroliz (TEN), które mogą być zagrażające życiu lub śmiertelne, związane z leczeniem epoetyną (patrz sekcja „Działania niepożądane”). Cięższe przypadki obserwowano podczas stosowania epoetyn o długim działaniu. Przy przepisywaniu leku należy poinformować pacjentów o objawach takich reakcji oraz należy dokładnie monitorować stan pacjentów pod kątem rozwoju reakcji skórnych. W przypadku pojawienia się objawów wskazujących na rozwój takich reakcji należy natychmiast przerwać stosowanie metoksy polietylenoglikolu-epoetyny beta i rozważyć możliwość leczenia alternatywnego. W przypadku rozwoju ciężkiej reakcji skórnej, takiej jak SJS lub TEN, spowodowanej stosowaniem metoksy polietylenoglikolu-epoetyny beta, nie należy ponownie przepisywać pacjentowi leczenia środkami stymulującymi erytropoezę.
Stężenie hemoglobiny: u pacjentów z przewlekłą chorobą nerek stężenie hemoglobiny nie powinno przekraczać górnej granicy docelowego stężenia hemoglobiny zalecanego w sekcji „Sposób stosowania i dawki”. W badaniach klinicznych obserwowano zwiększony ryzyko zgonów i poważnych zdarzeń sercowo-naczyniowych, w tym zatorowości lub zdarzeń mózgowo-naczyniowych, w tym udaru, przy stosowaniu środków stymulujących erytropoezę w celu osiągnięcia docelowego poziomu hemoglobiny powyżej 12 g/dl (7,5 mmol/l) (patrz sekcja „Działania niepożądane”).
W kontrolowanych badaniach klinicznych nie wykazano istotnej korzyści z zastosowania epoetyn w przypadku wzrostu stężenia hemoglobiny powyżej poziomu niezbędnego do kontroli objawów anemii i uniknięcia transfuzji krwi.
Nie ustalono bezpieczeństwa i skuteczności leczenia metoksy polietylenoglikolem-epoetyną beta u pacjentów z hemoglobinopatiami, napadami padaczkowymi, krwotokami, w tym niedawnymi krwotokami w wywiadzie wymagającymi transfuzji krwi, oraz z liczbą płytek krwi powyżej 500 × 10⁹/l. Dlatego lek Mirzera® należy stosować u tej grupy pacjentów z ostrożnością.
Wpływ na wzrost nowotworu: metoksy polietylenoglikol-epoetyna beta, podobnie jak inne środki stymulujące erytropoezę, jest czynnikiem wzrostu, który głównie stymuluje produkcję erytrocytów. Receptory dla erytropoetyny mogą być obecne na powierzchni różnych komórek nowotworowych. Uważa się, że środki stymulujące erytropoezę, podobnie jak inne czynniki wzrostu, mogą stymulować wzrost dowolnego typu nowotworów złośliwych. W dwóch kontrolowanych badaniach klinicznych, w których epoetyny podawano pacjentom z różnymi nowotworami, w tym z rakiem głowy i szyi, rakiem piersi, zaobserwowano zwiększoną śmiertelność, której przyczyna nie jest znana.
Błędne stosowanie metoksy polietylenoglikolu-epoetyny beta przez osoby zdrowe może prowadzić do nadmiernego wzrostu poziomu hemoglobiny, co może wiązać się z powikłaniami sercowo-naczyniowymi zagrażającymi życiu.
Śledzenie: w celu poprawy śledzenia stosowania leków biologicznych nazwa i numer serii podanego leku powinny być dokładnie udokumentowane.
Lek zawiera mniej niż 1 mmol sodu (23 mg) w 1 ml, co oznacza, że lek jest uznawany za pozbawiony sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża
Brak danych dotyczących stosowania metoksy polietylenoglikolu-epoetyny beta u kobiet w ciąży.
W badaniach na zwierzętach nie stwierdzono bezpośredniego szkodliwego wpływu leku Mirzera® na ciążę, rozwój embrionalny, rozwój płodu, poród i rozwój poporodowy, jednak zaobserwowano odwracalne zmniejszenie masy płodu związane z zastosowaniem środków stymulujących erytropoezę. Lek Mirzera® należy stosować u kobiet w ciąży z ostrożnością.
Okres karmienia piersią
Nie wiadomo, czy metoksy polietylenoglikol-epoetyna beta wydzielana jest w mleku matki. W jednym z badań na zwierzętach wykazano, że metoksy polietylenoglikol-epoetyna beta wydzielana jest w mleku matki. Decyzję o kontynuacji lub zaprzestaniu karmienia piersią lub kontynuacji lub zaprzestaniu leczenia metoksy polietylenoglikolem-epoetyną beta należy podjąć, biorąc pod uwagę korzyści karmienia piersią dla dziecka i korzyści z leczenia metoksy polietylenoglikolem-epoetyną beta dla kobiety.
Plodność
W badaniach na zwierzętach nie stwierdzono zaburzeń płodności. Potencjalne ryzyko dla człowieka jest nieznane.
Wpływ na zdolność prowadzenia pojazdów i obsługi innych maszyn.
Metoksy polietylenoglikol-epoetyna beta nie wpływa lub ma nieznaczny wpływ na zdolność prowadzenia pojazdów i obsługi innych maszyn.
Sposób stosowania i dawki
Leczenie należy prowadzić pod nadzorem lekarza posiadającego doświadczenie w leczeniu pacjentów z niewydolnością nerek.
Lek Mirzera® można podawać podskórnie lub dożylnie. Mirzera® podaje się podskórnie w okolicy brzucha, ramienia lub uda. Wskazane okolice są jednakowo odpowiednie do podania podskórnego.
Leczenie anemii objawowej u pacjentów z przewlekłą chorobą nerek
Objawy anemii i ich konsekwencje mogą się różnić w zależności od wieku, płci pacjenta oraz ogólnej ciężkości choroby, dlatego wymagana jest ocena indywidualnego przebiegu choroby i stanu pacjenta przez lekarza.
Lek można stosować podskórnie lub dożylnie w celu podniesienia stężenia hemoglobiny do poziomu nie wyższego niż 12 g/dl (7,45 mmol/l). U pacjentów nie poddawanych hemodializie należy preferować podanie podskórne w celu uniknięcia nakłucia żył obwodowych.
Z powodu indywidualnej zmienności u poszczególnych pacjentów rzadko może występować stężenie hemoglobiny wyższe lub niższe niż pożądany poziom. Na zmienność stężenia hemoglobiny można wpływać poprzez dostosowanie dawki z uwzględnieniem docelowego zakresu stężenia hemoglobiny od 10 g/dl (6,21 mmol/l) do 12 g/dl (7,45 mmol/l). Należy unikać trwałego podniesienia stężenia hemoglobiny powyżej 12 g/dl (7,45 mmol/l); zalecenia dotyczące odpowiedniej korekty dawki w przypadku wzrostu stężenia hemoglobiny powyżej 12 g/dl (7,45 mmol/l) podano poniżej.
Należy unikać wzrostu stężenia hemoglobiny o więcej niż 2 g/dl (1,24 mmol/l) u dorosłych oraz o więcej niż 1 g/dl (0,62 mmol/l) u dzieci w ciągu czterotygodniowego okresu. W przypadku wystąpienia takiej sytuacji należy skorygować dawkę leku Mirzera®.
Stan pacjentów należy dokładnie monitorować w celu zapewnienia, że otrzymują najniższą zatwierdzoną skuteczną dawkę leku Mirzera® niezbędną do adekwatnej kontroli objawów anemii przy jednoczesnym utrzymaniu stężenia hemoglobiny na poziomie niższym lub równym 12 g/dl (7,45 mmol/l).
Należy zachować ostrożność przy zwiększaniu dawki leku Mirzera® u pacjentów z przewlekłą niewydolnością nerek. Jeśli pacjent słabo reaguje pod względem stężenia hemoglobiny na stosowanie leku Mirzera®, należy rozważyć inne możliwe przyczyny (patrz dział „Szczególne ostrzeżenia i środki ostrożności”).
Poziom hemoglobiny należy kontrolować co 2 tygodnie do momentu jego ustabilizowania, a następnie okresowo (patrz dział „Szczególne ostrzeżenia i środki ostrożności”).
Dorośli pacjenci aktualnie nie leczeni lekami stymulującymi erytropoetynę
U pacjentów nie poddawanych dializie zalecana dawka początkowa wynosi 1,2 µg/kg masy ciała raz na miesiąc podskórnie w celu osiągnięcia poziomu hemoglobiny powyżej 10 g/dl (6,21 mmol/l).
Alternatywnie można podać zalecaną dawkę początkową 0,6 µg/kg masy ciała raz na 2 tygodnie podskórnie lub dożylnie pacjentom poddawanym dializie oraz pacjentom nie poddawanym dializie.
W przypadku wzrostu stężenia hemoglobiny mniejszego niż 1 g/dl (0,621 mmol/l) w ciągu jednego miesiąca dawkę leku Mirzera® można zwiększyć o około 25% w stosunku do poprzedniej dawki. Dalsze zwiększanie dawki leku Mirzera® o około 25% można prowadzić w odstępach co miesiąc do osiągnięcia indywidualnego docelowego poziomu hemoglobiny.
W przypadku wzrostu stężenia hemoglobiny o więcej niż 2 g/dl (1,24 mmol/l) w pierwszym miesiącu leczenia lub wzrostu stężenia hemoglobiny do 12 g/dl (7,45 mmol/l) dawkę leku Mirzera® należy zmniejszyć o około 25%. Jeśli stężenie hemoglobiny nadal rośnie, leczenie należy przerwać do momentu spadku stężenia hemoglobiny, po czym należy wznowić podawanie leku Mirzera® w dawce o około 25% mniejszej niż poprzednia. Po przerwaniu leczenia oczekuje się spadku stężenia hemoglobiny o około 0,35 g/dl (0,22 mmol/l) na tydzień. Korekty dawki leku nie należy wykonywać częściej niż raz na miesiąc.
Pacjentom, którzy otrzymują metoksy polietylenoglikol-epoetynę beta z częstotliwością raz na 2 tygodnie i u których stężenie hemoglobiny przekracza 10 g/dl (6,21 mmol/l), można podawać lek Mirzera® raz na miesiąc w dawce równej podwojonej dawce stosowanej co 2 tygodnie.
Dorośli pacjenci aktualnie leczeni lekami stymulującymi erytropoetynę
Pacjentów aktualnie leczonych lekami stymulującymi erytropoetynę można przełożyć na terapię metoksy polietylenoglikol-epoetyną beta podawaną raz na miesiąc dożylnie lub podskórnie. Początkową dawkę metoksy polietylenoglikol-epoetyny beta ustala się na podstawie poprzedniej tygodniowej dawki darbepoetyny alfa lub epoetyny podawanej w momencie zmiany (tabela 1). Pierwsze podanie należy wykonać w dniu zaplanowanego podania wcześniej stosowanej darbepoetyny alfa lub epoetyny.
Tabela 1
Początkowe dawki metoksy polietylenoglikol-epoetyny beta dla dorosłych pacjentów aktualnie leczonych lekami stymulującymi erytropoetynę
| Tygodniowa dawka darbepoetyny alfa (µg/tydzień), stosowana wcześniej |
Tygodniowa dawka epoetyny (jednostek/tydzień), stosowana wcześniej |
Dawka miesięczna (µg/1 raz na miesiąc) metoksy polietylenu glikolu-epoetyny beta przy podaniu podskórnym lub dożylnym |
| < 40 |
< 8000 |
120 |
| 40–80 |
8000–16000 |
200 |
| > 80 |
> 16000 |
360 |
Jeśli konieczna jest korekta dawki w celu utrzymania docelowej stężenia hemoglobiny powyżej 10 g/dl (6,21 mmol/l), miesięczna dawka leku może zostać zwiększona o około 25 %.
W przypadku wzrostu stężenia hemoglobiny o więcej niż 2 g/dl (1,24 mmol/l) w ciągu miesiąca lub wzrostu stężenia hemoglobiny do 12 g/dl (7,45 mmol/l), dawkę Mirzera® należy zmniejszyć o około 25 %. Jeśli stężenie hemoglobiny nadal rośnie, leczenie należy przerwać do momentu spadku stężenia hemoglobiny, po czym возобновить podawanie leku Mirzera® w dawce o około 25 % niższej niż poprzednia. Po przerwaniu leczenia stężenie hemoglobiny obniża się o około 0,35 g/dl (0,22 mmol/l) tygodniowo. Korekty dawki leku nie należy wykonywać częściej niż raz na miesiąc.
Z powodu ograniczonego doświadczenia w stosowaniu u pacjentów poddawanych dializie otrzewnowej, zaleca się regularne monitorowanie stężenia hemoglobiny oraz ścisłe przestrzeganie zaleceń dotyczących korekty dawki.
Dzieci w wieku od 3 miesięcy do 18 lat aktualnie leczone lekami stymulującymi erytropoetynę
Dzieci, u których stężenie hemoglobiny ustabilizowało się dzięki leczeniu lekiem stymulującym erytropoetynę, można przełożyć na terapię metoksy polietylenoglikolem-epoetyinem beta, podawaną co 4 tygodnie dożylnie lub podskórnie, pod warunkiem zachowania tej samej drogi podania. Początkową dawkę metoksy polietylenoglikolu-epoetyenu beta należy ustalić na podstawie poprzedniej tygodniowej dawki leku stymulującego erytropoetynę w momencie przejścia (tabela 2).
Tabela 2
Początkowe dawki metoksy polietylenoglikolu-epoetyenu beta dla dzieci w wieku od 3 miesięcy do 18 lat aktualnie leczonych lekami stymulującymi erytropoetynę
| Tygodniowa dawka darbepoetyny alfa (mkg/tydzień), która była stosowana wcześniej |
Tygodniowa dawka epoetyny (jednostek/tydzień), która była stosowana wcześniej |
Dawka (mkg) metoksy polietylen glikolu-epoetyny beta z częstotliwością podawania co 4 tygodnie |
| 9 – < 12 |
2000 – < 2700 |
30 |
| 12 – < 15 |
2700 – < 3500 |
50 |
| 15 – < 24 |
3500 – < 5500 |
75 |
| 24 – < 30 |
5500 – < 6500 |
100 |
| 30 – < 35 |
6500 – < 8000 |
120 |
| 35 – < 47 |
8000 – < 10000 |
150 |
| 47 – < 60 |
10000 – < 13000 |
200 |
| 60 – < 90 |
13000 – < 20000 |
250 |
| ≥ 90 |
≥ 20000 |
360 |
Wstępnie napełnione strzykawki nie są przeznaczone do podawania częściowych dawek. Ze względu na dostępne dawki wstępnie napełnionych strzykawek dzieci otrzymujące lek stymulujący erytropoezę w dawce < 9 μg/tydzień (darbepoetyne alfa) lub < 2000 MI/tydzień epoetyny nie powinny być przekładane na metoksy polietylenoglikol-epoetyne beta.
Jeśli konieczna jest korekta dawki w celu utrzymania docelowej stężenia hemoglobiny powyżej 10 g/dl, dawkę leku podawanego co 4 tygodnie można skorygować o około 25%.
W przypadku wzrostu poziomu hemoglobiny o więcej niż 1 g/dl (0,62 mmol/l) w ciągu 4 tygodni lub wzrostu i osiągnięcia poziomu hemoglobiny 12 g/dl (7,45 mmol/l) dawkę metoksy polietylenoglikol-epoetyny beta należy zmniejszyć o około 25%.
Jeśli poziom hemoglobiny nadal rośnie po zmniejszeniu dawki, leczenie należy wstrzymać do czasu spadku poziomu hemoglobiny, po czym należy wznowić terapię w dawce o około 25% mniejszej od poprzedniej.
Korekty dawki leku nie należy wykonywać częściej niż raz na 4 tygodnie.
Przestanie leczenia
Leczenie jest zazwyczaj długotrwałe. W razie potrzeby leczenie można przerwać w dowolnym momencie.
Pominięta dawka
Jeśli pominięto jedną dawkę Mirzera® należy ją podać jak najszybciej. Należy wznowić podawanie z częstotliwością stosowaną wcześniej.
Zastosowanie u dzieci
Skuteczność i bezpieczeństwo stosowania metoksy polietylenoglikol-epoetyny beta u dzieci poniżej 3. miesiąca życia nie zostały ustalone. Brak danych.
Specjalne zalecenia dawkowania
Pacjenci starsi
W badaniach klinicznych wiek 24% pacjentów otrzymujących leczenie Mirzerą® mieścił się w przedziale 65–74 lat, a 20% pacjentów miało 75 lat lub więcej. Pacjentom starszym powyżej 65. roku życia nie wymaga się korekty dawki.
Pacjenci z niewydolnością wątroby
Nie wymaga się korekty dawki początkowej ani schematu dawkowania metoksy polietylenoglikol-epoetyny beta u pacjentów z niewydolnością wątroby.
Zasady przechowywania roztworu
Wstępnie napełniona strzykawka jest gotowa do użycia. Sterylna, wstępnie napełniona strzykawka nie zawiera substancji konserwujących i każdą strzykawkę można użyć tylko raz. Jedną dawkę należy podać za pomocą jednej wstępnie napełnionej strzykawki. Wstępnie napełnione strzykawki nie są przeznaczone do podawania częściowych dawek. Dzieci (poniżej 18 roku życia) nie powinny samodzielnie podawać Mirzery® – podanie powinien wykonać personel medyczny lub wykwalifikowany dorosły opiekun. Można stosować wyłącznie przezroczysty, bezbarwny lub lekko żółtawy roztwór, który nie zawiera widocznych cząstek.
Nie wstrząsać.
Przed zastosowaniem należy doprowadzić roztwór do temperatury pokojowej. W tym celu należy wyjąć z lodówki opakowanie kartonowe zawierające Mirzerę®. Nie wyjmując strzykawki z opakowania kartonowego (w celu ochrony przed światłem), należy pozostawić strzykawkę i igłę na 30 minut, aby temperatura leku osiągnęła poziom pokojowy. Strzykawkę i igłę należy trzymać w suchym miejscu.
- Nie doprowadzenie leku do temperatury pokojowej może powodować dyskomfort podczas wstrzyknięcia oraz trudności z naciskaniem tłoka.
- Nie należy podgrzewać strzykawki żadnym innym sposobem.
Instrukcja obsługi wstępnie napełnionej strzykawki
- Wyjąć folię bąbelkową zawierającą Mirzerę® z opakowania kartonowego, nie usuwając folii ochronnej.
- Dokładnie umyć ręce ciepłą wodą z mydłem.
- Usunąć folię ochronną z folii bąbelkowej, wyjąć wstępnie napełnioną strzykawkę oraz pojemnik plastikowy z igłą.
- Trzymając pojemnik z igłą, przed sprawdzeniem jej pod kątem uszkodzeń, odkręcić nakrywkę ruchem obrotowym zgodnym z ruchem wskazówek zegara, jak pokazano na rysunku. Igielka jest krucha – należy z nią obchodzić się ostrożnie.
Nie należy używać igły, jeśli:
- Casually upuściłeś igłę.
- Jakakolwiek część igły wygląda na uszkodzoną.
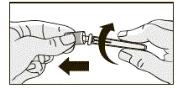
- Trzymając wstępnie napełnioną strzykawkę, usunąć gumowy nakrywacz, zginając i pociągając, jak pokazano na rysunku.
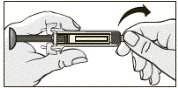
- Trzymając przezroczysty pojemnik z igłą, włożyć ją dokładnie i szczelnie do wstępnie napełnionej strzykawki, jak pokazano na rysunku.
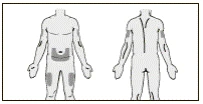
Podanie podskórne:
- W celu podania podskórnego Mirzery® wybrać jedną z zalecanych części ciała: przednią ścianę brzucha, przednią powierzchnię środkowej części uda lub zewnętrzną powierzchnię ramienia. Nie podawać leku w znamiona, blizny, siniaki, obrzęki, zaczerwienienia, zgrubienia lub inne zmiany skórne, ani w obszary, które mogą być drażnione przez pasek lub pasek odzieży.
- Dokładnie przetrzeć skórę w miejscu wstrzyknięcia watą nasączoną alkoholem. Poczekaj, aż przetarte miejsce wyschnie. Natychmiast wyrzuć watę nasączoną alkoholem po użyciu.
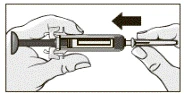
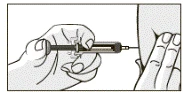
- Ostrożnie trzymając wstępnie napełnioną strzykawkę, nie naciskając tłoka, ostrożnie usunąć pojemnik z igłą. Trzymaj strzykawkę tylko za korpus, ponieważ każdy kontakt z zaciskami może spowodować przedwczesne zwolnienie mechanizmu zabezpieczającego.
- Dwoma palcami zebrać skórę w fałd w miejscu wstrzyknięcia. Wprowadzić igłę w fałd skóry pod kątem prostym.
- Powoli podać cały lek, płynnie naciskając tłok. Nie przestawaj naciskać tłoka wstępnie napełnionej strzykawki, dopóki nie wyjmiesz igły ze skóry!
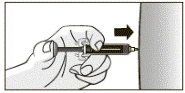
- Po podaniu całej dawki wyjąć igłę ze skóry, nie zwalniając tłoka wstępnie napełnionej strzykawki, jak pokazano na rysunku.
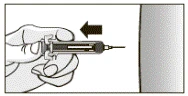
- Po zwolnieniu tłoka aktywuje się mechanizm ochronny i zakryje igłę.
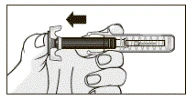
- Przycisnąć watą miejsce wstrzyknięcia Mirzery®. W razie potrzeby zakleić miejsce wstrzyknięcia plasterkiem. Natychmiast wyrzuć watę nasączoną alkoholem po użyciu.
Podanie dożylne:
- W celu podania dożylnego przygotuj strzykawkę zgodnie z punktami 1–6.
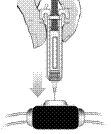
- Przetrzyj port żylny rury dializacyjnej watą nasączoną alkoholem zgodnie z instrukcjami dostawcy lub producenta. Natychmiast wyrzuć watę nasączoną alkoholem po użyciu.
- Włóż igłę wstępnie napełnionej strzykawki do oczyszczonego portu żylnego (jak pokazano na poniższym rysunku). Nie dotykaj miejsca wstrzyknięcia portu żylnego.
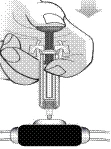
- Naciśnij tłok dużym palcem, trzymając strzykawkę palcem wskazującym i środkowym, aż cały lek zostanie podany (jak pokazano na poniższym rysunku).
- Wyjmij wstępnie napełnioną strzykawkę z portu żylnego, NIE zwalniając tłoka.
- Po zwolnieniu tłoka aktywuje się mechanizm ochronny i zakryje igłę.
Unieszkodliwienie strzykawki
- Zużyte strzykawki należy wyrzucać do pojemnika na ostre przedmioty lub do odpornego na przebicie pojemnika.
- Nie wolno ponownie używać ani sterylizować strzykawki i/lub igły.
- Nie próbuj ponownie założyć nasadki na igłę używanej strzykawki.
- Nie wyrzucaj zużytych strzykawek ani pojemnika na ostre przedmioty/odpornego na przebicie do śmieci domowych ani nie poddawaj recyklingowi.
- Unieszkodliwiaj pełen pojemnik na ostre przedmioty lub odporny na przebicie pojemnik.
Wszelkie nieużywane leki lub odpady należy unieszkodliwiać zgodnie z lokalnymi przepisami.
Dzieci.
Skuteczność i bezpieczeństwo stosowania metoksy polietylenoglikol-epoetyny beta u dzieci poniżej 3. miesiąca życia nie zostały ustalone. Brak danych.
Przedawkowanie.
Metoksy polietylenoglikol-epoetyna beta charakteryzuje się szerokim zakresem terapeutycznym. Przy wdrażaniu terapii należy wziąć pod uwagę indywidualną odpowiedź na leczenie. Przedawkowanie może prowadzić do nasilenia efektu farmakodynamicznego, tj. nadmiernego erytropoezy. W przypadku nadmiernego wzrostu poziomu hemoglobiny należy tymczasowo przerwać leczenie metoksy polietylenoglikol-epoetyną beta (patrz sekcja „Sposób stosowania i dawki”). W przypadku wskazań klinicznych można rozważyć flebotomię.
Efekty uboczne.
Baza danych dotyczących bezpieczeństwa, oparta na wynikach badań klinicznych, obejmuje 3042 pacjentów dorosłych z przewlekłą chorobą nerek, w tym 1939 dorosłych pacjentów leczonych metoksy polietylenoglikolem-epoetyną beta oraz 1103 pacjentów leczonych innym środkiem stymulującym erytropoetynę. W przypadku stosowania metoksy polietylenoglikolu-epoetyny beta wystąpienie działań niepożądanych jest możliwe u około 6% dorosłych pacjentów. Najczęstszym działaniem niepożądanym była nadciśnienie tętnicze (częste działanie niepożądane).
Do opisu częstości działań niepożądanych stosuje się następujące kryteria: bardzo częste (≥ 1/10); częste (≥ 1/100 i < 1/10); rzadkie (≥ 1/1000 i < 1/100); pojedyncze (≥ 1/10000 i < 1/1000); rzadkie (< 1/10000); częstość nieznana (nie można określić na podstawie dostępnych danych).
Działania niepożądane związane z leczeniem metoksy polietylenoglikolem-epoetyną beta u dorosłych pacjentów z przewlekłą chorobą nerek
Działania niepożądane obserwowane wyłącznie podczas stosowania po rejestracji leku oznaczono gwiazdką (*).
Ze strony krwi i układu chłonnego: rzadkie: trombocytopenia*; częstość nieznana: prawdziwa aplazja czerwonych komórek*.
Ze strony układu odpornościowego: pojedyncze: reakcje nadwrażliwości; częstość nieznana: reakcja anafilaktyczna*.
Ze strony układu nerwowego: rzadkie: ból głowy; pojedyncze: encefalopatia nadciśnieniowa.
Ze strony układu sercowo-naczyniowego: częste: nadciśnienie tętnicze; rzadkie: zakrzepica*; pojedyncze: naparzanie, zakrzepica tętnicy płucnej*.
Ze strony skóry i tkanki podskórnej: pojedyncze: wysypka makularno-papularna; częstość nieznana: zespół Stevensa-Johnsona/toksyczny epidermalny nekroliz*.
Urazy, zatrucia i komplikacje procedur: rzadkie: zakrzepica miejsca dostępu do naczyń.
Opis poszczególnych działań niepożądanych
Dorośli
Podczas stosowania po rejestracji zgłaszano przypadki trombocytopenii. W badaniach klinicznych zaobserwowano pewne obniżenie liczby płytek krwi w granicach normy.
W badaniach klinicznych trombocytopenia (liczba płytek krwi mniejsza niż 100 × 109/l) występowała u 7% dorosłych pacjentów leczonych metoksy polietylenoglikolem-epoetyną beta oraz u 4% dorosłych pacjentów leczonych innymi środkami stymulującymi erytropoetynę. W badaniu po rejestracji dotyczącego bezpieczeństwa z długotrwałym leczeniem do 8,4 roku liczba płytek krwi mniejsza niż 100 × 109/l na poziomie wyjściowym występowała u 2,1% dorosłych pacjentów w grupie metoksy polietylenoglikolu-epoetyny beta oraz u 2,4% dorosłych pacjentów w grupie innych środków stymulujących erytropoetynę. W trakcie tego badania liczba płytek krwi mniejsza niż 100 × 109/l występowała corocznie u 1,5–3,0% dorosłych pacjentów leczonych metoksy polietylenoglikolem-epoetyną beta oraz u 1,6–2,5% dorosłych pacjentów leczonych innymi środkami stymulującymi erytropoetynę.
Zgodnie z danymi z kontrolowanych badań klinicznych epoetyny alfa lub darbepoetyny alfa udar był powszechnym działaniem niepożądanym. Badanie po rejestracji dotyczące bezpieczeństwa wykazało podobną częstość udarów w grupie metoksy polietylenoglikolu-epoetyny beta (6,3%) i w grupie referencyjnych środków stymulujących erytropoetynę (epoetyna alfa, darbepoetyna alfa i epoetyna beta) (7%).
Tak jak w przypadku innych środków stymulujących erytropoetynę, podczas stosowania po rejestracji zgłaszano przypadki zakrzepicy, w tym zakrzepicy tętnicy płucnej (patrz sekcja „Szczególne środki ostrożności podczas stosowania”).
Stwierdzono przypadki prawdziwej aplazji czerwonych komórek spowodowanej wytwarzaniem przeciwciał neutralizujących erytropoetynę, których częstość jest nieznana. W przypadku rozpoznania prawdziwej aplazji czerwonych komórek leczenie metoksy polietylenoglikolem-epoetyną beta należy przerwać. Pacjentów nie należy przekładać na leczenie innymi rekombinowanymi erytropoetynami (patrz sekcja „Szczególne środki ostrożności podczas stosowania”).
Dzieci
W dwóch badaniach wzięło udział łącznie 104 pacjentów pediatrycznych, w tym 12 w wieku do 5 lat, 36 w wieku od 5 do 11 lat oraz 56 w wieku od 12 do 17 lat. Profil bezpieczeństwa metoksy polietylenoglikolu-epoetyny beta u dzieci włączonych do tych dwóch badań był ogólnie zgodny z profilem bezpieczeństwa u dorosłych, na podstawie niskiego nasycenia u pacjentów w tych badaniach.
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Personel medyczny i farmaceutyczny, a także pacjenci lub ich ustawowi przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez krajowy system raportowania (https://aisf.dec.gov.ua).
Okres ważności.
3 lata.
Warunki przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci. Przechowywać w temperaturze od 2 do 8 °C w oryginalnym opakowaniu w celu ochrony przed światłem. Nie zamrażać.
Preparat może być wyjęty z lodówki i przechowywany jednorazowo w temperaturze pokojowej (nie wyższej niż 30 °C). Preparat należy zużyć w ciągu tego okresu, który nie powinien przekraczać 1 miesiąca.
Niekompatybilność.
W przypadku braku badań kompatybilności nie należy mieszać preparatu Mirzera® z innymi lekami.
Opakowanie.
50 μg/0,3 ml lub 75 μg/0,3 ml w strzykawce wstępnie napełnionej. 1 strzykawka wstępnie napełniona razem z igłą do wstrzykiwań w opakowaniu tekturowym.
Kategoria wydania.
Na receptę.
Producent.
Roche Diagnostics GmbH
Adres producenta i miejsce prowadzenia działalności.
Sandhoferstrasse 116, 68305 Mannheim, Niemcy