Imunat
UcrainaIndice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE IMUNAT (IMMUNATE)
Composizione:
Principi attivi: fattore della coagulazione del sangue umano VIII, fattore di von Willebrand (vWF:RCo);
1 flaconcino con polvere contiene:
| Sostanza attiva |
Contenuto della sostanza per flaconcino |
||
| 250/190 UI |
500/375 UI |
1000/750 UI |
|
| Fattore della coagulazione del sangue umano VIII (corretto per il contenuto di albumina)* |
250 UI (50 UI/ml) |
500 UI (100 UI/ml) |
1 000 UI (100 UI/ml) |
| Attività specifica** |
(70 ± 30) UI/mg di proteine |
||
| Fattore di von Willebrand (vWF:RCo)*** |
190 UI (38 UI/ml) |
375 UI (75 UI/ml) |
750 UI (75 UI/ml) |
solvente: acqua per preparazioni iniettabili – 5 ml (per 250/190 UI e 500/375 UI) e 10 ml (per 1000/750 UI);
* sostanze ausiliarie: albumina umana, glicina, cloridrato di lisina, cloruro di sodio, citrato trisodico diidrato, cloruro di calcio diidrato.
____________________________________________________________________
* L'attività del fattore VIII è stata determinata in base allo standard internazionale OMS per i concentrati di fattore VIII.
** Senza stabilizzante (albumina).
L'attività specifica massima nel rapporto tra fattore VIII e antigene del fattore di von Willebrand di 1:1 è di 100 UI per mg di proteina.
*** L'attività del fattore di von Willebrand è stata determinata in base allo standard internazionale OMS per i concentrati di fattore VIII e fattore di von Willebrand nel plasma.
Forma farmaceutica. Polvere e solvente per soluzione iniettabile.
Principali proprietà fisico-chimiche: polvere o sostanza friabile di colore bianco o giallo pallido.
Gruppo farmacoterapeutico. Agenti emostatici. Fattori della coagulazione sanguigna. Fattore di von Willebrand in combinazione con il fattore della coagulazione VIII.
Codice ATC B02B D06.
Proprietà immunologiche e biologiche.
Farmacodinamica.
Il complesso fattore VIII/fattore di von Willebrand è costituito da due molecole (fattore della coagulazione VIII e fattore di von Willebrand) che svolgono diverse funzioni fisiologiche.
Il fattore VIII attivato agisce come cofattore per il fattore IX attivato, accelerando la trasformazione del fattore X in fattore X attivato. Il fattore X attivato converte la protrombina in trombina. Successivamente, la trombina converte il fibrinogeno in fibrina, formando un coagulo di fibrina. L'emofilia A è un disturbo ereditario legato al sesso della coagulazione sanguigna causato da livelli ridotti di attività del fattore VIII, che determina emorragie abbondanti nelle articolazioni, nei muscoli e negli organi interni, insorgenti spontaneamente o in seguito a traumi accidentali o chirurgici. I livelli plasmatici di fattore VIII aumentano mediante terapia sostitutiva, che corregge temporaneamente la carenza del fattore e la tendenza all'emorragia.
Oltre al ruolo di proteina protettiva per il fattore VIII, il fattore di von Willebrand (FVW) media l'adesione delle piastrine ai siti di danno vascolare e svolge un ruolo nell'aggregazione piastrinica ed è essenziale per la terapia sostitutiva nei pazienti affetti da malattia di von Willebrand.
Farmacocinetica.
I parametri farmacocinetici ottenuti da uno studio farmacocinetico in pazienti di età superiore ai 12 anni sono riportati nelle tabelle 1 e 2.
La tabella 1 descrive le proprietà farmacocinetiche relative al fattore VIII della coagulazione.
| Parametro |
|||||
| Numero di pazienti |
Valore medio |
Deviazione standard |
Mediana |
IC 90 % |
|
| AUC0–48 h ([MO × h]/ml) |
18 |
11,4 |
2,8 |
11,6 |
10,9–12,7 |
| AUC0–∞ h ([MO × h]/ml) |
18 |
12,2 |
3,1 |
12,4 |
11,1–13,2 |
| Cmax (MO/ml) |
18 |
1,0 |
0,3 |
0,9 |
0,8–1,0 |
| Tmax (h) |
18 |
0,3 |
0,1 |
0,3 |
0,3–0,3 |
| Periodo di emivita terminale (h) |
18 |
12,7 |
3,2 |
12,2 |
10,8–15,3 |
| Clearance (ml/h) |
18 |
283 |
146 |
232 |
199–254 |
| Tempo medio di permanenza del farmaco (h) |
18 |
15,3 |
3,6 |
15,3 |
12,1–17,2 |
| Vss (ml) |
18 |
4166 |
2021 |
3613 |
2815–4034 |
| Incremento di recupero del livello di attività ([MO/ml]/[MO/kg]) |
18 |
0,020 |
0,006 |
0,019 |
0,016–0,020 |
Tabella 1
Nella tabella 2 sono descritte le proprietà farmacocinetiche rispetto all'antigene FB.
Tabella 2
| Parametro |
|||||
| Numero di pazienti |
Mediana |
IC 90 % |
|||
| AUC0–∝([MO × h]/ml) |
15 |
24,6 |
12,8–48,3 |
||
| Cmax (MO/ml) |
17 |
1,40 |
1,15–1,51 |
||
| Tmax (h) |
17 |
0,28 |
0,25–1,00 |
||
| Periodo di emivita terminale (h) |
16 |
13,6 |
10,5–47,2 |
||
| Clearance (ml/h) |
15 |
136 |
68–178 |
||
| Tempo medio di permanenza (h) |
15 |
23,1 |
12,4–57,1 |
||
| Vss (ml) |
15 |
3156 |
2391–4672 |
||
| Incremento di recupero dell'attività ([MO/ml]/[MO/kg]) |
17 |
0,028 |
0,024–0,030 |
||
Caratteristiche cliniche.
Indicazioni.
Terapia e profilassi delle emorragie in pazienti con emofilia A (deficit congenito del fattore VIII, emofilia A con inibitore del fattore VIII, deficit acquisito del fattore VIII dovuto alla comparsa spontanea di inibitori del fattore VIII).
Malattia di von Willebrand con deficit del fattore VIII.
Nota. L'efficacia e la sicurezza del medicinale Imunat nel sindrome di von Willebrand-Jürgens sono state studiate clinicamente solo su un numero ridotto di pazienti. Ciò va particolarmente considerato per il tipo 3 di questa malattia.
Controindicazioni.
Ipersensibilità alla sostanza attiva o a qualsiasi eccipiente del medicinale.
Interazioni con altri medicinali e altre forme di interazione.
Non sono stati effettuati studi di interazione con il medicinale Imunat. Non sono stati riportati casi specifici di interazione tra il fattore di coagulazione del sangue umano VIII e altri medicinali.
Caratteristiche di impiego.
Come per tutti i principi attivi somministrati per via endovenosa, possono verificarsi reazioni di ipersensibilità di tipo allergico. I pazienti devono essere informati sui segni precoci di reazioni di ipersensibilità, in particolare ipotensione, tachicardia, dolore toracico, dispnea, edema (in particolare edema del viso e delle palpebre), orticaria, eruzioni cutanee, iperemia e prurito, nonché anafilassi fino allo shock anafilattico.
In caso di comparsa di sintomi di ipersensibilità, ai pazienti deve essere raccomandato di interrompere immediatamente la somministrazione del medicinale e di rivolgersi al proprio medico. In caso di insorgenza di shock, deve essere applicato il trattamento medico standard per lo shock.
Sono state segnalate anche altre reazioni correlate all'infusione di Imunat, come brividi, febbre o nausea.
Poiché la quantità di sodio nella dose giornaliera massima può superare i 200 mg, ciò deve essere tenuto in considerazione da persone che seguono una dieta a basso contenuto di sodio.
250 e 500 UI in flaconcino
Imunat contiene circa 9,8 mg di sodio per flaconcino. Ciò corrisponde allo 0,5% del consumo giornaliero massimo di sodio di 2 g raccomandato dall'OMS per gli adulti.
1000 UI in flaconcino
Imunat contiene circa 19,6 mg di sodio per flaconcino. Ciò corrisponde all'1% del consumo giornaliero massimo di sodio di 2 g raccomandato dall'OMS per gli adulti.
La formazione di anticorpi neutralizzanti (inibitori) contro il fattore VIII è una complicanza nota durante il trattamento dei pazienti con emofilia A. Tali inibitori sono generalmente immunoglobuline IgG dirette contro l'attività anticoagulante del fattore VIII, quantificate in unità Bethesda (UB) per 1 ml di plasma sanguigno mediante analisi modificato. Il rischio di sviluppare inibitori è correlato alla gravità della malattia e all'esposizione al fattore VIII. Questo rischio è massimo nei primi 50 giorni di trattamento, ma permane per tutta la vita, sebbene sia raro.
I pazienti sottoposti a trattamento con medicinali contenenti fattore della coagulazione VIII devono essere attentamente monitorati per la comparsa di inibitori mediante opportune osservazioni cliniche e analisi di laboratorio (vedere anche la sezione «Effetti indesiderati»).
Il rischio di sviluppare inibitori dipende da diversi fattori legati alle caratteristiche specifiche del singolo paziente. I principali fattori di rischio includono, in particolare, il tipo di mutazione del gene del fattore VIII, l'anamnesi familiare e l'appartenenza etnica del paziente.
La comparsa di inibitori è stata principalmente segnalata in pazienti precedentemente non trattati.
Imunat è prodotto a partire dal plasma umano. Le misure standard per prevenire l'infezione derivante dall'uso di medicinali prodotti da sangue o plasma umano comprendono la selezione dei donatori, lo screening delle singole unità e dei pool di plasma per specifici marcatori infettivi e fasi di produzione efficaci per l'inattivazione/rimozione dei virus. Nonostante ciò, con l'uso di medicinali prodotti da sangue o plasma umano, non può essere esclusa completamente la possibilità di trasmissione di agenti infettivi. Ciò vale anche per virus sconosciuti o nuovi e altri agenti infettivi.
Le misure adottate sono considerate efficaci nei confronti dei virus a membrana, come il virus dell'immunodeficienza umana (HIV), i virus dell'epatite B e dell'epatite C, nonché nei confronti del virus dell'epatite A, che è privo di membrana. Le misure adottate potrebbero avere un valore limitato nei confronti dei virus privi di membrana, come il parvovirus B19. L'infezione da parvovirus B19 può essere pericolosa per le donne in gravidanza (infezione fetale) e per soggetti con immunodeficienza o con aumentata eritropoiesi (ad esempio con anemia emolitica).
Nel trattamento prolungato della malattia di von Willebrand con un medicinale contenente fattore VIII, il livello di VIII:C può aumentare eccessivamente. Esiste un rischio di manifestazioni trombotiche durante il trattamento di pazienti con sindrome di von Willebrand, specialmente in pazienti con noti fattori di rischio clinici o di laboratorio. Pertanto, i pazienti devono essere monitorati per rilevare precocemente segni di trombosi. Nei pazienti con anamnesi di tromboembolia venosa, un elevato livello endogeno di fattore VIII è stato associato a un aumentato rischio di eventi trombotici successivi.
Nei pazienti che ricevono un medicinale contenente fattore von Willebrand e fattore VIII, devono essere controllati i livelli plasmatici di fattore VIII:C, per evitare un livello plasmatico persistente ed eccessivo di fattore VIII:C, che potrebbe aumentare il rischio di eventi trombotici.
Deve essere effettuata la profilassi della tromboembolia venosa in conformità con le raccomandazioni vigenti.
Nei pazienti con malattia di von Willebrand, specialmente nei pazienti di tipo 3, possono svilupparsi anticorpi neutralizzanti (inibitori) contro il fattore von Willebrand.
Tali anticorpi possono manifestarsi in stretta associazione con reazioni anafilattiche. Per questo motivo, i pazienti con reazione anafilattica devono essere sottoposti a controllo per la presenza di tale inibitore. Se non si raggiungono i livelli attesi di attività di vWF:RCo nel plasma o se il sanguinamento non è controllato con la dose appropriata, deve essere effettuato un test per determinare la presenza di inibitore del fattore von Willebrand. Nei pazienti con alto livello di inibitore, la terapia con fattore von Willebrand può risultare inefficace; pertanto, devono essere considerate altre opzioni terapeutiche. Il trattamento deve essere effettuato sotto la supervisione di un medico esperto nel trattamento dei disturbi dell'emostasi.
Ai pazienti che ricevono ripetutamente o regolarmente medicinali contenenti fattore VIII ottenuto dal plasma umano, deve essere valutata l'opportunità di una vaccinazione adeguata (epatite A e B).
Si raccomanda di registrare, nel diario del paziente, il nome e il numero di lotto del medicinale ad ogni utilizzo di Imunat, al fine di stabilire un collegamento tra il paziente e il lotto del prodotto.
Imunat contiene isoagglutinine dei gruppi sanguigni (anti-A e anti-B). Nei pazienti con gruppo sanguigno A, B o AB può verificarsi emolisi dopo somministrazioni ripetute a brevi intervalli o dopo somministrazione di dosi molto elevate. Dosaggi molto elevati in breve tempo potrebbero essere utilizzati nell'ambito della terapia di induzione della tolleranza immunitaria per il trattamento dell'emofilia con inibitori del fattore VIII.
Prima di prescrivere Imunat, è necessario accertare che il disturbo della coagulazione sia effettivamente dovuto a carenza di fattore VIII (emofilia A) o a carenza di fattore von Willebrand (sindrome di von Willebrand).
Uso durante la gravidanza e l’allattamento.
Non sono stati condotti studi sull'effetto del fattore VIII sulla fertilità negli animali. A causa della rarità dell'emofilia A nelle donne, non esiste esperienza clinica sull'uso del fattore VIII durante la gravidanza e l’allattamento. Pertanto, il fattore VIII deve essere usato durante la gravidanza e l’allattamento solo in caso di stretta necessità.
Informazioni sulla infezione da parvovirus B19 vedere nella sezione «Caratteristiche di impiego».
Capacità di influire sulla capacità di guidare veicoli o di usare macchinari.
Non sono disponibili informazioni sull'effetto del medicinale Imunat sulla capacità di guidare veicoli o di usare macchinari.
Modalità e posologia
Il trattamento deve essere effettuato sotto la supervisione di un medico esperto nella terapia dell'emofilia A.
Dosi standard
La dose e la durata della terapia sostitutiva dipendono dal grado di carenza del fattore VIII, dalla sede e dall'entità dell'emorragia, nonché dallo stato clinico del paziente.
La quantità somministrata di fattore VIII è espressa in unità internazionali (UI), secondo l'attuale standard dell'OMS per i medicinali a base di fattore VIII. L'attività del fattore VIII nel plasma è espressa in percentuale (rispetto al valore normale del plasma umano) oppure in unità internazionali (rispetto allo standard internazionale per il fattore VIII nel plasma sanguigno).
Un'unità internazionale (UI) di attività del fattore VIII corrisponde alla quantità di fattore VIII presente in 1 ml di plasma normale umano.
Dosi nell'emofilia A
Il calcolo della dose richiesta di fattore VIII si basa sulla relazione empirica seguente: 1 unità internazionale (UI) di fattore VIII per kg di peso corporeo aumenta l'attività del fattore VIII nel plasma di circa l'1,5-2% rispetto all'attività normale. La dose richiesta può essere calcolata mediante la seguente formula:
Numero richiesto di unità (UI) = peso corporeo (kg) × aumento desiderato
del fattore VIII (%) × 0,5
La dose e la frequenza di somministrazione devono sempre essere adattate per garantire l'efficacia clinica in ciascun paziente.
Emorragie e interventi chirurgici
In caso di episodi emorragici, l'attività del fattore VIII non deve scendere al di sotto del livello minimo richiesto nel plasma (espresso in % della norma oppure in UI/dl) durante il periodo appropriato. La tabella 3 riportata di seguito può essere utilizzata per determinare la dose necessaria in caso di episodi emorragici e durante interventi chirurgici.
Tabella 3
| Grado dell'emorragia/tipo di procedura chirurgica |
Livello richiesto del fattore VIII (% del normale) (UI/dl) |
Frequenza di somministrazione del dosaggio (ore)/durata del trattamento (giorni) |
| Emorragia |
||
| Emartrosi precoce, emorragia muscolare o ematoma orale |
20–40 |
Ripetere ogni 12–24 ore per almeno 1 giorno fino alla cessazione dell'emorragia, attestata dall'assenza di dolore e dal raggiungimento della guarigione della ferita. |
| Emartrosi più grave, emorragia muscolare o ematoma |
30–60 |
Ripetere l'infusione ogni 12–24 ore per 3–4 giorni o più fino alla scomparsa del dolore e alla risoluzione dei gravi disturbi funzionali. |
| Emorragia pericolosa per la vita |
60–100 |
Ripetere l'infusione ogni 8–24 ore fino alla scomparsa del pericolo. |
| Intervento chirurgico |
||
| Intervento chirurgico minore, inclusa l'estrazione dentaria |
30–60 |
Ogni 24 ore per almeno 1 giorno fino alla guarigione della ferita. |
| Intervento chirurgico maggiore |
80–100 (prima e dopo l'intervento chirurgico) |
Ripetere l'infusione ogni 8–24 ore fino a un adeguato rimarginamento della ferita; successivamente continuare il trattamento per almeno altri 7 giorni per mantenere il livello di attività del fattore VIII tra il 30–60% (UI/dl). |
La dose e la frequenza di somministrazione devono essere adattate alla risposta clinica in ciascun caso individuale. In determinate circostanze (ad esempio, in presenza di un titolo basso di inibitore del fattore VIII), specialmente all'inizio del trattamento, potrebbero essere necessarie dosi superiori a quelle calcolate secondo la formula.
Se il sanguinamento non può essere controllato con la dose calcolata, si raccomanda di determinare il livello di fattore VIII nel plasma e di somministrare una dose adeguata del medicinale Imunat per ottenere una risposta clinica soddisfacente.
Durante il trattamento si raccomanda di valutare l'attività del fattore VIII al fine di stabilire la necessità di aggiustamenti della dose e della frequenza delle infusioni ripetute. In caso di interventi chirurgici maggiori, è estremamente importante garantire un controllo accurato durante la terapia sostitutiva mediante test quantitativi dell'attività del fattore VIII nel plasma. La risposta al fattore VIII può variare da paziente a paziente, con differenti valori di emivita e grado di recupero in vivo.
Profilassi a lungo termine
Per la profilassi prolungata del sanguinamento nei pazienti con emofilia A grave, le dosi abituali sono di 20–40 UI di fattore VIII per kg di peso corporeo, con un intervallo di somministrazione di 2–3 giorni. In alcuni casi, specialmente nei pazienti più giovani, potrebbe rendersi necessario un intervallo più breve tra le somministrazioni o l'uso di dosi più elevate del medicinale.
Pazienti con inibitori del fattore VIII
I pazienti devono essere monitorati regolarmente per la comparsa di inibitori del fattore VIII. Se non si raggiungono i livelli attesi di attività del fattore VIII nel plasma o se il sanguinamento non è controllato con una dose appropriata, si deve effettuare un test per la ricerca di inibitori del fattore VIII. Nei pazienti con livelli elevati di inibitore, la terapia con fattore VIII potrebbe risultare inefficace e si devono considerare altre opzioni terapeutiche. La gestione di tali pazienti deve essere effettuata sotto la supervisione di medici esperti nel trattamento dell'emofilia, in particolare per quanto riguarda la comparsa di inibitori del fattore VIII (vedere anche il paragrafo «Speciali avvertenze e precauzioni per l'uso»).
Eventi cardiovascolari
Nei pazienti con fattori di rischio cardiovascolari preesistenti, la terapia sostitutiva con fattore VIII potrebbe aumentare il rischio cardiovascolare.
Malattia di von Willebrand con deficit di fattore VIII
Imunat è indicato per il trattamento e la profilassi del deficit di fattore VIII nei pazienti con malattia di von Willebrand con deficit di fattore VIII e nei casi in cui il trattamento con desmopressina soltanto risulti inefficace o controindicato. La terapia sostitutiva con Imunat per il controllo del sanguinamento e per la profilassi perioperatoria del sanguinamento deve essere effettuata secondo le stesse raccomandazioni previste per l’emofilia A.
L'esperienza d'uso nei bambini è limitata.
I pazienti devono essere monitorati per la comparsa di inibitori del fattore von Willebrand se non si raggiungono i livelli attesi di attività del fattore von Willebrand nel plasma o se il sanguinamento non è controllato nonostante la somministrazione di una dose appropriata. Nei pazienti con livelli elevati di inibitore, la terapia con fattore von Willebrand potrebbe risultare inefficace e si devono considerare altre opzioni terapeutiche (vedere anche il paragrafo «Speciali avvertenze e precauzioni per l'uso»).
Nel caso di utilizzo di un medicinale contenente sia il fattore von Willebrand che il fattore VIII, il medico curante deve essere consapevole che un trattamento continuativo può causare un aumento eccessivo del livello di fattore VIII:C. Si deve considerare la possibilità di ridurre la dose e/o prolungare gli intervalli tra le somministrazioni dopo un trattamento della durata di 24-48 ore, al fine di evitare livelli troppo elevati di fattore VIII:C.
Bambini
Il medicinale deve essere usato con cautela nei bambini di età inferiore a 6 anni che hanno avuto un'esposizione limitata ai preparati di fattore VIII, poiché i dati clinici in questo gruppo di pazienti sono limitati. Tuttavia, sulla base di casi clinici riportati, si può concludere che l'uso è efficace e sicuro.
Modalità di somministrazione
Per le istruzioni sulla ricostituzione del medicinale prima della somministrazione, vedere il paragrafo «Modalità di somministrazione e dosi».
Imunat deve essere somministrato per via endovenosa lentamente. La velocità massima di infusione non deve superare i 2 ml al minuto.
Preparazione della soluzione
Imunat deve essere ricostituito immediatamente prima della somministrazione. La soluzione deve essere utilizzata immediatamente, poiché il prodotto non contiene conservanti.
Prima dell'uso, i medicinali ricostituiti devono essere ispezionati visivamente per la presenza di particelle solide e variazioni di colore. Le soluzioni torbide o contenenti sedimenti non devono essere utilizzate.
Si raccomanda di lavare i dispositivi impiantabili per l'accesso venoso con soluzione fisiologica isotonica prima e dopo l'infusione del medicinale Imunat.
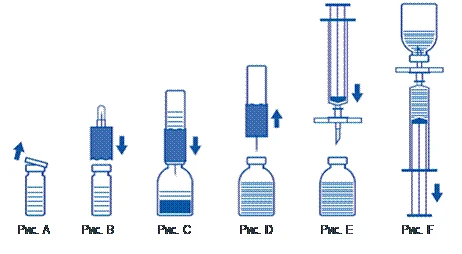
Ricostituzione della polvere: utilizzare una tecnica asettica, come descritto di seguito
- Riscaldare il flaconcino non aperto del solvente (acqua sterile per preparazioni iniettabili) a temperatura ambiente (non superiore a 37 °C).
- Rimuovere i tappi protettivi dal flaconcino della polvere e da quello del solvente (fig. A) e pulire i tappi di gomma di entrambi i flaconcini.
- Posizionare il bordo scanalato del dispositivo di trasferimento sul flaconcino del solvente e premere (fig. B).
- Rimuovere la protezione dall'altro estremo del set di trasferimento, cercando di non toccare l'estremità aperta.
- Capovolgere il set di trasferimento con il flaconcino del solvente attaccato sopra il flaconcino della polvere e inserire l'estremità libera dell'ago attraverso il tappo di gomma del flaconcino della polvere (fig. C). Il solvente verrà aspirato nel flaconcino della polvere per effetto del vuoto.
- Dopo circa un minuto, separare i due flaconcini rimuovendo il set di trasferimento con il flaconcino del solvente attaccato dal flaconcino della polvere (fig. D). Poiché il prodotto si ricostituisce facilmente, è sufficiente agitare leggermente il flaconcino del concentrato, se necessario. NON AGITARE IL FLACONCINO DELLA POLVERE. NON RIBALTARE IL FLACONCINO DELLA POLVERE FINO A QUANDO NON SI È PRONTI A ESTRARNE IL CONTENUTO.
- Dopo la ricostituzione, la soluzione preparata deve essere ispezionata visivamente per la presenza di inclusioni e variazioni di colore prima della somministrazione. Tuttavia, anche con un'accurata osservanza della procedura di ricostituzione, talvolta possono essere visibili alcune piccole particelle. Queste particelle verranno rimosse dal filtro incluso nel set, senza ridurre l'efficacia del prodotto.
Utilizzare una tecnica asettica, come descritto di seguito
- Per evitare l'introduzione di particelle di gomma provenienti dal tappo (rischio di microembolia), si deve utilizzare il set filtro incluso nel kit. Per estrarre il prodotto ricostituito, applicare il set filtro su una siringa monouso fornita e inserirla attraverso il tappo di gomma (fig. E).
- Staccare momentaneamente la siringa dal set filtro. L'aria entrerà nel flaconcino della polvere e qualsiasi schiuma scomparirà. Successivamente, aspirare la soluzione nella siringa attraverso il set filtro (fig. F).
- Staccare la siringa dal set filtro e somministrare lentamente la soluzione per via endovenosa (velocità massima di somministrazione: 2 ml al minuto) utilizzando il set per infusione con ali fornito (oppure un ago monouso fornito).
Qualsiasi medicinale non utilizzato o rifiuti derivanti dal suo utilizzo devono essere smaltiti in conformità con i requisiti locali.
Bambini
Il medicinale deve essere usato con cautela nei bambini di età inferiore a 6 anni che hanno avuto un'esposizione limitata ai preparati di fattore VIII, poiché i dati clinici in questo gruppo di pazienti sono limitati. Tuttavia, sulla base di casi clinici riportati, si può concludere che l'uso è efficace e sicuro.
Sovradosaggio
Non sono stati riportati sintomi associati al sovradosaggio di fattore VIII della coagulazione umana.
Generalmente, esiste il rischio di sviluppare fenomeni tromboembolici. Nei pazienti con gruppo sanguigno A, B o AB esiste il rischio di emolisi (vedere anche il paragrafo «Speciali avvertenze e precauzioni per l'uso»).
Effetti indesiderati.
Gli effetti indesiderati riportati di seguito sono stati osservati durante studi clinici e segnalati nel periodo post-registrazione dopo l’ottenimento dell’autorizzazione all’immissione in commercio del medicinale Imunat. La frequenza è indicata secondo la seguente scala: molto comune (≥ 10 %); comune (≥ 1 % – < 10 %); non comune (≥ 0,1 % – < 1 %); raro (≥ 0,01 % – < 0,1 %); molto raro (< 0,01 %).
Studi clinici
La frequenza di tutti gli effetti indesiderati elencati di seguito, riportati negli studi clinici, è stata «non comune» (≥ 0,1 % – < 1 %).
Malattie del sistema immunitario
Non comune: reazioni allergiche.
Segnalazioni spontanee dopo l’autorizzazione all’immissione in commercio
La frequenza di tutti gli effetti indesiderati elencati di seguito è stata «molto rara» (< 0,01 %).
Malattie del sangue e del sistema linfatico
Coagulopatia, inibizione del fattore VIII.
Disturbi psichiatrici
Ansia.
Malattie cardiache
Palpitazioni, tachicardia.
Disturbi gastrointestinali
Vomito, nausea.
Disturbi generali e condizioni in sede di somministrazione
Dolore toracico, disagio toracico, edema (in particolare edema periferico e edema facciale), irritazione in sede di iniezione (in particolare sensazione di bruciore), brividi, dolore, piressia.
Malattie del sistema immunitario
Ipersensibilità.
Malattie del sistema nervoso
Cefalea, capogiri, parestesie.
Malattie dell’apparato respiratorio, del torace e della pleura
Tosse, dispnea.
Malattie vascolari
Ipotensione arteriosa, emorragia, pallore.
Malattie dell’occhio
Congiuntivite, edema delle palpebre.
Malattie della cute e del tessuto sottocutaneo
Eritema, esantema, neurodermite, prurito, eruzione cutanea, eruzione eritematosa, eruzione cutanea generalizzata, orticaria, iperidrosi.
Malattie del sistema muscoloscheletrico e del tessuto connettivo
Mialgia.
Non sono ancora state segnalate le seguenti reazioni avverse, ma potrebbero verificarsi con l’uso del medicinale Imunat.
Malattie del sangue e del sistema linfatico
Emolisi nei pazienti con gruppo sanguigno A, B o AB.
Disturbi generali e condizioni in sede di somministrazione
Diminuzione della risposta terapeutica.
Descrizione di singoli effetti indesiderati
Reazioni di ipersensibilità o reazioni allergiche (che possono includere angioedema, sensazione di bruciore e formicolio in sede di infusione, brividi, eritema, orticaria generalizzata, cefalea, eruzione cutanea, ipotensione arteriosa, letargia, nausea, ansia, tachicardia, senso di costrizione toracica, formicolio, vomito, sibili respiratori) sono state osservate raramente e in alcuni casi possono progredire verso una grave anafilassi (incluso shock). Ai pazienti si raccomanda di rivolgersi immediatamente al medico in caso di comparsa di tali sintomi.
In rari casi è stata osservata piressia.
Nei pazienti affetti da emofilia A possono svilupparsi anticorpi neutralizzanti (inibitori) contro il fattore VIII. Se tali inibitori si sviluppano, la condizione si manifesta come mancata risposta clinica adeguata. In tali casi si raccomanda di rivolgersi a un centro specializzato per il trattamento dell’emofilia.
L’emolisi può verificarsi dopo la somministrazione di alte dosi (ad esempio quando è necessario raggiungere un livello di fattore VIII nel plasma superiore al 100 %) nei pazienti con gruppo sanguigno A, B o AB.
Per informazioni sulla sicurezza virale, si rimanda alla sezione «Particolari avvertenze e precauzioni per l’uso».
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse dopo l’immissione in commercio del medicinale è di fondamentale importanza. Permette di monitorare il rapporto beneficio/rischio del medicinale. Il personale medico e farmaceutico, nonché i pazienti o i loro rappresentanti legali, devono segnalare qualsiasi caso sospetto di reazione avversa e di mancata efficacia del medicinale attraverso il sistema informatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della validità
Medicinale: 2 anni.
Solvente (acqua per preparazioni iniettabili): 5 anni.
Condizioni di conservazione.
Conservare a una temperatura compresa tra 2 e 8 °C. Non congelare! Conservare nell’imballaggio originale per proteggere dal contatto con la luce. Tenere fuori dalla portata dei bambini. Entro il periodo di validità indicato, il medicinale può essere conservato a temperatura ambiente (non superiore a 25 °C) per un massimo di 6 mesi.
Dopo ricostituzione, il medicinale può essere conservato a temperatura ambiente (non superiore a 25 °C) per un massimo di 6 ore.
Incompatibilità
Imunat non deve essere miscelato con altri medicinali, poiché ciò potrebbe influire negativamente sulla sicurezza ed efficacia. Si devono utilizzare esclusivamente i set per infusione forniti, poiché l’adsorbimento del fattore di coagulazione VIII umano sulla superficie interna di alcuni dispositivi per infusione può portare a un’inefficacia del trattamento.
Confezione.
1 flacone contenente polvere (250/190 UI, 500/375 UI o 1000/750 UI) in combinazione con 1 flacone contenente solvente (acqua per preparazioni iniettabili, 5 ml o 10 ml) e un set per ricostituzione e somministrazione (1 dispositivo per il trasferimento/filtrazione, 1 siringa monouso (5 ml o 10 ml), 1 ago monouso, 1 sistema per infusione) in una confezione.
Categoria di prescrivibilità.
Sotto prescrizione medica.
Produttore.
Takeda Manufacturing Austria AG, Austria.
Sede del produttore e indirizzo del luogo di esercizio dell’attività.
Industriestrasse 67, 1221 Vienna, Austria.