Gliaf
UkrainaSpis treści
- INSTRUKCJA dotycząca stosowania leku Gliaf (Gliaf)
- Skład:
- Właściwości farmakodynamiczne
- Wstępnie określone jako jeden z dwóch kryteriów zatrzymania w zaplanowanej analizie pośredniej.
- Charakterystyki kliniczne
- Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
- Sposób stosowania i dawki
- Reakcje niepożądane
INSTRUKCJA dotycząca stosowania leku Gliaf (Gliaf)
Skład:
substancja czynna: empagliflozyna;
1 tabletka zawiera empagliflozyny 10 mg lub 25 mg;
substancje pomocnicze: laktoza monohydrat; celuloza mikrokryształowa (typ PH 101), hydroksypropyloceluloza, sodowa sól kroskarboksymetlocelulozy, celuloza mikrokryształowa (typ PH 102), dwutlenek krzemu koloidalny bezwodny, stearynian magnezu;
powłoka filmowa tabletek 10 mg: Opadry Yellow 03B220113 - hipromeloza (hydroksypropylo-metyloceluloza) (typ 2910), talk, polietylenoglikol (typ 400), dwutlenek tytanu (E 171), tlenek żelaza żółty (E 172);
powłoka filmowa tabletek 25 mg: Opadry Yellow 03B220114 - hipromeloza (hydroksypropylo-metyloceluloza) (typ 2910), talk, polietylenoglikol (typ 400), dwutlenek tytanu (E 171), tlenek żelaza żółty (E 172).
Postać leku. Tabletki powlekane.
Główne właściwości fizykochemiczne:
tabletki 10 mg: bladożółte, okrągłe (o średnicy około 9,1 mm), dwuwypukłe tabletki powlekane, z oznaczeniem tłoczonym „10” po jednej stronie;
tabletki 25 mg: bladożółte, owalne (około 11,1 mm × 5,6 mm), dwuwypukłe tabletki powlekane, z oznaczeniem tłoczonym „25” po jednej stronie.
Grupa farmakoterapeutyczna
Leki stosowane w cukrzycy, inhibitory współprzenoszenia glukozy typu 2 zależnego od sodu (iSGLT2). Kod ATC A10B K03.
Właściwości farmakodynamiczne
Mechanizm działania
Empagliflozyna jest odwracalnym, silnie działającym (IC50 1,3 nmol) i selektywnym inhibitorem konkurującym transportera glukozy zależnego od sodu 2 (SGLT2). Empagliflozyna nie hamuje innych transporterów glukozy, które odgrywają istotną rolę w dostarczaniu glukozy do tkanek obwodowych, a jej selektywność wobec SGLT2 jest 5000 razy wyższa niż wobec SGLT1, głównego transportera odpowiedzialnego za wchłanianie glukozy w jelitach. SGLT2 jest silnie ekspresyjny w nerkach, podczas gdy ekspresja w innych tkankach jest nieobecna lub bardzo niska. Jako główny transporter odpowiada za resorpcję glukozy z światła kanalików z powrotem do krwiobiegu. U pacjentów z cukrzycą typu 2 i hiperglikemią filtruje się i resorbuje większa ilość glukozy.
Empagliflozyna poprawia kontrolę glikemiczną u pacjentów z cukrzycą typu 2 poprzez zmniejszenie resorpcji glukozy przez nerki. Ilość glukozy wydalonej przez nerki za pomocą tego mechanizmu glukuretycznego zależy od stężenia glukozy we krwi i szybkości filtracji kłębuszkowej (eGFR). Hamowanie SGLT2 u pacjentów z cukrzycą typu 2 i hiperglikemią prowadzi do zwiększonej ekspresji glukozy z moczem. Ponadto empagliflozyna zwiększa wydalenie sodu, co prowadzi do osmotycznego diurezy i zmniejszenia objętości wewnątrznaczyniowej.
U pacjentów z cukrzycą typu 2 wydalanie glukozy zwiększało się natychmiast po pierwszej dawce empagliflozyny i utrzymywało się w ciągu 24-godzinnego okresu dawkowania. Zwiększone wydalanie glukozy z moczem utrzymywało się na końcu 4-tygodniowego okresu leczenia i średnio wynosiło około 78 g/dobę. Zwiększone wydalanie glukozy z moczem prowadziło do natychmiastowego obniżenia poziomów glukozy w osoczu u pacjentów z cukrzycą typu 2.
Empagliflozyna poprawia poziomy glukozy w osoczu zarówno na czczo, jak i po posiłku. Mechanizm działania empagliflozyny nie zależy od funkcji komórek β ani od drogi działania insuliny, co sprzyja zmniejszeniu ryzyka hipoglikemii. Zaobserwowano poprawę markerów funkcji komórek β, w tym wskaźnika homeostatycznego modelu oceny funkcji komórek β (HOMA-β). Ponadto wydalanie glukozy z moczem powoduje utratę kalorii, związaną ze zmniejszeniem tkanki tłuszczowej i masy ciała. Obserwowana glukozuria podczas stosowania empagliflozyny towarzyszy diurezie, która może sprzyjać długotrwałemu i umiarkowanemu obniżeniu ciśnienia tętniczego.
Empagliflozyna zmniejsza również resorpcję sodu i zwiększa dostarczanie sodu do kanalików odstających. Może to wpływać na kilka funkcji fizjologicznych, w tym zwiększenie sprzężenia zwrotnego kanalikowo-kłębuszkowego i obniżenie ciśnienia wewnątrzkłębuszkowego, zmniejszenie obciążenia wstępno- i postprzyładowego serca oraz zapobieganie aktywności współczulnej, a także zmniejszenie obciążenia ściany lewej komory, o czym świadczą niższe wartości NT-proBNP, co może mieć korzystny wpływ na remodelowanie serca, ciśnienie wypełnienia i funkcję rozkurczową, a także zachowanie struktury i funkcji nerek. Inne efekty, takie jak zwiększenie hematokrytu, zmniejszenie masy ciała i ciśnienia tętniczego, mogą dodatkowo sprzyjać korzyściom dla serca i nerek.
Skuteczność kliniczna i bezpieczeństwo
Cukrzyca typu 2
Poprawa kontroli glikemicznej oraz zmniejszenie ryzyka chorób sercowo-naczyniowych i śmiertelności są integralną częścią leczenia cukrzycy typu 2.
Leczenie empagliflozyną, zarówno samodzielnie, jak i w połączeniu z metformyną, pioglitazonem, sulfonylotkami, inhibitorami DPP-4 i insuliną, prowadziło do klinicznie istotnej poprawy poziomów HbA1c, glukozy w osoczu na czczo, masy ciała, ciśnienia tętniczego skurczowego i rozkurczowego. Stosowanie empagliflozyny w dawce 25 mg zwiększyło odsetek pacjentów osiągających docelowy poziom HbA1c poniżej 7% i zmniejszyło liczbę pacjentów wymagających terapii hipoglikemizującej w porównaniu z dawką 10 mg i placebo. Im wyższy był poziom HbA1c na początku leczenia, tym większe było jego obniżenie pod wpływem leku.
Ponadto empagliflozyna jako dodatek do standardowej terapii zmniejsza ryzyko śmierci sercowo-naczyniowej i wystąpienia chorób sercowo-naczyniowych u pacjentów z cukrzycą typu 2.
Prognostyczne skutki sercowo-naczyniowe
Podwójne ślepe, placebo-kontrolowane badanie EMPA-REG OUTCOME porównywało skuteczność empagliflozyny w dawkach 10 mg i 25 mg oraz placebo jako dodatek do standardowej terapii u pacjentów z cukrzycą typu 2 i potwierdzonymi chorobami sercowo-naczyniowymi.
Empagliflozyna była lepsza od placebo w zapobieganiu śmierci z przyczyn sercowo-naczyniowych, niezgonowego zawale serca lub niezgonowego udaru mózgu. Efekt wynikał z istotnego zmniejszenia ryzyka śmierci z przyczyn sercowo-naczyniowych bez istotnych zmian dotyczących niezgonowego zawale serca lub niezgonowego udaru mózgu. Obniżenie śmiertelności z przyczyn sercowo-naczyniowych było porównywalne przy stosowaniu empagliflozyny w dawkach 10 mg i 25 mg (patrz rys. 1–4 poniżej) i potwierdzono poprawę ogólnej przeżywalności (patrz tabela 1).
Wpływ empagliflozyny na podstawową złożoną punkt końcowy, tj. śmierć z przyczyn sercowo-naczyniowych, niezgonowy zawał serca lub niezgonowy udar mózgu, w znacznym stopniu nie zależał od kontroli glikemicznej ani od funkcji nerek, która w badaniu EMPA-REG OUTCOME u wszystkich grup pacjentów charakteryzowała się wartością eGFR ≥30 ml/min/1,73 m².
Skuteczność w zapobieganiu śmierci sercowo-naczyniowej nie została ostatecznie potwierdzona u pacjentów stosujących empagliflozynę jednocześnie z inhibitorami DPP-4 oraz u pacjentów nierasy czarnej, ponieważ reprezentacja tych grup w badaniu EMPA-REG OUTCOME była ograniczona.
Tabela 1
Efekt leczenia według głównych kryteriów oceny, ich składowych i poziomu śmiertelności a
| Wskaźnik skuteczności |
Placebo N = 2333 |
Empagliflozynb N = 4687 |
| Czas do wystąpienia pierwszego przypadku śmiertelnego wyniku z powodu chorób układu sercowo-naczyniowego, nieuległego zawału mięśnia sercowego lub nieuległego udaru mózgu, N (%) |
282 (12,1) |
490 (10,5) |
| Stosunek ryzyka w porównaniu z placebo (95,02 % przedział ufności (PU))* |
0,86 (0,74; 0,99) |
|
| Wartość p dla korzyści |
0,0382 |
|
| Śmiertelność z powodu chorób układu sercowo-naczyniowego, N (%) |
137 (5,9) |
172 (3,7) |
| Stosunek ryzyka w porównaniu z placebo (95 % PU) |
0,62 (0,49; 0,77) |
|
| Wartość p |
< 0,0001 |
|
| Nieuległy zawał mięśnia sercowego, N (%) |
121 (5,2) |
213 (4,5) |
| Stosunek ryzyka w porównaniu z placebo (95 % PU) |
0,87 (0,70; 1,09) |
|
| Wartość p |
0,2189 |
|
| Nieuległy udar mózgu, N (%) |
60 (2,6) |
150 (3,2) |
| Stosunek ryzyka w porównaniu z placebo (95 % PU) |
1,24 (0,92; 1,67) |
|
| Wartość p |
0,1638 |
|
| Całkowita śmiertelność, N (%) |
194 (8,3) |
269 (5,7) |
| Stosunek ryzyka w porównaniu z placebo (95 % PU) |
0,68 (0,57; 0,82) |
|
| Wartość p |
< 0,0001 |
|
| Śmiertelność niezwiązana z chorobami układu sercowo-naczyniowego, N (%) |
57 (2,4) |
97 (2,1) |
| Stosunek ryzyka w porównaniu z placebo (95 % PU) |
0,84 (0,60; 1,16) |
a Dane uzyskane u pacjentów leczonych (tj. pacjentów, którzy otrzymali co najmniej jedną dawkę badanego leku).
b Połączone dawki empagliflozyny 10 mg i 25 mg.
* Ponieważ wyniki badania zostały uwzględnione w analizie pośredniej, zastosowano dwustronny przedział ufności 95,02 %, co odpowiada wartościom p < 0,0498 dla istotności.
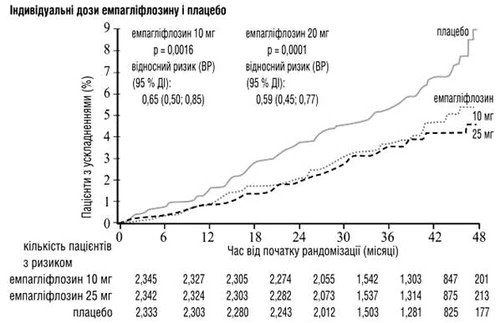
Rys. 1. Czas do wystąpienia śmierci z przyczyn sercowo-naczyniowych w badaniu EMPA-REG OUTCOME
Niewydolność serca wymagająca hospitalizacji
W trakcie badania EMPA-REG OUTCOME empagliflozyna zmniejszała ryzyko rozwoju niewydolności serca wymagającej hospitalizacji w porównaniu z placebo (grupa empagliflozyny – 2,7 %; grupa placebo – 4,1 %; HR 0,65; 95 % CI 0,50; 0,85).
Neuropatia
W trakcie badania EMPA-REG OUTCOME, czas do pierwszego epizodu neuropatii wynosił HR 0,61 (95 % CI 0,53; 0,70) w grupie empagliflozyny (12,7 %) w porównaniu z grupą placebo (18,8 %).
Dodatkowo, empagliflozyna zwiększała ryzyko (HR 1,82; 95 % CI 1,40; 2,37) trwałej normo- lub mikroalbuminurii (49,7 %) u pacjentów z makroalbuminurią na początku badania w porównaniu z placebo (28,8 %).
Niewydolność serca
Stosowanie empagliflozyny u pacjentów z niewydolnością serca i zmniejszoną frakcją wyrzutową
Przeprowadzono randomizowane, podwójne ślepe, kontrolowane placebo badanie (EMPEROR-reduced) z udziałem 3730 pacjentów z przewlekłą niewydolnością serca (klasy II–IV wg klasyfikacji Nowojorskiej Asocjacji Kardiologicznej (NYHA)) oraz zmniejszoną frakcją wyrzutową lewej komory (LVEF ≤ 40 %) w celu oceny skuteczności i bezpieczeństwa stosowania empagliflozyny w dawce 10 mg raz dziennie jako dodatek do standardowej terapii niewydolności serca. Pierwotnym punktem końcowym był czas do pierwszego potwierdzonego epizodu śmierci sercowo-naczyniowej (CV) lub hospitalizacji z powodu niewydolności serca. W analizie potwierdzającej uwzględniono również nachylenie zmiany potwierdzonych hospitalizacji (pierwszych i powtarzalnych) oraz eGFR (CKD-EPI) w odniesieniu do wartości wyjściowych. Leczenie niewydolności serca na początku badania obejmowało stosowanie inhibitorów ACE / blokerów receptora angiotensyny II / inhibitorów receptora angiotensyny-neprylizyny (88,3 %), blokerów β (94,7 %), antagonistów receptora mineralokortykoidowego (71,3 %) oraz diuretyków (95,0 %).
1863 pacjentów zostało randomizowanych do grupy empagliflozyny 10 mg (placebo – 1867). Mediana czasu trwania leczenia wyniosła 15,7 miesiąca. 76,1 % populacji badawczej stanowili mężczyźni, a 23,9 % – kobiety, których średni wiek wynosił 66,8 roku (zakres 25–94 lata); 26,8 % pacjentów miało powyżej 75 lat. 70,5 % populacji badawczej stanowili pacjenci rasy europejskiej, 18,0 % – azjatyckiej oraz 6,9 % – czarnoskórej / Afroamerykanie. W momencie randomizacji 75,1 % pacjentów miało niewydolność serca klasy II wg klasyfikacji NYHA, 24,4 % – klasy III wg klasyfikacji NYHA oraz 0,5 % – klasy IV wg klasyfikacji NYHA. Średnia wartość LVEF wyniosła 27,5 %. Na początku badania średnie eGFR wynosiło 62,0 ml/min/1,73 m², a średnie stężenie albumin/creatinina w moczu (UACR) – 22 mg/g. U około połowy pacjentów (51,7 %) eGFR wynosiło ≥ 60 ml/min/1,73 m², u 24,1 % – od 45 do < 60 ml/min/1,73 m², u 18,6 % – od 30 do < 45 ml/min/1,73 m² oraz u 5,3 % – od 20 do < 30 ml/min/1,73 m².
Empagliflozyna wykazała największą skuteczność w redukcji ryzyka pierwotnego złożonego punktu końcowego – śmierci CV lub hospitalizacji z powodu niewydolności serca w porównaniu z placebo. Ponadto, empagliflozyna istotnie zmniejszała ryzyko hospitalizacji z powodu niewydolności serca (pierwszych i powtarzalnych), a także istotnie spowalniała tempo spadku eGFR (tabela 2).
Tabela 2
Efekt leczenia w odniesieniu do pierwotnego złożonego punktu końcowego, jego składowych oraz dwóch kluczowych wtórnych punktów końcowych, włączonych do wcześniej zdefiniowanego testu potwierdzającego
| Wskaźnik |
Placebo |
Gliaf 10 mg |
| N |
1867 |
1863 |
| Czas do potwierdzonego pierwszego epizodu zgonu z przyczyn sercowo-naczyniowych lub hospitalizacji z powodu niewydolności serca, N (%) |
462 (24,7) |
361 (19,4) |
| Stosunek ryzyka w porównaniu z placebo (95 % CI)* |
0,75 (0,65, 0,86) |
|
| Wartość p dla największej skuteczności |
< 0,0001 |
|
| Zgon z przyczyn sercowo-naczyniowych, N (%) * |
202 (10,8) |
187 (10,0) |
| Stosunek ryzyka w porównaniu z placebo (95 % CI) |
0,92 (0,75, 1,12) |
|
| Hospitalizacja z powodu niewydolności serca (pierwszy epizod), N (%) |
342 (18,3) |
246 (13,2) |
| Stosunek ryzyka w porównaniu z placebo (95 % CI) |
0,69 (0,59, 0,81) |
|
| Hospitalizacja z powodu niewydolności serca (pierwsza i powtarzalna), liczba zdarzeń |
553 |
388 |
| Stosunek ryzyka w porównaniu z placebo (95 % CI)* |
0,70 (0,58, 0,85) |
|
| Wartość p |
0,0003 |
|
| Pochylenie zmiany eGFR (wg formuły CKD-EPIcr), szybkość spadku (ml/min/1,73 m²/rok) |
-2,28 |
-0,55 |
| Różnica między metodami leczenia w porównaniu z placebo (95 % CI) |
1,73 (1,10, 2,37) |
|
| Wartość p |
p < 0,0001 |
SS – układ sercowo-naczyniowy, SN – niewydolność serca, rZFK – rozszerzone klirensu kłębuszkowego, XXN EPI – epidemiologiczne wygładzenie przy przewlekłej chorobie nerek
* Śmiertelność z powodu choroby sercowo-naczyniowej oraz hospitalizacja z powodu niewydolności serca były ustalane przez niezależny komitet zdarzeń klinicznych i monitorowane na podstawie zestawu randomizacyjnego.
** Rozszerzone klirensu kłębuszkowego (rZFK) analizowano na podstawie zestawu analitycznego. Spadek wynosił -0,95 ml/min/1,73 m² przy podawaniu placebo oraz -3,02 ml/min/1,73 m² przy podawaniu empagliflozyny. Spadek odzwierciedla ostry efekt na rZFK, natomiast nachylenie odzwierciedla efekt długoterminowy.
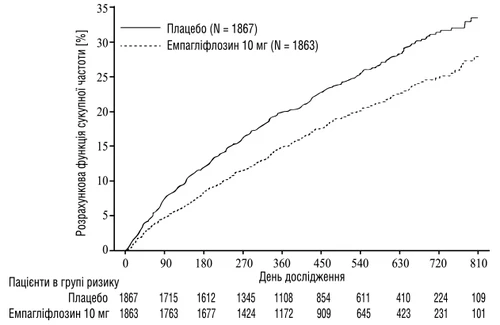
Rys. 2. Czas do pierwszego potwierdzonego zdarzenia sercowo-naczyniowego lub hospitalizacji z powodu niewydolności serca
Wyniki dotyczące pierwotnego punktu końcowego łączonego w ogólnym rozrachunku odpowiadały stosunkowi ryzyka (HR) poniżej 1 we wszystkich wcześniej określonych podgrupach, w tym u pacjentów z niewydolnością serca z cukrzycą typu 2 lub bez niej oraz z zaburzeniem funkcji nerek lub bez niej (nie poniżej rozszerzonego klirensu kłębuszkowego (rZFK) 20 ml/min/1,73 m²).
Stosowanie empagliflozyny u pacjentów z niewydolnością serca i zachowaną frakcją wyrzutu
Zrandomizowane, podwójnie ślepe, kontrolowane placebo badanie (EMPEROR-Preserved) przeprowadzono wśród 5 988 pacjentów z przewlekłą niewydolnością serca (NYHA II–IV) i zachowaną frakcją wyrzutu (LVEF > 40 %) w celu oceny skuteczności i bezpieczeństwa stosowania empagliflozyny w dawce 10 mg raz dziennie jako uzupełnienia standardowej terapii. Pierwotnym punktem końcowym był czas do pierwszego przypadku śmierci z powodu choroby sercowo-naczyniowej lub potwierdzonej hospitalizacji z powodu niewydolności serca (SN). Potwierdzona hospitalizacja z powodu SN (pierwsza i ponowna) oraz nachylenie zmiany rZFK (CKD-EPI) od poziomu wyjściowego zostały uwzględnione w testowaniu potwierdzającym. Standardowa terapia obejmowała inhibitory ACE / blokery receptora angiotensyny / inhibitor receptora angiotensyny/neprylizyny (80,7 %), beta-blokery (86,3 %), antagonistów receptora mineralokortykoidowego (37,5 %) oraz diuretyki (86,2 %).
Empagliflozyna wykazała istotną skuteczność w zmniejszaniu ryzyka pierwotnego punktu końcowego – śmierci z powodu choroby sercowo-naczyniowej lub hospitalizacji z powodu niewydolności serca w porównaniu z placebo. Ponadto empagliflozyna istotnie zmniejszała ryzyko hospitalizacji z powodu SN (pierwszej i ponownej) oraz istotnie spowalniała tempo spadku rZFK.
Tabela 3
Efekt leczenia na pierwotny punkt końcowy łączony, jego składowe oraz dwa kluczowe wtórne punkty końcowe, włączone do wcześniej określonego potwierdzającego badania
| Wskaźnik |
Placebo |
Empagliflozyna, 10 mg |
| N |
2991 |
2997 |
| Czas do potwierdzonego pierwszego epizodu zgonu z przyczyn sercowo-naczyniowych lub hospitalizacji z powodu niewydolności serca, N (%) |
511 (17,1) |
415 (13,8) |
| Stosunek ryzyka w porównaniu z takim przy stosowaniu placebo (95 % CI)* |
0,79 (0,69, 0,90) |
|
| Wartość p dla największej skuteczności |
0,0003 |
|
| Zgon z przyczyn sercowo-naczyniowych, N (%) * |
244 (8,2) |
219 (7,3) |
| Stosunek ryzyka w porównaniu z takim przy stosowaniu placebo (95 % CI) |
0,91 (0,76, 1,09) |
|
| Hospitalizacja z powodu niewydolności serca (pierwszy epizod), N (%) |
352 (11,8) |
259 (8,6) |
| Stosunek ryzyka w porównaniu z takim przy stosowaniu placebo (95 % CI) |
0,71 (0,60, 0,83) |
|
| Hospitalizacja z powodu niewydolności serca (pierwsza i ponowna), liczba zdarzeń |
541 |
407 |
| Stosunek ryzyka w porównaniu z takim przy stosowaniu placebo (95 % CI)* |
0,73 (0,61, 0,88) |
|
| Wartość p |
0,0009 |
|
| Szereg zmiany eGFR (krw EPI), tempo spadku (ml/min/1,73 m2/rok) |
-2,62 |
-1,25 |
| Różnica między metodami leczenia w porównaniu z placebo (95 % CI) |
1,36 (1,06, 1,66) |
|
| Wartość p |
p < 0,0001 |
SS – choroba sercowo-naczyniowa, CHN – niewydolność serca, eGFR – oszacowane tempo filtracji kłębuszkowej, CKD EPI – epidemiologiczne ujednolicenie przewlekłej choroby nerek.
* Śmierć z powodu choroby sercowo-naczyniowej oraz hospitalizacja z powodu niewydolności serca były oceniane przez niezależny komitet zdarzeń klinicznych i monitorowane na podstawie zestawu randomizacyjnego.
** Oszacowane tempo filtracji kłębuszkowej analizowano na podstawie zestawu analitycznego. Opóźnienie wynosiło -0,95 ml/min/1,73 m² przy stosowaniu placebo oraz -3,02 ml/min/1,73 m² przy stosowaniu empagliflozyny. Opóźnienie oznacza ostry efekt na oszacowane tempo filtracji kłębuszkowej, podczas gdy nachylenie oznacza efekt długoterminowy.
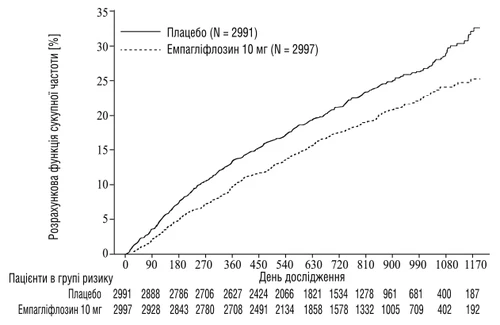
Rys. 3 Czas do pierwszego potwierdzonego zdarzenia śmierci sercowo-naczyniowej lub hospitalizacji z powodu niewydolności serca
Wyniki pierwotnego punktu końcowego złożonego były zgodne w każdej z wcześniej określonych podgrup, sklasyfikowanych np. według EFWS, statusu cukrzycy lub funkcji nerek (do 20 ml/min/1,73 m²).
Przewlekła choroba nerek
Przeprowadzono randomizowane, podwójne ślepe, placebo-kontrolowane badanie stosowania empagliflozyny w dawce 10 mg raz dziennie (EMPA-KIDNEY) jako dodatek do standardowej terapii u 6609 pacjentów z przewlekłą chorobą nerek (eGFR ≥ 20 – < 45 ml/min/1,73 m² lub eGFR ≥ 45 – < 90 ml/min/1,73 m² przy stosunku albumin do kreatyniny w moczu (UACR) ≥ 200 mg/g) w celu oceny wyników sercowo-nerekowych. Pierwotnym punktem końcowym był czas do pierwszego przypadku postępu choroby nerek (trwałe zmniejszenie ≥ 40 % eGFR od momentu randomizacji, trwałe eGFR < 10 ml/min/1,73 m², nerek w stadium zaawansowanym lub śmierć z powodu choroby nerek) lub śmierci z powodu choroby sercowo-naczyniowej. Pierwszy przypadek hospitalizacji z powodu niewydolności serca lub śmierci z powodu choroby sercowo-naczyniowej, hospitalizacja z dowolnego powodu (pierwsza i powtarzana) oraz śmierć z dowolnego powodu były uwzględnione w testowaniu potwierdzającym.
Leczenie na początku badania obejmowało stosowanie inhibitora UKR (85,2 % inhibitora ACE / blokera receptora angiotensyny II).
3304 pacjentów zostało zrandomizowanych do grupy empagliflozyny 10 mg (placebo – 3305). Mediana czasu trwania leczenia wynosiła 24,3 miesiąca. 66,8 % populacji badanej stanowili mężczyźni, a 33,2 % – kobiety, średnia wieku wynosiła 63,3 roku (zakres 18–94 lata), 23,0 % pacjentów miało ponad 75 lat. 58,4 % populacji badanej stanowili pacjenci rasy europejskiej, 36,2 % – rasy mongolskiej oraz 4,0 % – rasy negroidalnej (afroamerykanie).
Na początku badania średnie eGFR wynosiło 37,3 ml/min/1,73 m², u 21,2 % pacjentów eGFR wynosiło ≥ 45 ml/min/1,73 m², u 44,3 % – od 30 do < 45 ml/min/1,73 m² oraz u 34,5 % – < 30 ml/min/1,73 m², w tym 254 pacjentów z eGFR < 20 ml/min/1,73 m². Średni stosunek albumin do kreatyniny w moczu (UACR) wynosił 329 mg/g, u 20,1 % pacjentów UACR wynosiło < 30 mg/g, u 28,2 % – od 30 do ≤ 300 mg/g, u 51,7 % – > 300 mg/g oraz u 41,1 % – < 200 mg/g. Głównymi przyczynami PChN były nefropatia cukrzycowa / choroba nerek spowodowana cukrzycą (31 %), choroba kłębuszkowa (25 %), choroba nadciśnieniowa / choroba naczyniowo-nerek (22 %) oraz inne/nieznane przyczyny (22 %).
Empagliflozyna była lepsza od placebo w zmniejszaniu ryzyka pierwotnego złożonego punktu końcowego obejmującego postęp choroby nerek lub śmierć z powodu choroby sercowo-naczyniowej (patrz tabela 4). Ponadto empagliflozyna istotnie zmniejszała ryzyko hospitalizacji z dowolnego powodu (pierwszej i powtarzanej).
Tabela 4
Efekty leczenia dla pierwotnego złożonego punktu końcowego i kluczowych wtórnych punktów końcowych, włączonych do wcześniej określonego potwierdzającego badania, oraz ich składowych
| Placebo |
Empagliflozyna, 10 mg |
|
| N |
3 305 |
3 304 |
| Czas do pierwszego wystąpienia postępu choroby nerek (trwałe zmniejszenie eGFR ≥ 40 % od momentu randomizacji, trwałe eGFR < 10 ml/min/1,73 m², zaawansowana choroba nerek* (ESKD) lub zgon z powodu choroby nerek) lub zgon z powodu chorób układu sercowo-naczyniowego, N (%) |
558 (16,9) |
432 (13,1) |
| Stosunek ryzyka w porównaniu do placebo (99,83 % CI) |
0,72 (0,59; 0,89) |
|
| Wartość p dla potwierdzenia nieprzewagania skuteczności |
< 0,0001 |
|
| Trwałe zmniejszenie eGFR ≥ 40 % od momentu randomizacji, N (%) |
474 (14,3) |
359 (10,9) |
| Stosunek ryzyka w porównaniu do placebo (95 % CI) |
0,70 (0,61; 0,81) |
|
| Wartość p |
< 0,0001 |
|
| ESKD* lub trwałe eGFR < 10 ml/min/1,73 m², N (%) |
221 (6,7) |
157 (4,8) |
| Stosunek ryzyka w porównaniu do placebo (95 % CI) |
0,69 (0,56; 0,84) |
|
| Wartość p |
0,0003 |
|
| Zgon z powodu choroby nerek, N (%)** |
4 (0,1) |
4 (0,1) |
| Stosunek ryzyka w porównaniu do placebo (95 % CI) |
||
| Wartość p |
||
| Zgon z powodu chorób układu sercowo-naczyniowego, N (%) |
69 (2,1) |
59 (1,8) |
| Stosunek ryzyka w porównaniu do placebo (95 % CI) |
0,84 (0,60; 1,19) |
|
| Wartość p |
0,3366 |
|
| ESKD* lub zgon z powodu chorób układu sercowo-naczyniowego, N (%)# |
217 (6,6) |
163 (4,9) |
| Stosunek ryzyka w porównaniu do placebo (95 % CI) |
0,73 (0,59; 0,89) |
|
| Wartość p |
0,0023 |
|
| Częstość hospitalizacji ze wszystkich przyczyn (pierwsze i powtarzające się), N zdarzeń |
1895 |
1611 |
| Stosunek ryzyka w porównaniu do placebo (99,03 % CI) |
0,86 (0,75; 0,98) |
|
| Wartość p |
0,0025 |
SS – układ sercowo-naczyniowy, eGFR – oszacowana szybkość filtracji kłębuszkowej.
* Ostateczny etap choroby nerek (OEN) definiuje się jako początek dializy wspomagającej lub przeszczepienie nerki.
** Zanotowano zbyt mało przypadków zgonów z powodu choroby nerek, aby obliczyć odpowiednie współczynniki ryzyka.
Wstępnie określone jako jeden z dwóch kryteriów zatrzymania w zaplanowanej analizie pośredniej.
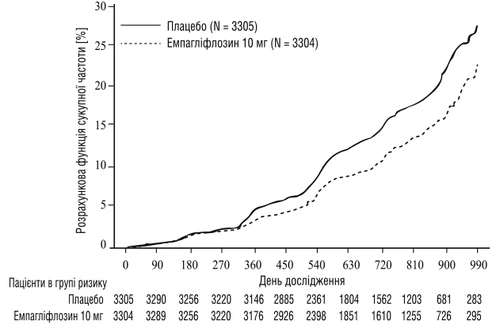
Rys. 4. Czas do pierwszego epizodu postępu choroby nerek lub przewidywanej śmierci z przyczyn SS, oszacowana funkcja skumulowanego zachorowania
Wyniki pierwotnego punktu końcowego złożonego były zgodne w każdej z wstępnie określonych podgrup, sklasyfikowanych według eGFR, głównej przyczyny choroby nerek, statusu cukrzycy lub stosowania inhibitorów układu renina-angiotensyna (RAS). Korzyści z leczenia były bardziej widoczne u pacjentów z wysokim poziomem albuminurii.
Podczas leczenia eGFR zmniejszała się wolniej w grupie leczonej empagliflozyną w porównaniu z grupą placebo. Empagliflozyna spowolniła roczną szybkość spadku eGFR o 1,37 ml/min/1,73 m²/rok (95 % przedział ufności 1,16, 1,59), na podstawie wyników analizy wcześniejszej wszystkich pomiarów eGFR wykonanych od wizyty po 2 miesiącach do końcowej wizyty kontrolnej. U pacjentów leczonych empagliflozyną zaobserwowano początkowy spadek wartości eGFR, który powrócił do poziomu wyjściowego po zakończeniu leczenia, co zostało wykazane w kilku badaniach empagliflozyny, potwierdzając, że zmiany hemodynamiczne odgrywają rolę w ostrym wpływie empagliflozyny na eGFR.
Dzieci
Cukrzyca typu 2
Skuteczność kliniczną i bezpieczeństwo stosowania empagliflozyny (10 mg z możliwością zwiększenia dawki do 25 mg) oraz linagliptyny (5 mg) raz dziennie badano u dzieci i nastolatków w wieku od 10 do 17 lat z cukrzycą typu 2 w badaniu kontrolowanym placebo (DINAMO) trwającym 26 tygodni, z przedłużonym okresem bezpieczeństwa do 52 tygodni.
Podstawowe leczenie jako uzupełnienie diety i aktywności fizycznej obejmowało metforminę (51 %), kombinację metforminy i insuliny (40,1 %), insulinę (3,2 %) lub nie obejmowało żadnego z nich (5,7 %).
Skorygowana średnia zmiana HbA1c po 26 tygodniach między empagliflozyną (N=52) a placebo (N=53), wynosząca -0,84 %, była klinicznie i statystycznie istotna (95 % przedział ufności -1,50, -0,19; p = 0,0116).
Dodatkowo, leczenie empagliflozyną w porównaniu z placebo doprowadziło do klinicznie istotnej skorygowanej średniej zmiany poziomu glukozy we krwi na czczo o -35,2 mg/dl (95 % przedział ufności -58,6, -11,7) (-1,95 mmol/l (-3,25, -0,65)).
Niewydolność serca i przewlekła choroba nerek
Europejska Agencja Leków odmówiła obowiązku przedstawienia wyników badań leku referencyjnego we wszystkich podgrupach dzieci z przewlekłą chorobą nerek i niewydolnością serca (informacje dotyczące stosowania u dzieci znajdują się w sekcji „Sposób stosowania i dawki”).
Farmakokinetyka
Wchłanianie
Farmakokinetyka empagliflozyny została szczegółowo opisana u zdrowych ochotników oraz u pacjentów z cukrzycą typu 2. Po podaniu doustnym empagliflozyna była szybko wchłaniana, a maksymalne stężenie w osoczu obserwowano przy średnim czasie tmax wynoszącym 1,5 godziny po podaniu leku. Następnie stężenie w osoczu obniżało się dwufazowo, z szybką fazą rozproszenia i stosunkowo wolną fazą końcową. Średnie wartości pola pod krzywą „stężenie-czas” (AUC) oraz maksymalnego stężenia (Cmax) w osoczu w stanie stacjonarnym wynosiły 1870 nmol·h/l i 259 nmol/l po podaniu empagliflozyny w dawce 10 mg oraz 4740 nmol·h/l i 687 nmol/l po podaniu empagliflozyny w dawce 25 mg raz dziennie. Działanie systemowe empagliflozyny wzrastało proporcjonalnie do dawki. Parametry farmakokinetyczne empagliflozyny w stanie stacjonarnym po podaniu dawki pojedynczej były podobne, co wskazuje na kinetykę liniową w odniesieniu do czasu. Nie stwierdzono klinicznie istotnej różnicy w farmakokinetyce empagliflozyny między zdrowymi ochotnikami a pacjentami z cukrzycą typu 2.
Podanie 25 mg empagliflozyny po posiłku o wysokiej zawartości kalorii i tłuszczu spowodowało pewne zmniejszenie jej działania: AUC zmniejszyła się o około 16 %, a Cmax – o około 37 % – w porównaniu z podaniem na czczo. Ten wpływ posiłku na farmakokinetykę empagliflozyny nie jest uważany za klinicznie istotny. Empagliflozynę można przyjmować niezależnie od posiłku.
Rozkład
Objętość rozkładu w stanie stacjonarnym wynosi 73,8 l. Po podaniu doustnego roztworu [14C]-empagliflozyny zdrowym ochotnikom rozkład do erytrocytów wynosił około 37 %, a wiązanie z białkami osocza – 86 %.
Biotransformacja
Główne metabolity empagliflozyny w osoczu krwi człowieka nie były wykrywane. Najczęstsze metabolity to trzy koniugaty glukuronidowe (2-, 3- i 6-O-glukuronid). Ekspozycja systemowa każdego metabolitu była mniejsza niż 10 % całkowitej ekspozycji na lek. Badania in vitro wskazują, że główną drogą metabolizmu empagliflozyny u człowieka jest glukuronidacja przez uridylo-5’-difosfoglikuronosylotransferazy UGT2B7, UGT1A3, UGT1A8 oraz UGT1A9.
Eliminacja
Okres półtrwania eliminacji empagliflozyny wynosi 12,4 godziny, a pozorny klirens doustny – 10,6 l/h. Wewnątrzosobnicza i pozostała zmienność klirensu empagliflozyny doustnej wynosiła odpowiednio 39,1 % i 35,8 %. Po podawaniu raz dziennie stężenia empagliflozyny w osoczu krwi w stanie stacjonarnym osiągane były do podania 5. dawki. Zgodnie z okresem półtrwania, do 22 % akumulacji (w odniesieniu do AUC w osoczu krwi) obserwowano w stanie stacjonarnym. Po podaniu doustnego roztworu [14C]-empagliflozyny zdrowym ochotnikom około 96 % znacznika wydalało się z kałem (41 %) i moczem (54 %). Większość znacznika w niezmienionej formie wydalała się z kałem, a około połowa znacznika w niezmienionej formie – z moczem.
Specjalne kategorie pacjentów
Pacjenci z zaburzeniami czynności nerek
U pacjentów z łagodnymi, umiarkowanymi lub ciężkimi zaburzeniami czynności nerek (eGFR < 30–< 90 ml/min/1,73 m²) oraz u pacjentów z niewydolnością nerek / ostatecznym etapem choroby nerek (OEN) AUC empagliflozyny wzrosła odpowiednio o około 18 %, 20 %, 66 % i 48 % w porównaniu z podmiotami z normalną czynnością nerek. Maksymalne stężenia empagliflozyny w osoczu krwi były podobne u pacjentów z umiarkowanymi zaburzeniami czynności nerek oraz u pacjentów z niewydolnością nerek / OEN w porównaniu z podmiotami z normalną czynnością nerek. Maksymalne stężenia empagliflozyny w osoczu krwi były o około 20 % wyższe u pacjentów z łagodnymi i ciężkimi zaburzeniami czynności nerek w porównaniu z podmiotami z normalną czynnością nerek. Analiza farmakokinetyki populacyjnej wykazała, że pozorny klirens doustny empagliflozyny zmniejszał się ze spadkiem eGFR, co prowadzi do nasilenia działania leku.
Pacjenci z zaburzeniami czynności wątroby
U pacjentów z łagodnymi, umiarkowanymi i ciężkimi zaburzeniami czynności wątroby zgodnie z klasyfikacją Childa-Pugha AUC empagliflozyny wzrosła odpowiednio o około 23 %, 47 % i 75 %, a Cmax – odpowiednio o około 4 %, 23 % i 48 % w porównaniu z podmiotami z normalną czynnością wątroby.
Wskaźnik masy ciała
Wskaźnik masy ciała nie miał klinicznie istotnego wpływu na farmakokinetykę empagliflozyny. AUC była o 5,82 %, 10,4 % i 17,3 % niższa u pacjentów z BMI 30, 35 i 45 kg/m² odpowiednio w porównaniu z pacjentami z wskaźnikiem masy ciała 25 kg/m².
Płeć
Płeć nie miała klinicznie istotnego wpływu na farmakokinetykę empagliflozyny.
Rasa
AUC była o 13,5 % wyższa u pacjentów rasy mongolskiej z wskaźnikiem masy ciała 25 kg/m² w porównaniu z pacjentami innych ras z wskaźnikiem masy ciała 25 kg/m².
Pacjenci w wieku podeszłym
Wiek pacjenta nie miał klinicznie istotnego wpływu na farmakokinetykę empagliflozyny.
Dzieci
Rozpoczęto badania kliniczne stosowania empagliflozyny u dzieci w wieku 10–18 lat z cukrzycą typu 2. Dotychczas uzyskane dane farmakokinetyczne i farmakodynamiczne są porównywalne z danymi u dorosłych.
Badanie pediatryczne fazy 3 badało farmakokinetykę i farmakodynamikę (zmiana HbA1c od wartości wyjściowej) 10 mg empagliflozyny z możliwością zwiększenia dawki do 25 mg u dzieci i nastolatków w wieku od 10 do 17 lat z cukrzycą typu 2. Obserwowane zależności ekspozycja-odpowiedź były ogólnie porównywalne u dorosłych, dzieci i nastolatków. Podanie doustne empagliflozyny prowadziło do ekspozycji w zakresie obserwowanym u dorosłych pacjentów.
Obserwowane średnie geometryczne stężenia minimalne oraz średnie geometryczne stężenia po 1,5 godziny po podaniu w stanie stacjonarnym wynosiły 26,6 nmol/l i 308 nmol/l po przyjmowaniu 10 mg empagliflozyny raz dziennie oraz 67,0 nmol/l i 525 nmol/l z empagliflozyną 25 mg raz dziennie.
Charakterystyki kliniczne
Wskazania
Cukrzyca typu 2
Gliaf jest wskazany u dorosłych i dzieci w wieku od 10 lat w leczeniu nieodpowiednio kontrolowanej cukrzycy typu 2 jako uzupełnienie diety i aktywności fizycznej:
- jako monoterapia, gdy zastosowanie metformyny jest niemożliwe z powodu nietolerancji leku;
- jako dodatek do innych leków stosowanych w leczeniu cukrzycy typu 2.
Informacje dotyczące terapii skojarzonej, wpływu na kontrolę glikemii, zdarzeń sercowo-naczyniowych i nerkowych oraz badanych populacji znajdują się w sekcjach „Szczególne wytyczne dotyczące stosowania”, „Interakcje z innymi lekami i inne formy interakcji” oraz „Farmakodynamika”.
Niewydolność serca
Gliaf jest wskazany u dorosłych w leczeniu przewlekłej niewydolności serca o charakterze objawowym.
Przewlekła choroba nerek
Gliaf jest wskazany u dorosłych w leczeniu przewlekłej choroby nerek.
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub dowolny składnik pomocniczy.
Interakcje z innymi lekami i inne formy interakcji
Interakcje farmakodynamiczne
Diuretyki
Empagliflozyna może nasilać działanie moczopędne diuretyków tiazydowych i pętlowych oraz zwiększać ryzyko odwodnienia i hipotensji (patrz sekcja „Szczególne wytyczne dotyczące stosowania”).
Inzulina i leki stymulujące wydzielanie insuliny
Inzulina oraz leki stymulujące wydzielanie insuliny, takie jak pochodne sulfoniliomocznika, mogą zwiększać ryzyko hipoglikemii. W celu zmniejszenia ryzyka hipoglikemii może być konieczne obniżenie dawki insuliny lub leku stymulującego wydzielanie insuliny podczas stosowania w połączeniu z empagliflozyną (patrz sekcje „Sposób dawkowania i stosowania” oraz „Działania niepożądane”).
Interakcje farmakokinetyczne
Wpływ innych leków na empagliflozynę
Dane in vitro wskazują, że główną drogą metabolizmu empagliflozyny u ludzi jest glukuronidacja przez urdyno-5'-difosfo-glukuronilotransferazy UGT1A3, UGT1A8, UGT1A9 i UGT2B7. Empagliflozyna jest substytutem transporterów wchłaniania u ludzi OAT3, OATP1B1 i OATP1B3, ale nie OAT1 i OCT2. Empagliflozyna jest substytutem glikoproteiny P (P-gp) oraz białka oporności na raka piersi (BCRP).
Stosowanie empagliflozyny razem z probenecydem, inhibitorem enzymów urydyno-difosfo-glukuronilotransferazy (uridine-diphospho-glucuronyltransferase (UGT)) i OAT3, spowodowało wzrost stężenia wierzchołkowego empagliflozyny we krwi o 26% i wzrost AUC o 53%. Zmiany te nie były uważane za klinicznie istotne.
Wpływ indukcji UGT (w tym indukcji ryfampicyną lub fenytoiną) na empagliflozynę nie był badany. Nie zaleca się jednoczesnego leczenia znanymi induktorami enzymów UGT ze względu na potencjalne ryzyko zmniejszenia skuteczności. Jeśli konieczne jest jednoczesne podanie induktora enzymów UGT, zaleca się monitorowanie kontroli glikemii w celu oceny odpowiedzi na Gliaf.
Badanie interakcji z gemfibrozylem, inhibitorem transporterów OAT3 i OATP1B1/1B3 in vitro, wykazało, że po jednoczesnym stosowaniu Cmax empagliflozyny wzrosło o 15%, a AUC wzrosło o 59%. Zmiany te nie były uważane za klinicznie istotne.
Inhibicja transporterów OATP1B1/1B3 przy jednoczesnym stosowaniu z ryfampicyną spowodowała wzrost Cmax o 75% i wzrost AUC empagliflozyny o 35%. Zmiany te nie były uważane za klinicznie istotne.
Wpływ empagliflozyny przy jednoczesnym stosowaniu z werapamylem, inhibitorem P-gp, był podobny. Wskazuje to, że inhibicja P-gp nie ma klinicznie istotnego wpływu na empagliflozynę.
Badania interakcji przeprowadzone u zdrowych ochotników wskazują, że farmakokinetyka empagliflozyny nie jest wpływowana przez jednoczesne podanie metforminy, glipekamidu, pioglitazonu, sitagliptyny, linagliptyny, warfaryny, werapamilu, ramiprylu, symwatatyny, torasemidu i hydrochlorotiazydu.
Wpływ empagliflozyny na inne leki
Empagliflozyna może zwiększać wydalanie litu przez nerki i obniżać stężenie litu we krwi. Po rozpoczęciu stosowania empagliflozyny oraz po zmianie dawki należy częściej kontrolować stężenie litu w osoczu. Pacjenta należy skierować do lekarza, który przepisał leki zawierające lit, w celu monitorowania stężenia litu w osoczu.
Dane badań in vitro wskazują, że empagliflozyna nie hamuje, nie dezaktywuje ani nie indukuje izoform CYP450. Empagliflozyna nie hamuje UGT1A1, UGT1A3, UGT1A8, UGT1A9 ani UGT2B7. Interakcje lek-lek związane z głównymi izoformami CYP450 lub UGT z empagliflozyną i współpodawanymi substancjami będącymi substratami tych enzymów są mało prawdopodobne.
Empagliflozyna nie hamuje P-gp w dawkach terapeutycznych. Dane badań in vitro wskazują, że empagliflozyna mało prawdopodobnie spowoduje interakcję z lekami będącymi substratami P-gp. Jednoczesne stosowanie derywatu, substratu P-gp, i empagliflozyny spowodowało wzrost AUC o 6% i wzrost Cmax derywatu o 14%. Zmiany te nie były uważane za klinicznie istotne.
Empagliflozyna nie hamuje transporterów wchłaniania u ludzi, takich jak OAT3, OATP1B1 i OATP1B3, in vitro w klinicznie istotnych stężeniach w osoczu, co oznacza, że interakcje lek-lek z substratami tych transporterów wchłaniania są mało prawdopodobne.
Badania interakcji przeprowadzone u zdrowych ochotników wskazują, że empagliflozyna nie wywiera klinicznie istotnego wpływu na farmakokinetykę metforminy, glipekamidu, pioglitazonu, sitagliptyny, linagliptyny, symwatatyny, warfaryny, ramiprylu, derywatu, diuretyków i doustnych środków antykoncepcyjnych.
Dzieci
Badania interakcji przeprowadzono wyłącznie u dorosłych.
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
Empagliflozyna nie powinna być stosowana u pacjentów z cukrzycą typu 1 (patrz poniżej „Kwacidoza ketonowa”).
Kwacidoza ketonowa
U pacjentów z cukrzycą stosujących inhibitory SGLT-2 (w tym empagliflozynę) zgłaszano przypadki kwacidozy ketonowej, w tym przypadki zagrażające życiu oraz śmiertelne. W wielu przypadkach kwacidoza ketonowa przebiegała atypowo, z tylko umiarkowanym wzrostem stężenia glukozy we krwi (poniżej 14 mmol/l (250 mg/dl)). Nie wiadomo, czy zwiększenie dawki empagliflozyny wpływa na ryzyko wystąpienia kwacidozy ketonowej. Choć ryzyko kwacidozy ketonowej jest mniejsze u pacjentów bez cukrzycy, przypadki te odnotowano również u takich pacjentów.
Należy brać pod uwagę ryzyko kwacidozy ketonowej w przypadku wystąpienia niestosunkowych objawów, takich jak nudności, wymioty, brak apetytu, ból brzucha, nadmierne pragnienie, trudności w oddychaniu, dezorientacja, nietypowe zmęczenie lub senność. W przypadku wystąpienia tych objawów pacjentów należy natychmiast zbadać pod kątem rozwoju kwacidozy ketonowej, niezależnie od stężenia glukozy we krwi.
Jeśli u pacjenta podejrzewa się lub stwierdza kwacidozę ketonową, należy natychmiast przerwać stosowanie empagliflozyny.
W przypadku hospitalizacji pacjenta w celu przeprowadzenia znaczących zabiegów chirurgicznych lub w przypadku wystąpienia ciężkich chorób ostrych należy przerwać leczenie empagliflozyną. Pacjentom tym zaleca się monitorowanie poziomu ciał ketonowych. Pomiar stężenia ciał ketonowych w moczu jest bardziej priorytetowy niż ich oznaczenie we krwi. Leczenie empagliflozyną można wznowić, gdy poziom ciał ketonowych wróci do normy i stan pacjenta ustabilizuje się.
Przed rozpoczęciem stosowania empagliflozyny należy przeanalizować wywiad pacjenta pod kątem czynników, które mogą wskazywać na skłonność do kwacidozy ketonowej.
Podczas stosowania empagliflozyny obserwowano trwającą kwacidozę ketonową i trwającą glukozurię.
Ze względu na okres półtrwania empagliflozyny kwacidoza ketonowa może trwać również po przerwaniu przyjmowania tego leku. Czynniki niezależne od przyjmowania empagliflozyny (takie jak niedobór insuliny) mogą wydłużać okresy kwacidozy ketonowej.
Pacjenci z wysokim ryzykiem kwacidozy ketonowej to osoby z niską funkcją komórek β (np. w przypadku cukrzycy typu 2 z niskim stężeniem peptydu C, ukrytego autoimmunologicznego cukrzycy dorosłych lub zapalenia trzustki w wywiadzie); pacjenci ze stanami prowadzącymi do ograniczenia przyjmowania pokarmu lub silnego odwodnienia; pacjenci, u których dawkę insuliny zmniejszono, oraz pacjenci z zwiększonego zapotrzebowania na insulinę z powodu choroby ostrej, zabiegu chirurgicznego lub nadużywania alkoholu. Inhibitory SGLT-2 należy stosować tym pacjentom z ostrożnością.
Wznowienie terapii inhibitorami SGLT-2 u pacjentów, u których wcześniej wystąpiła kwacidoza ketonowa podczas leczenia inhibitorami SGLD-2, nie jest zalecane, jeśli nie ustalono i nie wyeliminowano innego czynnika wywołującego kwacidozę ketonową.
Gliaf nie powinien być stosowany u pacjentów z cukrzycą typu 1. Dane z programu badań klinicznych u pacjentów z cukrzycą typu 1 wykazały zwiększoną częstość występowania kwacidozy ketonowej przy stosowaniu empagliflozyny w dawkach 10 mg i 25 mg jako leku wspomagającego insulinę w porównaniu z placebo.
Niewydolność nerek
Z uwagi na ograniczone doświadczenie stosowania nie zaleca się rozpoczynania leczenia empagliflozyną u pacjentów z eGFR < 20 ml/min/1,73 m².
U pacjentów z eGFR < 60 ml/min/1,73 m² zalecana dawka dobową empagliflozyny wynosi 10 mg (patrz sekcja „Sposób stosowania i dawki”).
Skuteczność empagliflozyny w obniżaniu poziomu glukozy zależy od funkcji nerek i zmniejsza się u pacjentów z eGFR < 45 ml/min/1,73 m² oraz prawdopodobnie brakuje jej u pacjentów z eGFR < 30 ml/min/1,73 m² (patrz sekcje „Sposób stosowania i dawki”, „Farmakodynamika” oraz „Farmakokinetyka”).
Monitorowanie funkcji nerek
Zaleca się przeprowadzanie oceny funkcji nerek w następujący sposób:
- przed rozpoczęciem stosowania empagliflozyny oraz okresowo w trakcie leczenia, co najmniej raz w roku (patrz sekcje „Sposób stosowania i dawki”, „Działania niepożądane”, „Farmakodynamika” oraz „Farmakokinetyka”);
- przed rozpoczęciem stosowania każdego leku współbieżnego, który może niekorzystnie wpływać na funkcje nerek.
Stosowanie u pacjentów z ryzykiem zmniejszenia objętości płynu międzykomórkowego
Ze względu na mechanizm działania inhibitorów SGLT-2, osmotyczne działanie moczopędne towarzyszące glukozurii może prowadzić do nieznacznego obniżenia ciśnienia tętniczego (patrz sekcja „Farmakodynamika”). Lek należy stosować z ostrożnością u pacjentów, u których obniżenie ciśnienia tętniczego spowodowane przez empagliflozynę może stanowić zagrożenie, np. u pacjentów z chorobami układu sercowo-naczyniowego, u pacjentów stosujących leki przeciwhypertensyjne i z historią hipotensji lub u pacjentów w wieku 75 lat i starszych.
W przypadku rozwoju stanów, które mogą prowadzić do utraty płynu (np. choroby przewodu pokarmowego), pacjentom przyjmującym empagliflozynę zaleca się staranne monitorowanie stopnia zmniejszenia objętości płynu międzykomórkowego (np. badanie fizykalne, pomiar ciśnienia tętniczego, badania laboratoryjne, w tym poziom hematokrytu) oraz stosowanie elektrolitów. Należy rozważyć konieczność tymczasowego przerwania leczenia empagliflozyną do czasu usunięcia utraty płynu.
Pacjenci w starszym wieku
Wpływ empagliflozyny na wydalanie glukozy z moczem wiąże się z osmotycznym działaniem moczopędnym, które może wpływać na stan nawodnienia. Pacjenci w wieku 75 lat i starszych mają zwiększone ryzyko zmniejszenia objętości płynu międzykomórkowego. Większość takich pacjentów, którzy otrzymywali empagliflozynę, doświadczyła działań niepożądanych związanych ze zmniejszeniem objętości płynu międzykomórkowego w porównaniu z pacjentami z grupy placebo (patrz sekcja „Działania niepożądane”). Dlatego konieczna jest szczególna uwaga do objętości płynu międzykomórkowego w przypadku jednoczesnego stosowania leków, które mogą prowadzić do jej zmniejszenia (np. diuretyków, inhibitorów ACE).
Powikłane infekcje dróg moczowych
Zgłaszano przypadki powikłań w postaci infekcji dróg moczowych, w tym nefrytu i urosepsę, u pacjentów przyjmujących empagliflozynę (patrz sekcja „Działania niepożądane”). Należy rozważyć konieczność tymczasowego przerwania leczenia empagliflozyną u pacjentów z powikłanymi infekcjami dróg moczowych.
Uogólnione martwicze zapalenie powięzi (gangrena Fourniera)
Zgłoszono przypadki uogólnionego martwiczego zapalenia powięzi (znane również jako gangrena Fourniera) u kobiet i mężczyzn z cukrzycą, którzy przyjmowali inhibitory SGLT-2, w tym empagliflozynę. Gangrena Fourniera to rzadkie, ale poważne i potencjalnie zagrażające życiu zakażenie, wymagające natychmiastowego leczenia chirurgicznego i podania antybiotyków.
Pacjentów należy poinformować o konieczności natychmiastowego skontaktowania się z lekarzem w przypadku wystąpienia takich objawów jak ból, uczucie bolesności, zaczerwienienie lub obrzęk w okolicy narządów płciowych lub w okolicy międzypochwowej, towarzyszone gorączką lub niedyspozycją. Należy zauważyć, że infekcja dróg moczowo-płciowych lub ropień okolicy międzypochwowej może poprzedzać martwicze zapalenie powięzi. W przypadku podejrzenia gangreny Fourniera lek Gliaf należy odstawić i szybko rozpocząć leczenie (w tym podanie antybiotyków i leczenie chirurgiczne obszaru zmienionego chorobowo).
Amputacja kończyn dolnych
W badaniu z innym inhibitorem SGLT-2 zaobserwowano zwiększoną liczbę przypadków amputacji kończyn dolnych (głównie palców stóp). Nie wiadomo, czy ten efekt jest wspólny dla całej klasy leków. Pacjentom z cukrzycą należy zalecić profilaktyczne leczenie chorób stóp.
Uszkodzenie wątroby
W trakcie badań klinicznych zgłaszano przypadki uszkodzenia wątroby podczas stosowania empagliflozyny. Związek przyczynowo-skutkowy między stosowaniem empagliflozyny a uszkodzeniem wątroby nie został ustalony.
Zwiększony hematokryt
Zwiększenie hematokrytu obserwuje się podczas leczenia empagliflozyną (patrz sekcja „Działania niepożądane”). Pacjentów z wyraźnym zwiększeniem hematokrytu należy monitorować i badać pod kątem obecności podstawowego schorzenia hematologicznego.
Przewlekła choroba nerek
Pacjenci z albuminurią mogą odnieść większą korzyść z leczenia empagliflozyną.
Choroba naciekowa lub kardiomiopatia takotsubo
Stosowanie empagliflozyny u pacjentów z chorobą naciekową lub kardiomiopatią takotsubo nie było specjalnie badane. Dlatego skuteczność u tych pacjentów nie została ustalona.
Badania laboratoryjne moczu
U pacjentów przyjmujących Gliaf test glukozy w moczu będzie dodatni ze względu na mechanizm działania leku.
Interferencja z zawartością 1,5-anhydroglukitolu (1,5-AG)
Monitorowanie kontroli glikemicznej z wykorzystaniem 1,5-AG nie jest zalecane, ponieważ oznaczenia 1,5-AG są nieprecyzyjne przy ocenie kontroli glikemicznej u pacjentów przyjmujących inhibitory SGLT-2. Zaleca się stosowanie alternatywnych metod kontroli glikemicznej.
Laktoza
Ten lek zawiera laktozę. Pacjenci z rzadką dziedziczną nietolerancją galaktozy, całkowitym niedoborem laktozy lub zespołem złego wchłaniania glukozy-galaktozy nie powinni przyjmować tego leku.
Sód
Jedna tabletka leku zawiera mniej niż 1 mmol sodu (23 mg), co oznacza, że lek ten jest niemal pozbawiony sodu.
Stosowanie w okresie ciąży lub karmienia piersią
Ciąża
Brak danych dotyczących stosowania empagliflozyny u kobiet w ciąży. Badania na zwierzętach wykazały, że empagliflozyna przenika przez łożysko w późnych stadiach ciąży w bardzo ograniczonym stopniu, ale nie wskazują na bezpośredni lub pośredni szkodliwy wpływ na wczesny rozwój embrionalny. Jednak badania na zwierzętach wykazały niekorzystny wpływ na rozwój postnatalny. Jako środek ostrożności zaleca się unikanie stosowania leku Gliaf w czasie ciąży.
Karmienie piersią
Nie wiadomo, czy empagliflozyna przenika do mleka matki. Leku Gliaf nie należy stosować w czasie karmienia piersią.
Funkcja rozrodcza
Nie przeprowadzono badań wpływu leku Gliaf na płodność człowieka.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn
Lek Gliaf ma nieznaczny wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn. Jednak pacjentów należy informować o ryzyku hipoglikemii, jeśli Gliaf jest stosowany w połączeniu z lekami z grupy sulfonamidów lub/ i insuliną.
Sposób stosowania i dawki
Dawkowanie
Cukrzyca typu 2
Zalecana dawka początkowa to 10 mg empagliflozyny raz dziennie, stosowanej jako monoterapia lub w ramach terapii skojarzonej z innymi lekami stosowanymi w leczeniu cukrzycy. U pacjentów dobrze tolerujących empagliflozynę w dawce 10 mg raz dziennie, u których eSzCZ ≥ 60 ml/min/1,73 m² i którzy wymagają bardziej rygorystycznego kontroli glikemii, dawkę można zwiększyć do 25 mg raz dziennie. Maksymalna dzienna dawka wynosi 25 mg (patrz informacje poniżej i sekcja „Szczególne środki ostrożności”).
Niewydolność serca
Zalecana dawka to 10 mg empagliflozyny raz dziennie.
Przewlekła choroba nerek
Zalecana dawka to 10 mg empagliflozyny raz dziennie.
Wszystkie wskazania
Gdy empagliflozyna jest stosowana w połączeniu z sulfonilomocznikami lub insuliną, należy rozważyć zastosowanie niższych dawek sulfonilomoczników lub insuliny w celu zmniejszenia ryzyka hipoglikemii (patrz sekcje „Interakcje z innymi lekami i inne formy interakcji” oraz „Działania niepożądane”).
W przypadku opuszczenia dawki należy ją przyjąć tak szybko, jak pacjent sobie o niej przypomni, jednak nie należy przyjmować podwójnej dawki w tym samym dniu.
Grupy specjalne pacjentów
Pacjenci z niewydolnością nerek
Z uwagi na ograniczone doświadczenie nie zaleca się rozpoczynania leczenia empagliflozyną u pacjentów z eSzCZ < 20 ml/min/1,73 m².
U pacjentów z eSzCZ < 60 ml/min/1,73 m² zalecana dzienna dawka empagliflozyny wynosi 10 mg.
U pacjentów z cukrzycą typu 2 skuteczność empagliflozyny w obniżaniu poziomu glukozy maleje przy eSzCZ < 45 ml/min/1,73 m² i prawdopodobnie brakuje przy eSzCZ < 30 ml/min/1,73 m². W związku z tym, jeśli eSzCZ jest niższe niż 45 ml/min/1,73 m², należy rozważyć dodatkowe leczenie obniżające poziom cukru we krwi (patrz sekcje „Szczególne środki ostrożności”, „Działania niepożądane”, „Farmakodynamika” oraz „Farmakokinetyka”).
Pacjenci z niewydolnością wątroby
U pacjentów z niewydolnością wątroby nie jest wymagana korekta dawki. Działanie empagliflozyny może być nasilone u pacjentów z ciężką niewydolnością wątroby. Doświadczenie w stosowaniu empagliflozyny u pacjentów z ciężką niewydolnością wątroby jest ograniczone, dlatego nie zaleca się stosowania leku tej grupie pacjentów (patrz sekcja „Właściwości farmakologiczne”).
Pacjenci w wieku podeszłym
Nie jest wymagana korekta dawki ze względu na wiek pacjenta. U pacjentów w wieku powyżej 75 lat należy uwzględnić zwiększony ryzyko zmniejszenia objętości płynów międzykomórkowych (patrz sekcje „Szczególne środki ostrożności”, „Działania niepożądane”).
Sposób stosowania
Tabletki można przyjmować z posiłkiem lub bez, popijając wodą, nie żując.
Dzieci
Zalecana dawka początkowa to 10 mg empagliflozyny raz dziennie. U pacjentów dobrze tolerujących empagliflozynę w dawce 10 mg raz dziennie i wymagających dodatkowej kontroli glikemii dawkę można zwiększyć do 25 mg raz dziennie (patrz sekcje „Farmakodynamika” i „Farmakokinetyka”). Brak danych dotyczących stosowania u dzieci z eSzCZ < 60 ml/min/1,73 m² oraz u dzieci poniżej 10. roku życia.
Bezpieczeństwo i skuteczność stosowania empagliflozyny w leczeniu niewydolności serca lub przewlekłej choroby nerek u dzieci (do 18. roku życia) nie były badane. Leku nie stosuje się tej grupie pacjentów.
Przedawkowanie
Objawy
W przebiegu kontrolowanych badań klinicznych pojedyncze dawki empagliflozyny do 800 mg u zdrowych ochotników oraz wielokrotne dawki dziennie do 100 mg u pacjentów z cukrzycą typu 2 nie powodowały żadnych toksycznych skutków. Empagliflozyna zwiększała wydalanie glukozy z moczem, co prowadziło do zwiększenia objętości moczu. Stwierdzono zwiększenie objętości moczu niezależne od dawki, które nie miało znaczenia klinicznego. Nie ma doświadczenia w stosowaniu u ludzi dawek wyższych niż 800 mg.
Leczenie
W przypadku przedawkowania należy rozpocząć leczenie odpowiednio do stanu klinicznego pacjenta. Nie badano wyprowadzania empagliflozyny metodą hemodializy.
Reakcje niepożądane
Cukrzyca typu 2
Najczęstszą reakcją niepożądaną była hipoglikemia występująca podczas stosowania empagliflozyny z sulfonem amidowym lub insuliną.
Niewydolność serca
W badaniach EMPEROR uczestniczyli pacjenci z niewydolnością serca i zmniejszoną frakcją wyrzutu lewej komory (N = 3 726) oraz zachowaną frakcją wyrzutu (N = 5 985), którzy przyjmowali empagliflozynę w dawce 10 mg lub placebo. Około połowa tych pacjentów miała cukrzycę typu 2. Najczęstszą reakcją niepożądaną w połączonych danych z badań EMPEROR-Reduced i EMPEROR-Preserved była hipowolemia (empagliflozyna 10 mg – 11,4 %, placebo – 9,7 %).
Przewlekła choroba nerek
Badanie EMPA-KIDNEY objęło pacjentów z przewlekłą chorobą nerek (N = 6 609), którzy otrzymywali 10 mg empagliflozyny lub placebo. Około 44 % pacjentów miało cukrzycę typu 2.
Najczęstszymi reakcjami niepożadanymi w badaniu EMPA-KIDNEY były podagra (empagliflozyna – 7,0 %, placebo – 8,0 %) oraz ostra niewydolność nerek (empagliflozyna – 2,8 %, placebo – 3,5 %), o których częściej informowano u pacjentów przyjmujących placebo.
Ogólny profil bezpieczeństwa leku Gliaf odpowiadał zazwyczaj wszystkim badanym wskazaniom terapeutycznym.
Reakcje niepożądane sklasyfikowano według układów narządów (zgodnie z MedDRA) oraz częstości występowania. Częstość występowania określa się jako bardzo często (> 1/10), często (> 1/100 – < 1/10), rzadko (> 1/1000 – < 1/100), pojedynczo (> 1/10 000 – < 1/1000) lub rzadko (< 1/10000) oraz nieznane (nie można oszacować na podstawie dostępnych danych).
Reakcje niepożądane (na podstawie badań placebo-kontrolowanych)
Infekcje i inwazje
Często: kandydoza pochwy, zapalenie cewki i pochwy, balanit oraz inne infekcje narządów płciowycha, infekcje dróg moczowych (w tym zapalenie nerek i urosepsa)a.
Pojedynczo: martwicze zapalenie powięzi okolicy krocza (gangrena Fourniera)*.
Zaburzenia przemiany materii i układu pokarmowego
Bardzo często: hipoglikemia (w przypadku stosowania z lekami z grupy sulfonów amidowych lub insuliną)a.
Często: uczucie pragnienia.
Rzadko: kwasica ketonowa*.
Zaburzenia ze strony układu pokarmowego
Często: zaparcia.
Zaburzenia ze strony skóry i tkanki podskórnej
Często: świąd (ogólny), wysypka.
Rzadko: pokrzywka, obrzęk naczynioruchowy.
Zaburzenia ze strony naczyń
Bardzo często: zmniejszenie objętości płynu międzykomórkowegoa.
Zaburzenia ze strony nerek i dróg moczowych
Często: zwiększone wydalanie moczua.
Rzadko: dyzuria.
Rzadko: zapalenie naczyniowo-śródmiąższowe nerek.
Badania
Często: zwiększenie stężenia lipidów w surowicya.
Rzadko: podwyższenie stężenia kreatyniny we krwi / obniżenie szybkości filtracji kłębuszkoweja, zwiększenie hematokrytua.
a Dodatkowe informacje znajdują się w poniższych podrozdziałach.
* Zob. sekcję „Szczególne ostrzeżenia i środki ostrożności stosowania”.
Opis niektórych reakcji niepożądanych
Hipoglikemia
Częstość hipoglikemii zależała od terapii towarzyszącej podczas odpowiednich badań i była podobna w przypadku stosowania empagliflozyny i placebo jako monoterapii, jako dodatku do metformyny, jako dodatku do pioglitazonu z metformyną lub bez, jako dodatku do linagliptyny i metformyny, a także w przypadku stosowania kombinacji empagliflozyny z metformyną u pacjentów leczonych po raz pierwszy, w porównaniu z pacjentami, którzy wcześniej stosowali empagliflozynę i metformynę jako oddzielne składniki. Zwiększenie częstości łagodnej hipoglikemii obserwowano przy podawaniu empagliflozyny i placebo jako dodatku do metformyny i sulfonu amidowego (empagliflozyna w dawce 10 mg – 16,1 %, empagliflozyna w dawce 25 mg – 11,5 %, placebo – 8,4 %) oraz jako dodatku do insuliny bazalnej z metformyną lub bez oraz z lekiem z grupy sulfonów amidowych lub bez (empagliflozyna w dawce 10 mg – 19,5 %, empagliflozyna w dawce 25 mg – 28,4 %, placebo – 20,6 % w pierwszych 18 tygodniach leczenia, gdy dawkę insuliny nie można było dostosować; empagliflozyna w dawce 10 mg – 36,1 %, empagliflozyna w dawce 25 mg – 36,1 %, placebo – 35,3 % w trakcie 78-tygodniowego badania), a także jako dodatku do insuliny w strzykawce dozującej z metformyną lub bez (empagliflozyna w dawce 10 mg – 39,8 %, empagliflozyna w dawce 25 mg – 41,3 %, placebo – 37,2 % w pierwszych 18 tygodniach leczenia, gdy dawkę insuliny nie można było dostosować; empagliflozyna w dawce 10 mg – 51,1 %, empagliflozyna w dawce 25 mg – 57,7 %, placebo – 58 % w trakcie 52-tygodniowego badania).
W badaniach EMPEROR dotyczących niewydolności serca podobną częstość hipoglikemii obserwowano po dodaniu leku do sulfonu amidowego lub insuliny (empagliflozyna 10 mg – 6,5 %, placebo – 6,7 %).
Ciężka hipoglikemia (hipoglikemia wymagająca leczenia)
Zwiększenia częstości ciężkiej hipoglikemii nie obserwowano przy stosowaniu empagliflozyny w porównaniu z placebo jako monoterapii, jako dodatku do metformyny, jako dodatku do metformyny i leku z grupy sulfonów amidowych oraz jako dodatku do pioglitazonu z metformyną lub bez, jako dodatku do linagliptyny i metformyny, a także jako dodatku do standardowej terapii, w przypadku stosowania kombinacji empagliflozyny z metformyną u pacjentów leczonych po raz pierwszy, w porównaniu z pacjentami, którzy wcześniej stosowali empagliflozynę i metformynę jako oddzielne składniki. Zwiększenie częstości ciężkiej hipoglikemii obserwowano przy podawaniu empagliflozyny i placebo jako dodatku do insuliny bazalnej z metformyną lub bez oraz z lekiem z grupy sulfonów amidowych lub bez (empagliflozyna w dawce 10 mg – 0 %, empagliflozyna w dawce 25 mg – 1,3 %, placebo – 0 % w pierwszych 18 tygodniach leczenia, gdy dawkę insuliny nie można było dostosować; empagliflozyna w dawce 10 mg – 0 %, empagliflozyna w dawce 25 mg – 1,3 %, placebo – 0 % w trakcie 78-tygodniowego badania) oraz jako dodatku do insuliny w strzykawce dozującej z metformyną lub bez (empagliflozyna w dawce 10 mg – 0,5 %, empagliflozyna w dawce 25 mg – 0,5 %, placebo – 0,5 % w pierwszych 18 tygodniach leczenia, gdy dawkę insuliny nie można było dostosować; empagliflozyna w dawce 10 mg – 1,6 %, empagliflozyna w dawce 25 mg – 0,5 %, placebo – 1,6 % w trakcie 52-tygodniowego badania).
W badaniach EMPEROR dotyczących niewydolności serca obserwowano podobną częstość hipoglikemii u chorych na cukrzycę leczonych empagliflozyną i placebo jako dodatkiem do sulfonu amidowego lub insuliny (empagliflozyna 10 mg – 2,2 %, placebo – 1,9 %).
Kandydoza pochwy, zapalenie cewki i pochwy, balanit oraz inne infekcje narządów płciowych
Kandydoza pochwy, zapalenie cewki i pochwy, balanit oraz inne infekcje narządów płciowych występowały częściej przy stosowaniu empagliflozyny (empagliflozyna w dawce 10 mg – 4,0 %, empagliflozyna w dawce 25 mg – 3,9 %) w porównaniu z placebo (1,0 %). Infekcje te występowały częściej u kobiet przyjmujących empagliflozynę w porównaniu z kobietami z grupy placebo; różnica częstości u mężczyzn była mniej wyrażona. Infekcje narządów płciowych były łagodne lub umiarkowane pod względem nasilenia.
W badaniach EMPEROR dotyczących niewydolności serca częstość tych infekcji była wyższa u pacjentów z cukrzycą typu 2 (empagliflozyna 10 mg – 2,3 %, placebo – 0,8 %) niż u pacjentów bez cukrzycy (empagliflozyna 10 mg – 1,7 %, placebo – 0,7 %), przy leczeniu empagliflozyną w porównaniu z placebo.
Zgłaszano przypadki rozwinięcia się ciasnoty napletka / nabytej ciasnoty napletka, towarzyszące infekcjom narządów płciowych, w niektórych przypadkach konieczne było okoliczne usunięcie napletka.
Zwiększone wydalanie moczu
Zwiększone wydalanie moczu (w tym wcześniej zdefiniowane terminy – polakiuria, poliuria i nokturia) występowało częściej u pacjentów przyjmujących empagliflozynę (empagliflozyna w dawce 10 mg – 3,5 %, empagliflozyna w dawce 25 mg – 3,3 %) w porównaniu z pacjentami z grupy placebo (1,4 %). Zwiększone wydalanie moczu było głównie łagodne lub umiarkowane pod względem nasilenia. Częstość przypadków nokturii była podobna przy stosowaniu placebo i empagliflozyny (< 1 %).
W badaniach EMPEROR dotyczących niewydolności serca zwiększone oddawanie moczu obserwowano z podobną częstością u pacjentów przyjmujących empagliflozynę i placebo (empagliflozyna 10 mg – 0,9 %, placebo – 0,5 %).
Infekcje dróg moczowych
Ogólna częstość infekcji dróg moczowych zgłaszanych jako niepożądane zdarzenia była podobna u pacjentów przyjmujących empagliflozynę w dawce 25 mg i pacjentów z grupy placebo (7,0 % i 7,2 %) oraz wyższa przy stosowaniu empagliflozyny w dawce 10 mg (8,8 %). W porównaniu z placebo, infekcje dróg moczowych występowały częściej u pacjentów przyjmujących empagliflozynę z historią przewlekłych lub nawracających infekcji dróg moczowych. Nasilenie (łagodne, umiarkowane, ciężkie) infekcji dróg moczowych było podobne u pacjentów przyjmujących empagliflozynę i pacjentów z grupy placebo. Infekcje dróg moczowych występowały częściej u kobiet przyjmujących empagliflozynę w porównaniu z kobietami z grupy placebo; u mężczyzn różnica nie występowała.
Zmniejszenie objętości płynu międzykomórkowego
Ogólna częstość zmniejszenia objętości płynu międzykomórkowego (w tym wcześniej zdefiniowane terminy – obniżenie ciśnienia tętniczego [w warunkach ambulatoryjnych], obniżenie ciśnienia tętniczego skurczowego, odwodnienie, hipotensja tętnicza, hipowolemia, hipotonia ortostatyczna i omdlenia) była podobna u pacjentów przyjmujących empagliflozynę (empagliflozyna w dawce 10 mg – 0,6 %, empagliflozyna w dawce 25 mg – 0,4 %) i pacjentów z grupy placebo (0,3 %). Częstość przypadków zmniejszenia objętości płynu międzykomórkowego była zwiększona u pacjentów w wieku 75 lat i starszych przyjmujących empagliflozynę w dawce 10 mg (2,3 %) lub empagliflozynę w dawce 25 mg (4,3 %) w porównaniu z pacjentami przyjmującymi placebo (2,1 %).
Zwiększenie stężenia kreatyniny we krwi / obniżenie szybkości filtracji kłębuszkowej
Ogólna częstość zwiększenia stężenia kreatyniny we krwi i obniżenia szybkości filtracji kłębuszkowej była podobna przy stosowaniu empagliflozyny i placebo (zwiększenie stężenia kreatyniny we krwi: empagliflozyna w dawce 10 mg – 0,6 %, empagliflozyna w dawce 25 mg – 0,1 %, placebo – 0,5 %; obniżenie szybkości filtracji kłębuszkowej: empagliflozyna w dawce 10 mg – 0,1 %, empagliflozyna w dawce 25 mg – 0 %, placebo – 0,3 %).
Zazwyczaj u pacjentów przyjmujących empagliflozynę, początkowe zwiększenie stężenia kreatyniny i obniżenie szybkości filtracji kłębuszkowej podczas długotrwałego leczenia były tymczasowe lub odwracalne po przerwaniu terapii.
W trakcie badania EMPA-REG OUTCOME u pacjentów przyjmujących empagliflozynę obserwowano początkowe obniżenie eGFR (średnia wartość 3 ml/min/1,73 m²). Następnie eGFR pozostawało stabilne w trakcie ciągłego leczenia. Średnie eGFR wracało do wartości wyjściowych po zakończeniu leczenia, co wskazuje, że ciężkie zaburzenia hemodynamiki mogą odgrywać pewną rolę w tych zmianach funkcji nerek. Zjawisko to obserwuje się również w badaniu dotyczącym niewydolności serca EMPEROR oraz w badaniu EMPA-KIDNEY.
Zwiększenie stężenia lipidów w surowicy krwi
Średni procentowy wzrost od wartości wyjściowej przy stosowaniu empagliflozyny w dawce 10 mg i 25 mg w porównaniu z placebo wynosił odpowiednio: cholesterol ogólny 4,9 % i 5,7 % vs. 3,5 %; cholesterol LPWZ 3,3 % i 3,6 % vs. 0,4 %; cholesterol LPNZ 9,5 % i 10,0 % vs. 7,5 %; trójglicerydy 9,2 % i 9,9 % vs. 10,5 %.
Zwiększenie hematokrytu
Średnie zmiany hematokrytu od wartości wyjściowej wynosiły 3,4 % i 3,6 % przy stosowaniu empagliflozyny w dawce 10 mg i 25 mg w porównaniu z 0,1 % przy stosowaniu placebo. W badaniu EMPA-REG OUTCOME wartości hematokrytu wróciły do wartości wyjściowych w ciągu 30 dni po zakończeniu leczenia.
Dzieci
W badaniu DINAMO leczono 157 dzieci w wieku od 10 lat z cukrzycą typu 2, z których 52 pacjentów otrzymywało empagliflozynę, 52 pacjentów otrzymywało linagliptynę, a 53 pacjentów otrzymywało placebo (zob. sekcję „Farmakodynamika”). W trakcie fazy kontrolowanej placebo najczęściej występującą reakcją niepożądaną była hipoglikemia, z wyższym ogólnym wskaźnikiem u pacjentów z grupy empagliflozyny w porównaniu z pacjentami z grupy placebo (empagliflozyna 10 mg i 25 mg łącznie – 23,1 %, placebo – 9,4 %). Żaden z tych przypadków nie był ciężki i nie wymagał leczenia.
Ogólnie profil bezpieczeństwa u dzieci był podobny do tego obserwowanego u dorosłych z cukrzycą typu 2.
Zgłaszanie podejrzewanych reakcji niepożądanych
Zgłaszanie reakcji niepożądanych po rejestracji leku ma istotne znaczenie. Pozwala to na monitorowanie stosunku korzyści do ryzyka w stosowaniu tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawowe przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych reakcji niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 2 lata.
Warunki przechowywania
Lek nie wymaga specjalnych warunków przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie. Po 10 tabletek w blistrze; po 3 blisterach w tece kartonowej.
Kategoria wydania. Na receptę.
Producent. PharmaPath S.A.
Siedziba producenta i adres miejsca prowadzenia działalności
28 Oktovriu 1, Agia Varvara, 123 51, Grecja / 28is Oktovriou 1, Agia Varvara, 123 51, Greece.