Imunat
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku Imunat (IMMUNATE)
Skład:
substancje czynne: ludzki czynnik krzepnięcia krwi VIII, czynnik von Willebranda (vWF:RCo);
1 fiolka z proszkiem zawiera:
| Działająca substancja |
Zawartość substancji na fiolkę |
||
| 250/190 JM |
500/375 JM |
1000/750 JM |
|
| Czynnik krzepnięcia krwi człowieka VIII (z korektą zawartości albuminy)* |
250 JM (50 JM/ml) |
500 JM (100 JM/ml) |
1000 JM (100 JM/ml) |
| Aktywność specyficzna** |
(70 ± 30) JM/mg białka |
||
| Czynnik von Willebranda (vWF:RCo)*** |
190 JM (38 JM/ml) |
375 JM (75 JM/ml) |
750 JM (75 JM/ml) |
roztwornik: woda do wstrzykiwań – 5 ml (dla 250/190 JM i 500/375 JM) oraz 10 ml (dla 1000/750 JM);
substancje pomocnicze: albumina ludzka, glicyna, lizyny chlorowodorek, chlorek sodu, cytrynian trójsodowy dwuwodny, chlorek wapnia dwuwodny.
____________________________________________________________________
* Aktywność czynnika VIII była określana według międzynarodowym standardem WHO dla stężonych czynników VIII.
** Bez stabilizatora (albuminy).
Maksymalna aktywność specyficzna przy stosunku czynnika VIII do antygenu czynnika von Willebranda wynoszącym 1:1 wynosi 100 JM na mg białka.
*** Aktywność czynnika von Willebranda była określana według międzynarodowym standardem WHO dla stężonych czynników VIII i czynnika von Willebranda w osoczu.
Postać leku. Proszyk i roztwornik do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: proszek lub kruche białe lub bladożółte ciało stałe.
Grupa farmakoterapeutyczna. Środki hemostatyczne. Czynniki krzepnięcia krwi. Czynnik von Willebranda w połączeniu z czynnikiem krzepnięcia VIII.
Kod ATC B02B D06.
Właściwości immunologiczne i biologiczne.
Farmakodynamika.
Zespół czynnika VIII/czynnika von Willebranda składa się z dwóch cząsteczek (czynnika krzepnięcia krwi VIII i czynnika von Willebranda), które mają różne funkcje fizjologiczne.
Aktywowany czynnik VIII działa jako kofaktor dla aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w aktywowany czynnik X. Aktywowany czynnik X przekształca protrombinę w trombinę. Następnie trombina przekształca fibrynogen w fibrynę, tworząc się skrzele fibrynowe. Hemofilia A to uwarunkowana genetycznie, sprzężona z płcią choroba krzepnięcia krwi, spowodowana obniżonym poziomem aktywności czynnika VIII, prowadząca do obfitych krwawień w stawach, mięśniach i narządach wewnętrznych, które pojawiają się spontanicznie lub w wyniku urazów przypadkowych lub chirurgicznych. Poziomy czynnika VIII we krwi są podnoszone za pomocą terapii zastępczej, która tymczasowo koryguje niedobór czynnika i skłonność do krwawień.
Oprócz roli białka ochronnego czynnika VIII, czynnik von Willebranda (FV) pośredniczy w adhezji płytek krwi do miejsc uszkodzenia naczyń i odgrywa pewną rolę w agregacji płytek krwi, a jest niezbędny w terapii zastępczej u pacjentów z chorobą von Willebranda.
Farmakokinetyka.
Parametry farmakokinetyczne, uzyskane w wyniku badania farmakokinetycznego u pacjentów powyżej 12. roku życia, przedstawiono w tabelach 1 i 2.
W tabeli 1 opisano właściwości farmakokinetyczne według czynnika krzepnięcia VIII.
| Parametr |
|||||
| Liczba pacjentów |
Średnia wartość |
Odchylenie standardowe |
Mediana |
90 % CI |
|
| AUC0–48 godz. ([J.M. × godz.]/ml) |
18 |
11,4 |
2,8 |
11,6 |
10,9–12,7 |
| AUC0–∞ godz. ([J.M. × godz.]/ml) |
18 |
12,2 |
3,1 |
12,4 |
11,1–13,2 |
| Cmax (J.M./ml) |
18 |
1,0 |
0,3 |
0,9 |
0,8–1,0 |
| Tmax (godz.) |
18 |
0,3 |
0,1 |
0,3 |
0,3–0,3 |
| Okres półtrwania końcowego (godz.) |
18 |
12,7 |
3,2 |
12,2 |
10,8–15,3 |
| Clearance (ml/godz.) |
18 |
283 |
146 |
232 |
199–254 |
| Średni czas utrzymywania leku (godz.) |
18 |
15,3 |
3,6 |
15,3 |
12,1–17,2 |
| Vss (ml) |
18 |
4166 |
2021 |
3613 |
2815–4034 |
| Przyrost odbudowy poziomu aktywności ([J.M./ml]/ [J.M./kg]) |
18 |
0,020 |
0,006 |
0,019 |
0,016–0,020 |
Tabela 1
W tabeli 2 opisano właściwości farmakokinetyczne względem antygenu FV.
Tabela 2
| Parametr |
|||||
| Liczba pacjentów |
Mediana |
90 % przedział ufności |
|||
| AUC0–∝([MO × god]/ml) |
15 |
24,6 |
12,8–48,3 |
||
| Cmax (MO/ml) |
17 |
1,40 |
1,15–1,51 |
||
| Tmax (god) |
17 |
0,28 |
0,25–1,00 |
||
| Okres półtrwania końcowy (god) |
16 |
13,6 |
10,5–47,2 |
||
| Klirens (ml/god) |
15 |
136 |
68–178 |
||
| Średni czas utrzymywania leku (god) |
15 |
23,1 |
12,4–57,1 |
||
| Vss (ml) |
15 |
3156 |
2391–4672 |
||
| Przyrost odbudowy poziomu aktywności ([MO/ml]/ [MO/kg]) |
17 |
0,028 |
0,024–0,030 |
||
Dane kliniczne.
Wskazania.
Leczenie i profilaktyka krwawień u pacjentów z hemofilią A (wrodzony niedobór czynnika VIII, hemofilia A z inhibitorem czynnika VIII, nabyte niedobory czynnika VIII spowodowane spontanicznym wystąpieniem inhibitorów czynnika VIII).
Choroba von Willebranda z niedoborem czynnika VIII.
Uwaga. Skuteczność i bezpieczeństwo leku Imunat w zespole von Willebranda-Jürgens została oceniona klinicznie jedynie u niewielkiej liczby pacjentów. Należy szczególnie wziąć to pod uwagę w przypadku typu 3 tej choroby.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek ze składników pomocniczych leku.
Interakcje z innymi lekami i inne formy interakcji.
Nie przeprowadzono badań interakcji z lekiem Imunat. Nie odnotowano specyficznych interakcji ludzkiego czynnika krzepnięcia krwi VIII z innymi lekami.
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
Tak jak w przypadku wszystkich substancji podawanych dożylnie, możliwe są reakcje nadwrażliwości o charakterze alergicznym. Pacjentów należy poinformować o wczesnych objawach reakcji nadwrażliwości, w szczególności o hipotensji, tachykardii, bólu w klatce piersiowej, duszności, obrzęku (w tym obrzęku twarzy i powiek), pokrzywce, wysypce, zaczerwienieniu i swędzeniu, a także o reakcjach anafilaktycznych aż do wstrząsu anafilaktycznego.
W przypadku wystąpienia objawów nadwrażliwości należy zalecić pacjentom natychmiastowe przerwanie podania leku i skontaktowanie się z lekarzem. W przypadku wystąpienia wstrząsu należy zastosować standardowe leczenie wstrząsu.
Zgłaszano również inne reakcje związane z podaniem infuzyjnym leku Imunat, takie jak dreszcze, gorączka lub nudności.
Ponieważ ilość sodu w maksymalnej dobowej dawce może przekraczać 200 mg, należy to wziąć pod uwagę u osób przestrzegających diety o niskiej zawartości sodu.
250 i 500 JI w fiolce
Imunat zawiera około 9,8 mg sodu w jednej fiolce. Odpowiada to 0,5% z 2 g maksymalnego dziennego spożycia sodu zalecanego przez WHO dla dorosłych.
1000 JI w fiolce
Imunat zawiera około 19,6 mg sodu w jednej fiolce. Odpowiada to 1% z 2 g maksymalnego dziennego spożycia sodu zalecanego przez WHO dla dorosłych.
Powstawanie przeciwciał neutralizujących (inhibitorów) wobec czynnika VIII jest znanym powikłaniem leczenia pacjentów z hemofilią A. Te inhibitory są zazwyczaj immunoglobulinami klasy IgG skierowanymi przeciwko działaniu przeciwkrzepliwemu czynnika VIII, które ilościowo oznacza się w jednostkach Bethesda (JB) na 1 ml osocza krwi za pomocą zmodyfikowanego testu. Ryzyko powstania inhibitorów koreluje z ciężkością choroby oraz ekspozycją na czynnik VIII. Ryzyko to jest najwyższe w ciągu pierwszych 50 dni stosowania leku, ale utrzymuje się przez całe życie, choć jest rzadkie.
Pacjentów otrzymujących leczenie lekami zawierającymi czynnik krzepnięcia krwi VIII należy dokładnie monitorować pod kątem rozwoju inhibitorów poprzez odpowiednie obserwacje kliniczne i badania laboratoryjne (patrz również sekcja „Działania niepożądane”).
Ryzyko rozwoju inhibitorów zależy od różnych czynników związanych z indywidualnymi cechami danego pacjenta. Głównymi czynnikami ryzyka są m.in. typ mutacji genu czynnika VIII, wywiad rodzinny oraz przynależność etniczna pacjenta.
O inhibitorach zgłaszano głównie u pacjentów wcześniej nieleczonego.
Imunat wytwarza się z osocza krwi ludzkiej. Standardowe środki zapobiegające zakażeniu w wyniku stosowania leków pochodzących z krwi lub osocza ludzkiego obejmują dobór dawców, badanie indywidualnych porcji i pul osocza pod kątem konkretnych markerów infekcji oraz skuteczne etapy produkcji mające na celu inaktywację/usunięcie wirusów. Pomimo tego, przy stosowaniu leków pochodzących z krwi lub osocza ludzkiego, nie można całkowicie wykluczyć możliwości przeniesienia patogenów zakaźnych. Dotyczy to również nieznanych lub nowych wirusów oraz innych patogenów.
Podjęte środki uważane są za skuteczne wobec wirusów otoczkowych, takich jak wirus HIV (ludzkiego wirusa niedoboru odporności), wirusy zapalenia wątroby typu B i C, a także wobec wirusa zapalenia wątroby typu A, który nie ma otoczki. Środki te mogą mieć ograniczoną skuteczność wobec wirusów bez otoczki, takich jak parwowirus B19. Infekcja wywołana parwowirusem B19 może stanowić zagrożenie dla kobiet w ciąży (infekcja płodu) oraz dla osób z niedoborem odporności lub zwiększonym erytropoezą (np. z anemią hemolityczną).
Przy długotrwałym leczeniu choroby von Willebranda lekiem zawierającym czynnik VIII, poziom VIII:C może być nadmiernie podwyższony. Istnieje ryzyko wystąpienia zjawisk zakrzepowych podczas leczenia pacjentów z zespołem von Willebranda, szczególnie u pacjentów z znanymi czynnikami ryzyka klinicznymi lub laboratoryjnymi. Dlatego pacjentów należy monitorować pod kątem wczesnych objawów zakrzepicy. U pacjentów z wywiadem wczesnych zdarzeń zakrzepowo-zatorowych żylno-żylnych, wysoki poziom endogennego czynnika VIII był powiązany ze zwiększonym ryzykiem kolejnych zdarzeń zakrzepowych.
U pacjentów otrzymujących lek zawierający czynnik von Willebranda i czynnik VIII należy kontrolować stężenia czynnika VIII:C w osoczu, aby uniknąć trwałego nadmiernego poziomu czynnika VIII:C we krwi, który może zwiększyć ryzyko zdarzeń zakrzepowych.
Należy stosować zapobieganie zdarzeniom zakrzepowo-zatorowym zgodnie z obowiązującymi wytycznymi.
U pacjentów z chorobą von Willebranda, szczególnie u pacjentów z typem 3, mogą rozwijać się przeciwciała neutralizujące (inhibitory) wobec czynnika von Willebranda.
Takie przeciwciała mogą występować w ścisłym związku z reakcjami anafilaktycznymi. Z tego powodu pacjentów z reakcją anafilaktyczną należy przebadać pod kątem obecności takiego inhibitora. Jeśli oczekiwane poziomy aktywności vWF:RCo w osoczu krwi nie są osiągane lub jeśli krwawienie nie ustępuje po odpowiedniej dawce, należy przeprowadzić badanie w celu wykrycia obecności inhibitora czynnika von Willebranda. U pacjentów z wysokim poziomem inhibitora terapia czynnikiem von Willebranda może być nieskuteczna, dlatego należy rozważyć inne opcje leczenia. Leczenie powinno być prowadzone pod nadzorem lekarza z doświadczeniem w leczeniu zaburzeń hemostazy.
Pacjentom, którym regularnie/wielokrotnie podaje się leki zawierające czynnik VIII pochodzący z osocza krwi ludzkiej, należy rozważyć odpowiednie szczepienia (zapalenie wątroby typu A i B).
Zaleca się, aby przy każdym stosowaniu leku Imunat zapisywać w dzienniku pacjenta nazwę i numer serii leku, aby umożliwić powiązanie pacjenta z serią produktu.
Imunat zawiera izoaglutyniny grupowe (anty-A i anty-B). U pacjentów z grupą krwi A, B lub AB może wystąpić hemoliza po wielokrotnym podaniu w krótkich odstępach czasu lub po podaniu bardzo dużych dawek. Bardzo duże dawki w krótkim czasie mogą być stosowane w ramach terapii indukcji tolerancji immunologicznej w leczeniu hemofilii z inhibitorami czynnika VIII.
Przed zastosowaniem leku Imunat należy upewnić się, że zaburzenie krzepnięcia krwi rzeczywiście dotyczy niedoboru czynnika VIII (hemofilia A) lub niedoboru czynnika von Willebranda (zespół von Willebranda).
Stosowanie w okresie ciąży i karmienia piersią.
Badania wpływu czynnika VIII na funkcje rozrodcze u zwierząt nie były prowadzone. Ze względu na rzadkie występowanie hemofilii A u kobiet, brak jest doświadczeń z zastosowania czynnika VIII w okresie ciąży i karmienia piersią. Dlatego czynnik VIII należy stosować w okresie ciąży i karmienia piersią tylko w przypadku wyraźnych wskazań.
Informacje dotyczące infekcji parwowirusem B19 znajdują się w sekcji „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Brak informacji dotyczących wpływu leku Imunat na szybkość reakcji podczas prowadzenia pojazdów lub obsługiwanie innych maszyn.
Sposób stosowania i dawki.
Leczenie należy przeprowadzać pod nadzorem lekarza z doświadczeniem w leczeniu hemofilii A.
Dawkowanie standardowe
Dawka i długość trwania terapii zastępczej zależą od stopnia niedoboru czynnika VIII, lokalizacji i objętości krwawienia oraz stanu klinicznego pacjenta.
Podaną ilość jednostek czynnika VIII wyraża się w jednostkach międzynarodowych (j.m.), zgodnie z obowiązującym aktualnie standardem WHO dla leków opartych na czynniku VIII. Aktywność czynnika VIII we krwi wyraża się albo w procentach (w stosunku do normy dla osocza krwi człowieka), albo w jednostkach międzynarodowych (w stosunku do międzynarodowego standardu dla czynnika VIII w osoczu krwi).
Jedna jednostka międzynarodowa (j.m.) aktywności czynnika VIII odpowiada ilości czynnika VIII zawartemu w 1 ml normalnego osocza człowieka.
Dawkowanie w hemofilii A
Obliczenie potrzebnej dawki czynnika VIII opiera się na empirycznie ustalonej zależności: 1 jednostka międzynarodowa (j.m.) czynnika VIII na 1 kg masy ciała zwiększa aktywność czynnika VIII we krwi o około 1,5–2% w stosunku do normy. Potrzebną dawkę ustala się według następującego wzoru:
Potrzebna ilość jednostek (j.m.) = masa ciała (kg) × pożądane zwiększenie
czynnika VIII (%) × 0,5
Dawkę i częstotliwość stosowania należy zawsze dostosować tak, aby zapewnić skuteczność terapii u każdego konkretnego pacjenta.
Krwiawienia i zabiegi chirurgiczne
W przypadku wystąpienia kolejnych epizodów krwawienia aktywność czynnika VIII nie powinna opadać poniżej ustalonego poziomu aktywności we krwi (w % normy lub j.m./dl) w odpowiednim okresie. Poniższą tabelę 3 można wykorzystać do ustalenia dawki w epizodach krwawień oraz podczas zabiegów chirurgicznych.
Tabela 3
| Stopień krwawienia/rodzaj zabiegu chirurgicznego |
Wymagany poziom czynnika VIII (% normy) (j.m./dL) |
Częstotliwość podawania dawek (godziny)/czas trwania leczenia (dni) |
| Krwawienie |
||
| Wczesny hemartroza, krwawienia do mięśni lub jamy ustnej |
20–40 |
Powtarzać co 12–24 godziny przez co najmniej 1 dzień do ustania krwawienia, o czym świadczy brak bólu i gojenie się rany. |
| Bardziej nasilona hemartroza, krwawienia do mięśni lub hematoma |
30–60 |
Infuzję powtarzać co 12–24 godziny przez 3–4 dni lub dłużej, aż do ustąpienia bólu i usunięcia znacznego zaburzenia funkcji. |
| Krwawienia zagrażające życiu |
60–100 |
Infuzję powtarzać co 8–24 godziny do ustąpienia zagrożenia. |
| Zabieg chirurgiczny |
||
| Mały zabieg chirurgiczny, w tym ekstrakcja zębów |
30–60 |
Co 24 godziny przez co najmniej 1 dzień do gojenia się rany. |
| Duży zabieg chirurgiczny |
80–100 (przed i po zabiegu chirurgicznym) |
Infuzję powtarzać co 8–24 godziny do odpowiedniego gojenia się rany, po czym należy kontynuować leczenie przez co najmniej kolejne 7 dni, utrzymując aktywność czynnika VIII na poziomie 30–60 % (j.m./dL). |
Dawkę i częstotliwość podawania należy dostosować do odpowiedzi klinicznej w każdym przypadku. W niektórych okolicznościach (np. przy obecności niskiego miana inhibitora czynnika VIII) mogą być wymagane dawki większe niż obliczone według formuły, szczególnie na początku leczenia.
Jeśli krwawienia nie można kontrolować za pomocą dawki obliczonej, należy określić poziom czynnika VIII we krwi i podać odpowiednią dawkę leku Imunat w celu osiągnięcia satysfakcjonującej odpowiedzi klinicznej.
W trakcie leczenia zaleca się ocenę aktywności czynnika VIII w celu ustalenia potrzeby dostosowania dawki i częstotliwości powtórzonych infuzji. W przypadku dużych zabiegów chirurgicznych konieczne jest zapewnienie dokładnego monitorowania w trakcie terapii zastępczej poprzez ilościowe oznaczenie aktywności czynnika VIII we krwi. Odpowiedź na czynnik VIII może różnić się u każdego pacjenta, z różnym okresem półtrwania i różnym stopniem odbudowy in vivo.
Długoterminowa profilaktyka
Dla długotrwałej profilaktyki krwawień u pacjentów z ciężką formą hemofilii A typowe dawki wynoszą 20–40 JМ czynnika VIII na 1 kg masy ciała, podawane co 2–3 dni. W niektórych przypadkach, szczególnie u młodszych pacjentów, może być konieczne krótsze odstępy między dawkami lub stosowanie wyższych dawek leku.
Pacjenci z inhibitorami czynnika VIII
Pacjentów należy regularnie monitorować pod kątem rozwoju inhibitorów czynnika VIII. Jeśli oczekiwane poziomy aktywności czynnika VIII we krwi nie zostaną osiągnięte lub jeśli krwawienie nie poddaje się kontroli mimo odpowiedniej dawki, należy przeprowadzić test na obecność inhibitora czynnika VIII. U pacjentów z wysokim mianem inhibitora terapia czynnikiem VIII może być nieskuteczna, dlatego należy rozważyć inne opcje leczenia. Leczenie takich pacjentów powinno odbywać się pod nadzorem lekarzy z doświadczeniem w leczeniu hemofilii, w szczególności w przypadku występowania inhibitorów czynnika VIII (patrz również sekcja „Szczególne wskazania stosowania”).
Zdarzenia sercowo-naczyniowe
U pacjentów z istniejącymi czynnikami ryzyka sercowo-naczyniowego terapia zastępcza czynnikiem VIII może zwiększać ryzyko sercowo-naczyniowe.
Choroba von Willebranda z niedoborem czynnika VIII
Imunat jest wskazany w leczeniu i profilaktyce niedoboru czynnika VIII u pacjentów z chorobą von Willebranda z niedoborem czynnika VIII oraz w przypadkach, gdy leczenie wyłącznie desmopresyną jest nieskuteczne lub jest przeciwwskazane. Terapia zastępcza lekiem Imunat w celu kontrolowania krwawień oraz profilaktyki krwawień w okresie okołochirurgicznym powinna być prowadzona zgodnie z tymi samymi zaleceniami, co w przypadku hemofilii A.
Doświadczenie stosowania u dzieci jest ograniczone.
Pacjentów należy monitorować pod kątem rozwoju inhibitorów czynnika von Willebranda, jeśli oczekiwane poziomy aktywności czynnika von Willebranda we krwi nie zostaną osiągnięte lub jeśli krwawienie nie poddaje się kontroli mimo odpowiedniej dawki. U pacjentów z wysokimi poziomami inhibitora terapia czynnikiem von Willebranda może być nieskuteczna, należy rozważyć inne opcje terapeutyczne (patrz również sekcja „Szczególne wskazania stosowania”).
W przypadku stosowania leku zawierającego czynnik von Willebranda i czynnik VIII lekarz prowadzący powinien wiedzieć, że ciągłe leczenie może prowadzić do nadmiernego wzrostu poziomu czynnika VIII:C. Należy rozważyć możliwość zmniejszenia dawki i/lub wydłużenia odstępów między dawkami po leczeniu trwającym od 24 do 48 godzin, aby uniknąć zbyt wysokich poziomów czynnika VIII:C.
Dzieci
Lek należy stosować z ostrożnością u dzieci poniżej 6. roku życia, które miały ograniczone doświadczenie z lekami zawierającymi czynnik VIII, ponieważ dane kliniczne dla tej grupy pacjentów są ograniczone. Jednak na podstawie doniesień o przypadkach klinicznych można stwierdzić skuteczność i bezpieczeństwo stosowania.
Sposób stosowania
Instrukcje dotyczące rozcieńczania leku przed podaniem znajdują się w sekcji „Sposób stosowania i dawki”.
Imunat należy podawać powoli dożylnie. Maksymalna szybkość infuzji nie powinna przekraczać 2 ml na minutę.
Przygotowanie roztworu
Imunat należy rozpuszczać bezpośrednio przed podaniem. Roztwór należy użyć natychmiast, ponieważ lek nie zawiera środków konserwujących.
Przed zastosowaniem rozcieńczonego leku należy wizualnie sprawdzić obecność cząstek stałych i zmiany barwy. Nie należy stosować roztworów, które są mętne lub zawierają osad.
Zaleca się płukanie wszczepialnych urządzeń do dostępu do żył roztworem izotonicznym chlorku sodu przed i po infuzji leku Imunat.
Rozcieńczanie proszku: stosować technikę bezpylną, jak opisano poniżej
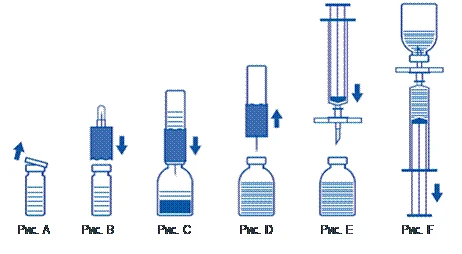
- Nieotwierany fiolę z rozpuszczalnikiem (woda do wstrzykiwań sterylna) należy podgrzać do temperatury pokojowej (nie wyższej niż 37 °C).
- Zdjąć ochronne kapturki z fiolki z proszkiem i fiolki z rozpuszczalnikiem (rys. A) oraz oczyścić gumowe przeciwciski obu fiol.
- Umieścić falisty brzeg urządzenia do przenoszenia na fiolkę z rozpuszczalnikiem i przycisnąć (rys. B).
- Zdjąć ochronne przykrycie z drugiego końca zestawu do przenoszenia, starając się nie dotykać odkrytego końca.
- Odwrócić zestaw do przenoszenia z przymocowaną fiolką z rozpuszczalnikiem nad fiolką z proszkiem i włożyć wolny koniec igły w gumowy przycisk fiolki z proszkiem (rys. C). Rozpuszczalnik zostanie wciągnięty do fiolki z proszkiem dzięki działaniu próżni.
- Po około jednej minucie rozłączyć dwie fiolki, wyciągając zestaw do przenoszenia z przymocowaną fiolką z rozpuszczalnikiem z fiolki z proszkiem (rys. D). Ponieważ lek łatwo się rozpuszcza, należy jedynie delikatnie, jeśli w ogóle, wymieszać zawartość fiolki z koncentratem. NIE WSTRZĄSAĆ FIOLKĄ Z PROSZKIEM. NIE OBRACAĆ FIOLKI Z PROSZKIEM DO CHWILI OSTATECZNEGO ODBIERANIA ZAWARTOŚCI.
- Po rozcieńczeniu przygotowany roztwór należy wizualnie sprawdzić pod kątem obecności zanieczyszczeń i zmiany barwy przed podaniem. Jednak nawet przy starannym przestrzeganiu procedury odtwarzania czasem mogą być widoczne pojedyncze drobne cząstki. Cząstki te zostaną usunięte za pomocą filtra zawartego w zestawie, bez obniżania skuteczności leku.
Stosować technikę bezpylną, jak opisano poniżej
- Aby zapobiec podaniu cząstek gumy pochodzących z przycisku (ryzyko mikroembolii), należy korzystać z zestawu z filtrem dostarczanym w opakowaniu. Aby odciągnąć rozpuszczony lek, założyć zestaw z filtrem na jednorazowy strzykawka dołączony do opakowania i włożyć go przez gumowy przycisk (rys. E).
- Na chwilę odłączyć strzykawkę od zestawu z filtrem. Powietrze dostanie się do fiolki z proszkiem, a ewentualna piana zniknie. Następnie odciągnąć roztwór do strzykawki przez zestaw z filtrem (rys. F).
- Odłączyć strzykawkę od zestawu z filtrem i powoli podać roztwór dożylnie (maksymalna szybkość podania: 2 ml na minutę) za pomocą zestawu do infuzji z motylkami dołączonym do opakowania (lub jednorazowej igły dołączanej do opakowania).
Nie wykorzystany lek lub odpady należy zutylizować zgodnie z lokalnymi przepisami.
Dzieci
Lek należy stosować z ostrożnością u dzieci poniżej 6. roku życia, które miały ograniczone doświadczenie z lekami zawierającymi czynnik VIII, ponieważ dane kliniczne dla tej grupy pacjentów są ograniczone. Jednak na podstawie doniesień o przypadkach klinicznych można stwierdzić skuteczność i bezpieczeństwo stosowania.
Przedawkowanie
Nie opisano objawów związanych z przedawkowaniem ludzkiego czynnika krzepnięcia VIII.
Zazwyczaj istnieje ryzyko wystąpienia zjawisk tromboembolicznych. U pacjentów z grupą krwi A, B lub AB istnieje ryzyko hemolizy (patrz również sekcja „Szczególne wskazania stosowania”).
Działania niepożądane.
Poniższe działania niepożądane zostały wykryte podczas badań klinicznych oraz zgłoszone w okresie po rejestracji po wydaniu certyfikatu rejestracyjnego leku Imunat. Częstość podana jest według następującej skali: bardzo często (≥ 10 %); często (≥ 1 % — < 10 %), rzadko (≥ 0,1 % – < 1 %), bardzo rzadko (≥ 0,01 % – < 0,1 %), niezwykle rzadko (< 0,01 %).
Badania kliniczne
Częstość wszystkich poniżej wymienionych działań niepożądanych zgłoszonych w wyniku badań klinicznych była „rzadko” (≥ 0,1 % — < 1 %).
Choroby układu odpornościowego
Rzadko: reakcje alergiczne.
Doniesienia spontaniczne po wydaniu certyfikatu rejestracyjnego
Częstość wszystkich poniżej wymienionych działań niepożądanych była „niezwykle rzadko” (< 0,01 %).
Choroby układu krwiotwórczego i chłonnego
Koagulopatia, hamowanie czynnika VIII.
Zaburzenia psychiczne
Niekoić
Choroby serca
Przyspieszone bicie serca, tachykardia.
Choroby przewodu pokarmowego
Wymioty, nudności.
Ogólne choroby i dyskomfort w miejscu podania
Ból w klatce piersiowej, dyskomfort w klatce piersiowej, obrzęk (w tym obrzęk obwodowy i obrzęk twarzy), podrażnienie w miejscu wstrzyknięcia (w tym uczucie pieczenia), dreszcze, ból, gorączka.
Choroby układu odpornościowego
Podwyższona wrażliwość.
Choroby układu nerwowego
Bóle głowy, zawroty głowy, parestezje.
Choroby dróg oddechowych, narządów klatki piersiowej i jamy śródpiersia
Kaszel, duszność.
Choroby układu naczyniowego
Hipotensja tętnicza, zaczerwienienie, bladość.
Choroby oczu
Zapalenie spojówek, obrzęk powiek.
Choroby skóry i tkanek podskórnych
Erytem, egzantema, neurodermitis, swędzenie, wysypka, wysypka rumieniowa, wysypka uogólniona, pokrzywka, nadpotliwość.
Choroby mięśni szkieletowych, tkanki łącznej i kości
Mialgia.
Nie otrzymano jeszcze zgłoszeń dotyczących poniższych działań niepożądanych, ale mogły one wystąpić podczas stosowania leku Imunat.
Choroby układu krwiotwórczego i chłonnego
Hemoliza u pacjentów z grupą krwi A, B lub AB.
Ogólne choroby i dyskomfort w miejscu podania
Obniżenie odpowiedzi terapeutycznej.
Opis poszczególnych działań niepożądanych
Reakcje nadwrażliwości lub reakcje alergiczne (które mogą obejmować obrzęk naczynioruchowy, uczucie pieczenia i mrowienia w miejscu infuzji, dreszcze, erytem, uogólnioną pokrzywkę, ból głowy, wysypkę, hipotensję tętniczą, osłabienie, nudności, niepokój, tachykardię, uczucie ucisku w klatce piersiowej, mrowienie, wymioty, świsty) występowały rzadko i w niektórych przypadkach mogły postępować do ciężkiej reakcji anafilaktycznej (w tym szok). Pacjentom należy zalecić skontaktowanie się z lekarzem w przypadku wystąpienia tych objawów.
W rzadkich przypadkach obserwowano gorączkę.
U pacjentów z hemofilią A mogą rozwijać się przeciwciała neutralizujące (inhibitory) wobec czynnika VIII. Jeśli takie inhibitory powstają, stan objawia się niewystarczającą odpowiedzią kliniczną. W takich przypadkach zaleca się skontaktowanie się ze specjalistycznym ośrodkiem leczenia hemofilii.
Hemoliza może wystąpić po podaniu dużych dawek (np. gdy konieczne jest osiągnięcie poziomu czynnika VIII w osoczu powyżej 100 %) u pacjentów z grupą krwi A, B lub AB.
Informacje dotyczące bezpieczeństwa wirusowego znajdują się w sekcji „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie.
Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego ze stosowaniem tego leku. Personel medyczny i farmaceutyczny, a także pacjenci lub ich uprawnieni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez zautomatyzowany system informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
Lek – 2 lata.
Roztwórnik (woda do wstrzykiwań) – 5 lat.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8 °C. Nie zamarzać! Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem. Przechowywać w miejscu niedostępnym dla dzieci. W ramach podanego okresu ważności lek może być przechowywany w temperaturze pokojowej (nie wyższej niż 25 °C) przez okres do 6 miesięcy.
Po rozcieńczeniu lek może być przechowywany w temperaturze pokojowej (nie wyższej niż 25 °C) przez 6 godzin.
Niezgodność
Imunat nie może być mieszany z innymi lekami, ponieważ może to negatywnie wpłynąć na bezpieczeństwo i skuteczność. Należy używać wyłącznie dostarczonych zestawów infuzyjnych, ponieważ adsorpcja czynnika krzepnięcia krwi człowieka VIII przez wewnętrzną powierzchnię niektórych urządzeń infuzyjnych może prowadzić do nieskutecznego leczenia.
Opakowanie.
1 fiolka z proszkiem (250/190 MI, 500/375 MI lub 1000/750 MI) w zestawie z 1 fiolką z rozpuszczalnikiem (woda do wstrzykiwań 5 ml lub 10 ml) oraz zestawem do rozpuszczenia i podania (1 urządzenie do przetaczania/filtracji, 1 jednorazowy strzykawka (5 ml lub 10 ml), 1 jednorazowa igła, 1 zestaw do infuzji) w pudełku.
Kategoria wydawania.
Na receptę.
Producent.
Takeda Manufacturing Austria AG, Austria.
Lokalizacja producenta i adres miejsca prowadzenia działalności.
Industriestraße 67, 1221 Wiedeń, Austria.