Gliaph
UcraniaContenido
- INSTRUCCIONES DE USO PARA USO MÉDICO DEL MEDICAMENTO Gliaf (Gliaf)
- Composición:
- Propiedades farmacodinámicas
- Predefinido como uno de los dos criterios de interrupción en un análisis intermedio previamente planificado.
- Características clínicas
- Características de uso
- Vía de administración y dosis
- Reacciones adversas
INSTRUCCIONES DE USO PARA USO MÉDICO DEL MEDICAMENTO Gliaf (Gliaf)
Composición:
Sustancia activa: empagliflozina;
Cada comprimido contiene 10 mg o 25 mg de empagliflozina;
Sustancias auxiliares: lactosa monohidrato, celulosa microcristalina (tipo PH 101), hidroxipropilcelulosa, croscarmelosa sódica, celulosa microcristalina (tipo PH 102), dióxido de silicio coloidal anhidro, estearato de magnesio;
Recubrimiento filmogénico para comprimidos de 10 mg: Opadry Yellow 03B220113 - hipromelosa (hidroxipropilmetilcelulosa) (tipo 2910), talco, polietilenglicol (tipo 400), dióxido de titanio (E 171), óxido de hierro amarillo (E 172);
Recubrimiento filmogénico para comprimidos de 25 mg: Opadry Yellow 03B220114 - hipromelosa (hidroxipropilmetilcelulosa) (tipo 2910), talco, polietilenglicol (tipo 400), dióxido de titanio (E 171), óxido de hierro amarillo (E 172).
Forma farmacéutica. Comprimidos recubiertos con película.
Propiedades físico-químicas principales:
Comprimidos de 10 mg: comprimidos redondos, de color amarillo pálido (diámetro aproximado de 9,1 mm), biconvexos, recubiertos con película, con la impresión «10» en una cara;
Comprimidos de 25 mg: comprimidos ovales, de color amarillo pálido (aproximadamente 11,1 mm × 5,6 mm), biconvexos, recubiertos con película, con la impresión «25» en una cara.
Grupo farmacoterapéutico
Medicamentos utilizados en la diabetes, inhibidores del cotransportador de sodio y glucosa tipo 2 (SGLT2). Código ATC A10B K03.
Propiedades farmacodinámicas
Mecanismo de acción
Empagliflozina es un inhibidor competitivo selectivo y potente (IC50 1,3 nmol) del cotransportador de sodio y glucosa 2 (SGLT2) reversible. Empagliflozina no inhibe otros transportadores de glucosa que desempeñan un papel importante en la entrega de glucosa a los tejidos periféricos, y es 5000 veces más selectivo frente al SGLT2 que frente al SGLT1, el principal transportador responsable de la absorción de glucosa en el intestino. El SGLT2 se expresa a niveles elevados en los riñones, mientras que su expresión en otros tejidos es ausente o muy baja. Como transportador principal, es responsable de la reabsorción de glucosa desde la luz tubular de nuevo a la circulación sanguínea. En pacientes con diabetes mellitus tipo 2 e hiperglucemia, se filtra y reabsorbe una mayor cantidad de glucosa.
Empagliflozina mejora el control glucémico en pacientes con diabetes mellitus tipo 2 al reducir la reabsorción renal de glucosa. La cantidad de glucosa excretada por los riñones mediante este mecanismo glucurético depende de la concentración de glucosa en sangre y de la tasa de filtración glomerular (TFG). La inhibición del SGLT2 en pacientes con diabetes mellitus tipo 2 e hiperglucemia conduce a un aumento de la excreción urinaria de glucosa. Además, empagliflozina aumenta la excreción de sodio, lo que provoca un diuresis osmótica y reduce el volumen intravascular.
En pacientes con diabetes mellitus tipo 2, la excreción de glucosa aumentó inmediatamente después de la primera dosis de empagliflozina y se mantuvo durante el intervalo de 24 horas entre dosis. El aumento de la excreción urinaria de glucosa se mantuvo al final del período de tratamiento de 4 semanas y fue en promedio de aproximadamente 78 g/día. El aumento de la excreción urinaria de glucosa provocó una reducción inmediata de los niveles de glucosa en plasma en pacientes con diabetes mellitus tipo 2.
Empagliflozina mejora los niveles de glucosa en plasma en ayunas y posprandiales. El mecanismo de acción de empagliflozina no depende de la función de las células β ni de la vía de acción de la insulina, lo que contribuye a reducir el riesgo de hipoglucemia. Se ha observado una mejora en los marcadores de función de las células β, incluyendo el modelo homeostático de evaluación de la función de las células β (HOMA-β). Además, la excreción urinaria de glucosa provoca una pérdida calórica asociada con la reducción de grasa corporal y disminución del peso corporal. La glucosuria observada con empagliflozina se acompaña de diuresis, lo que puede contribuir a una reducción sostenida y moderada de la presión arterial.
Empagliflozina también reduce la reabsorción de sodio y aumenta la entrega de sodio a los túbulos distales. Esto puede influir en varias funciones fisiológicas, incluyendo el aumento de la retroalimentación túbuloglomerular, la reducción de la presión intraglomerular, la disminución de la precarga/postcarga cardíaca y la prevención de la actividad simpática, así como la reducción de la carga en la pared del ventrículo izquierdo, evidenciado por valores más bajos de NT-proBNP, lo que puede tener un efecto beneficioso sobre el remodelado cardíaco, la presión de llenado y la función diastólica, así como la preservación de la estructura y función renales. Otros efectos, como el aumento del hematocrito, la reducción del peso corporal y la presión arterial, pueden contribuir adicionalmente a los beneficios cardio-renales.
Eficacia clínica y seguridad
Diabetes mellitus tipo 2
La mejora del control glucémico y la reducción del riesgo de enfermedades cardiovasculares y mortalidad son componentes esenciales del tratamiento de la diabetes mellitus tipo 2.
El tratamiento con empagliflozina, ya sea solo o en combinación con metformina, pioglitazona, sulfonilureas, inhibidores de la DPP-4 o insulina, condujo a una mejora clínicamente significativa en los niveles de HbA1c, glucosa en plasma en ayunas, peso corporal, presión arterial sistólica y diastólica. Con la dosis de empagliflozina de 25 mg, aumentó la proporción de pacientes que alcanzaron el objetivo de HbA1c inferior al 7 % y disminuyó el número de pacientes que requirieron terapia glucémica, en comparación con la dosis de 10 mg de empagliflozina y placebo. Cuanto mayor era el nivel basal de HbA1c, mayor fue su reducción con el tratamiento.
Además, empagliflozina como complemento a la terapia estándar reduce la mortalidad cardiovascular y la incidencia de eventos cardiovasculares en pacientes con diabetes mellitus tipo 2.
Pronóstico cardiovascular
El estudio EMPA-REG OUTCOME, doble ciego y controlado con placebo, comparó la eficacia de empagliflozina en dosis de 10 mg y 25 mg frente a placebo, como complemento a la terapia estándar en pacientes con diabetes mellitus tipo 2 y enfermedad cardiovascular establecida.
Empagliflozina fue superior al placebo en la prevención de muerte por enfermedad cardiovascular, infarto de miocardio no mortal o ictus no mortal. Este efecto se debió principalmente a una reducción significativa del riesgo de muerte cardiovascular, sin cambios sustanciales en cuanto al infarto de miocardio no mortal o al ictus no mortal. La reducción en la mortalidad por enfermedad cardiovascular fue comparable con las dosis de 10 mg y 25 mg de empagliflozina (ver figs. 1-4 más abajo) y se confirmó mediante una mejora en la supervivencia global (ver tabla 1).
El efecto de empagliflozina sobre el punto final primario combinado (muerte por enfermedad cardiovascular, infarto de miocardio no mortal o ictus no mortal) fue en gran medida independiente del control glucémico o de la función renal, que en general, en todos los grupos de pacientes del estudio EMPA-REG OUTCOME, se caracterizó por un valor de TFG ≥30 ml/min/1,73 m².
La eficacia en la prevención de la muerte cardiovascular no se estableció definitivamente en pacientes que recibieron empagliflozina junto con inhibidores de la DPP-4 ni en pacientes de raza no caucásica, debido a la representación limitada de estos grupos en el estudio EMPA-REG OUTCOME.
Tabla 1
Efecto del tratamiento sobre los principales criterios de evaluación, sus componentes y la mortalidad a
| Indicador de eficacia |
Placebo N = 2333 |
Empagliflozina N = 4687 |
| Tiempo hasta el primer evento de muerte por causas cardiovasculares, infarto de miocardio no mortal o ictus no mortal, N (%) |
282 (12,1) |
490 (10,5) |
| Relación de riesgos en comparación con placebo (intervalo de confianza del 95,02 % (IC))* |
0,86 (0,74; 0,99) |
|
| Valor p para beneficio |
0,0382 |
|
| Muerte por causas cardiovasculares, N (%) |
137 (5,9) |
172 (3,7) |
| Relación de riesgos en comparación con placebo (95 % IC) |
0,62 (0,49; 0,77) |
|
| Valor p |
< 0,0001 |
|
| Infarto de miocardio no mortal, N (%) |
121 (5,2) |
213 (4,5) |
| Relación de riesgos en comparación con placebo (95 % IC) |
0,87 (0,70; 1,09) |
|
| Valor p |
0,2189 |
|
| Ictus no mortal, N (%) |
60 (2,6) |
150 (3,2) |
| Relación de riesgos en comparación con placebo (95 % IC) |
1,24 (0,92; 1,67) |
|
| Valor p |
0,1638 |
|
| Mortalidad total, N (%) |
194 (8,3) |
269 (5,7) |
| Relación de riesgos en comparación con placebo (95 % IC) |
0,68 (0,57; 0,82) |
|
| Valor p |
< 0,0001 |
|
| Mortalidad no relacionada con causas cardiovasculares, N (%) |
57 (2,4) |
97 (2,1) |
| Relación de riesgos en comparación con placebo (95 % IC) |
0,84 (0,60; 1,16) |
a Datos de pacientes tratados (es decir, pacientes que recibieron al menos una dosis del medicamento en estudio).
b Dosis combinadas de empagliflozina 10 mg y 25 mg.
* Dado que los resultados del estudio se incluyeron en el análisis intermedio, se aplica un intervalo de confianza bilateral del 95,02 %, que corresponde a valores de p < 0,0498 para la significación.
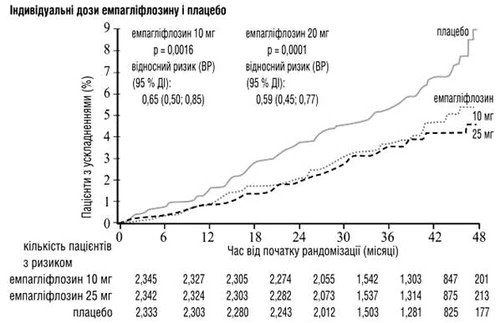
Fig. 1. Tiempo hasta la muerte por causas cardiovasculares en el estudio EMPA-REG OUTCOME
Insuficiencia cardíaca que requiere hospitalización
En el estudio EMPA-REG OUTCOME, la empagliflozina redujo el riesgo de insuficiencia cardíaca que requirió hospitalización en comparación con placebo (grupo empagliflozina – 2,7 %; grupo placebo – 4,1 %; HR 0,65; IC 95 % 0,50; 0,85).
Nefropatía
En el estudio EMPA-REG OUTCOME, respecto al tiempo hasta el primer episodio de nefropatía, la HR fue de 0,61 (IC 95 % 0,53; 0,70) en el grupo de empagliflozina (12,7 %) en comparación con el grupo placebo (18,8 %).
Además, la empagliflozina aumentó el riesgo (HR 1,82; IC 95 % 1,40; 2,37) de desarrollar normo- o microalbuminuria sostenida (49,7 %) en pacientes con macroalbuminuria al inicio del estudio, en comparación con placebo (28,8 %).
Insuficiencia cardíaca
Uso de empagliflozina en pacientes con insuficiencia cardíaca y fracción de eyección reducida
Se realizó un estudio aleatorizado, doble ciego, controlado con placebo (EMPEROR-reduced) con 3730 pacientes con insuficiencia cardíaca crónica (clases II-IV según la clasificación de la New York Heart Association (NYHA)) y fracción de eyección del ventrículo izquierdo reducida (FEVI ≤ 40 %) para evaluar la eficacia y seguridad del uso de empagliflozina 10 mg una vez al día como complemento a la terapia estándar para la insuficiencia cardíaca. El punto final primario fue el tiempo hasta el primer episodio de muerte cardiovascular (CV) confirmada o hospitalización por insuficiencia cardíaca. La pendiente del cambio en hospitalizaciones confirmadas (primera y repetidas) y la TFG (CKD-EPI) respecto a los valores basales se incluyeron en la prueba confirmatoria. El tratamiento para la insuficiencia cardíaca al inicio del estudio incluyó el uso de inhibidores de la ECA / bloqueadores de los receptores de la angiotensina II / inhibidores del receptor de angiotensina-neprilisina (88,3 %), betabloqueadores (94,7 %), antagonistas de los receptores de mineralocorticoides (71,3 %) y diuréticos (95,0 %).
Un total de 1863 pacientes fueron aleatorizados al grupo de empagliflozina 10 mg (placebo – 1867). La mediana de duración del tratamiento fue de 15,7 meses. El 76,1 % de la población estudiada eran hombres y el 23,9 % mujeres, con una edad media de 66,8 años (rango 25-94 años); el 26,8 % de los pacientes tenían más de 75 años. El 70,5 % de la población estudiada correspondía a pacientes de raza caucásica, el 18,0 % a raza asiática y el 6,9 % a raza negra / afroamericanos. En el momento de la aleatorización, el 75,1 % de los pacientes tenían insuficiencia cardíaca clase II según la clasificación NYHA, el 24,4 % insuficiencia cardíaca clase III según NYHA y el 0,5 % insuficiencia cardíaca clase IV según NYHA. El valor medio de la FEVI fue del 27,5 %. Al inicio del estudio, la TFG media fue de 62,0 ml/min/1,73 m² y la relación albúmina/creatinina en orina (UACR) media fue de 22 mg/g. En aproximadamente la mitad de los pacientes (51,7 %), la TFG fue ≥ 60 ml/min/1,73 m², en el 24,1 % fue de 45 a < 60 ml/min/1,73 m², en el 18,6 % fue de 30 a < 45 ml/min/1,73 m² y en el 5,3 % fue de 20 a < 30 ml/min/1,73 m².
La empagliflozina demostró una mayor eficacia en la reducción del riesgo del punto final combinado primario: muerte CV o hospitalización por insuficiencia cardíaca, en comparación con placebo. Además, la empagliflozina redujo significativamente el riesgo de hospitalización por insuficiencia cardíaca (primera y repetida), así como redujo significativamente la velocidad de disminución de la TFG (tabla 2).
Tabla 2
Efecto del tratamiento sobre el punto final combinado primario, sus componentes y dos puntos finales secundarios clave incluidos en la prueba confirmatoria predefinida
| Indicador |
Placebo |
Empagliflozina 10 mg |
| N |
1867 |
1863 |
| Tiempo hasta el primer episodio confirmado de muerte CV o hospitalización por IC, N (%) |
462 (24,7) |
361 (19,4) |
| Relación de riesgos frente al placebo (IC 95 %)* |
0,75 (0,65; 0,86) |
|
| Valor p para eficacia principal |
< 0,0001 |
|
| Muerte CV, N (%) * |
202 (10,8) |
187 (10,0) |
| Relación de riesgos frente al placebo (IC 95 %) |
0,92 (0,75; 1,12) |
|
| Primera hospitalización por IC, N (%) |
342 (18,3) |
246 (13,2) |
| Relación de riesgos frente al placebo (IC 95 %) |
0,69 (0,59; 0,81) |
|
| Primera y posteriores hospitalizaciones por IC, número de eventos |
553 |
388 |
| Relación de riesgos frente al placebo (IC 95 %)* |
0,70 (0,58; 0,85) |
|
| Valor p |
0,0003 |
|
| Pendiente del cambio en el FGRe (CKD-EPI)cr, velocidad de descenso (ml/min/1,73 m²/año) |
-2,28 |
-0,55 |
| Diferencia entre tratamientos frente al placebo (IC 95 %) |
1,73 (1,10; 2,37) |
|
| Valor p |
p < 0,0001 |
SS – cardiovascular, IC – insuficiencia cardíaca, TFGc – tasa de filtración glomerular calculada, EPI – ecuación de ajuste epidemiológico en enfermedad renal crónica
* La muerte por enfermedad cardiovascular y la hospitalización por insuficiencia cardíaca se definieron mediante un comité independiente de eventos clínicos y se evaluaron según el conjunto de datos aleatorizados.
** La tasa de filtración glomerular calculada (TFGc) se analizó según el conjunto de datos evaluado. El descenso fue de -0,95 ml/min/1,73 m² con placebo y de -3,02 ml/min/1,73 m² con empagliflozina. Este descenso representa un efecto agudo sobre la TFGc, mientras que la pendiente representa el efecto a largo plazo.
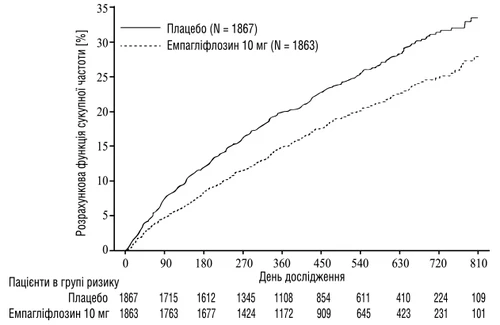
Fig. 2. Tiempo hasta el primer evento confirmado de muerte cardiovascular o hospitalización por insuficiencia cardíaca
Los resultados respecto al punto final combinado primario mostraron una relación de riesgos (RR) inferior a 1 en todos los subgrupos previamente definidos, incluyendo pacientes con insuficiencia cardíaca con o sin diabetes tipo 2 y con o sin disfunción renal (no inferior a una tasa de filtración glomerular calculada (TFGc) de 20 ml/min/1,73 m²).
Uso de empagliflozina en pacientes con insuficiencia cardíaca y fracción de eyección conservada
Se realizó un estudio aleatorizado, doble ciego y controlado con placebo (EMPEROR-Preserved) con 5 988 pacientes con insuficiencia cardíaca crónica (NYHA II–IV) y fracción de eyección conservada (FEVI > 40 %), con el fin de evaluar la eficacia y seguridad del uso de empagliflozina a una dosis de 10 mg una vez al día como complemento a la terapia estándar. El punto final primario fue el tiempo hasta el primer evento de muerte por enfermedad cardiovascular o hospitalización confirmada por insuficiencia cardíaca (IC). La hospitalización confirmada por IC (primera y recurrente) y la pendiente del cambio en la TFGc (según ecuación EPI-ERC) desde el valor basal se incluyeron en la evaluación confirmatoria preespecificada. El tratamiento basal incluyó inhibidores de la ECA / bloqueantes de los receptores de la angiotensina / inhibidor del receptor de angiotensina/neprilisina (80,7 %), betabloqueantes (86,3 %), antagonistas de los receptores de mineralocorticoides (37,5 %) y diuréticos (86,2 %).
La empagliflozina demostró una mayor eficacia en la reducción del riesgo del punto final primario combinado: muerte por enfermedad cardiovascular o hospitalización por insuficiencia cardíaca, en comparación con placebo. Además, la empagliflozina redujo significativamente el riesgo de hospitalización por IC (primera y recurrente) y ralentizó significativamente la velocidad de disminución de la TFGc.
Tabla 3
Efecto del tratamiento sobre el punto final combinado primario, sus componentes y dos puntos finales secundarios clave incluidos en la prueba confirmatoria preespecificada
| Indicador |
Placebo |
Empagliflozina, 10 mg |
| N |
2991 |
2997 |
| Tiempo hasta el primer episodio confirmado de muerte por CC o hospitalización por IC, N (%) |
511 (17,1) |
415 (13,8) |
| Relación de riesgos en comparación con el placebo (IC del 95 %)* |
0,79 (0,69; 0,90) |
|
| Valor de p para la mayor eficacia |
0,0003 |
|
| Muerte por CC, N (%) * |
244 (8,2) |
219 (7,3) |
| Relación de riesgos en comparación con el placebo (IC del 95 %) |
0,91 (0,76; 1,09) |
|
| Primera hospitalización por IC, N (%) |
352 (11,8) |
259 (8,6) |
| Relación de riesgos en comparación con el placebo (IC del 95 %) |
0,71 (0,60; 0,83) |
|
| Primera y repetidas hospitalizaciones por IC, número de eventos |
541 |
407 |
| Relación de riesgos en comparación con el placebo (IC del 95 %)* |
0,73 (0,61; 0,88) |
|
| Valor de p |
0,0009 |
|
| Pendiente del cambio en el FGRe (CKD EPI)cr, velocidad de disminución (ml/min/1,73 m²/año) |
-2,62 |
-1,25 |
| Diferencia entre tratamientos en comparación con el placebo (IC del 95 %) |
1,36 (1,06; 1,66) |
|
| Valor de p |
p < 0,0001 |
CC – cardiovascular, IC – insuficiencia cardíaca, TFG – tasa de filtración glomerular estimada, EPI de ERC – Estudio de Epidemiología de la Enfermedad Renal Crónica.
* La muerte por enfermedad cardiovascular y la hospitalización por insuficiencia cardíaca fueron determinadas por un comité independiente de eventos clínicos y evaluadas según el conjunto aleatorizado.
** La tasa de filtración glomerular estimada se analizó según el conjunto evaluado. La caída fue de -0,95 ml/min/1,73 m² con placebo y de -3,02 ml/min/1,73 m² con empagliflozina. La caída representa el efecto agudo sobre la tasa de filtración glomerular estimada, mientras que la pendiente representa el efecto a largo plazo.
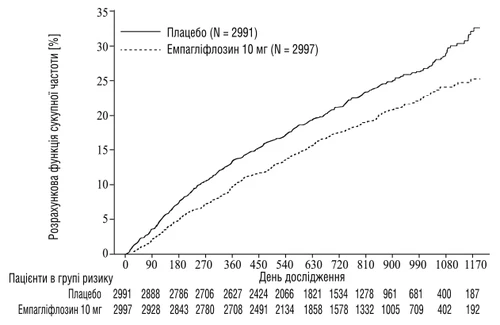
Fig. 3 Tiempo hasta el primer evento confirmado de muerte cardiovascular o hospitalización por insuficiencia cardíaca
Los resultados del punto final combinado primario fueron consistentes en cada uno de los subgrupos previamente definidos, clasificados, por ejemplo, según la fracción de eyección del ventrículo izquierdo, el estado de diabetes mellitus o la función renal (hasta 20 ml/min/1,73 m²).
Enfermedad renal crónica
Se realizó un estudio aleatorizado, doble ciego, controlado con placebo, sobre el uso de empagliflozina 10 mg una vez al día (EMPA-KIDNEY), como complemento a la terapia estándar, en 6609 pacientes con enfermedad renal crónica (TFG ≥ 20 ‒ < 45 ml/min/1,73 m² o TFG ≥ 45 ‒ < 90 ml/min/1,73 m² con una relación albúmina/creatinina en orina (UACR) ≥ 200 mg/g), para evaluar los resultados cardio-renales. El punto final primario fue el tiempo hasta el primer caso de progresión de la enfermedad renal (disminución sostenida ≥ 40 % de la TFG desde la aleatorización, TFG sostenida < 10 ml/min/1,73 m², enfermedad renal en etapa terminal o muerte por enfermedad renal) o muerte por enfermedad cardiovascular. La primera hospitalización por insuficiencia cardíaca o muerte por enfermedad cardiovascular, la hospitalización por cualquier causa (primera y repetida) y la muerte por todas las causas se incluyeron en la prueba de confirmación.
El tratamiento al inicio del estudio incluyó el uso de un inhibidor del SRA (85,2 % de inhibidores de la ECA / bloqueadores del receptor de angiotensina II).
Se aleatorizaron 3304 pacientes al grupo de empagliflozina 10 mg (placebo – 3305). La mediana de duración del tratamiento fue de 24,3 meses. El 66,8 % de la población estudiada eran hombres y el 33,2 % mujeres, con una edad media de 63,3 años (rango de 18 a 94 años), y el 23,0 % de los pacientes tenían más de 75 años. El 58,4 % de la población estudiada correspondía a raza caucásica, el 36,2 % a raza mongoloide y el 4,0 % a raza negra (afroamericanos).
Al inicio del estudio, la TFG media fue de 37,3 ml/min/1,73 m², en el 21,2 % de los pacientes la TFG fue ≥ 45 ml/min/1,73 m², en el 44,3 % fue de 30 a < 45 ml/min/1,73 m² y en el 34,5 % fue < 30 ml/min/1,73 m², incluyendo 254 pacientes con TFG < 20 ml/min/1,73 m². La relación media albúmina/creatinina en orina (UACR) fue de 329 mg/g, en el 20,1 % de los pacientes la UACR fue < 30 mg/g, en el 28,2 % fue de 30 a ≤ 300 mg/g, en el 51,7 % fue > 300 mg/g y en el 41,1 % fue < 200 mg/g. Las principales causas de ERC fueron nefropatía diabética/enfermedad renal diabética (31 %), enfermedad glomerular (25 %), enfermedad hipertensiva/enfermedad reno-vascular (22 %) y otras/causas desconocidas (22 %).
Empagliflozina fue superior al placebo en la reducción del riesgo del punto final combinado primario, que incluyó la progresión de la enfermedad renal o muerte por enfermedad cardiovascular (véase la tabla 4). Además, empagliflozina redujo significativamente el riesgo de hospitalización por cualquier causa (primera y repetida).
Tabla 4
Efectos del tratamiento sobre el punto final combinado primario y los puntos finales secundarios clave incluidos en la prueba confirmatoria previamente definida, y sus componentes
| Placebo |
Empagliflozina, 10 mg |
|
| N |
3 305 |
3 304 |
| Tiempo hasta la primera manifestación de progresión de enfermedad renal (disminución persistente del FGeG ≥ 40 % desde la aleatorización, FGeG persistente < 10 ml/min/1,73 m², enfermedad renal en estadio terminal* (ERET) o muerte por enfermedad renal) o muerte por enfermedad cardiovascular, N (%) |
558 (16,9) |
432 (13,1) |
| Relación de riesgos frente al placebo (IC del 99,83 %) |
0,72 (0,59; 0,89) |
|
| Valor p para demostrar eficacia no inferior |
< 0,0001 |
|
| Disminución persistente del FGeG ≥ 40 % desde la aleatorización, N (%) |
474 (14,3) |
359 (10,9) |
| Relación de riesgos frente al placebo (IC del 95 %) |
0,70 (0,61; 0,81) |
|
| Valor p |
< 0,0001 |
|
| ERET* o FGeG persistente < 10 ml/min/1,73 m², N (%) |
221 (6,7) |
157 (4,8) |
| Relación de riesgos frente al placebo (IC del 95 %) |
0,69 (0,56; 0,84) |
|
| Valor p |
0,0003 |
|
| Muerte por enfermedad renal, N (%)** |
4 (0,1) |
4 (0,1) |
| Relación de riesgos frente al placebo (IC del 95 %) |
||
| Valor p |
||
| Muerte por enfermedad cardiovascular, N (%) |
69 (2,1) |
59 (1,8) |
| Relación de riesgos frente al placebo (IC del 95 %) |
0,84 (0,60; 1,19) |
|
| Valor p |
0,3366 |
|
| ERET* o muerte por enfermedad cardiovascular, N (%)# |
217 (6,6) |
163 (4,9) |
| Relación de riesgos frente al placebo (IC del 95 %) |
0,73 (0,59; 0,89) |
|
| Valor p |
0,0023 |
|
| Frecuencia de hospitalizaciones por cualquier causa (primera y repetidas), N de eventos |
1895 |
1611 |
| Relación de riesgos frente al placebo (IC del 99,03 %) |
0,86 (0,75; 0,98) |
|
| Valor p |
0,0025 |
SC – cardiovascular, TFGc – tasa de filtración glomerular calculada.
* La enfermedad renal en etapa terminal (ERET) se define como el inicio de diálisis de mantenimiento o trasplante renal.
** Hubo demasiados pocos eventos de muerte por enfermedad renal para calcular la razón de riesgos correspondiente.
Predefinido como uno de los dos criterios de interrupción en un análisis intermedio previamente planificado.
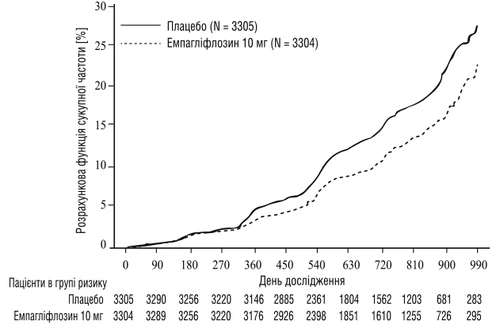
Fig. 4. Tiempo hasta el primer episodio de progresión de la enfermedad renal o eventos cardiovasculares fatales, función acumulativa estimada de morbilidad
Los resultados del punto final combinado primario fueron consistentes en cada uno de los subgrupos predefinidos, clasificados según el TFGc, la causa principal de la enfermedad renal, el estado de diabetes o el uso de inhibidores del SRA. Las ventajas del tratamiento fueron más evidentes en pacientes con niveles elevados de albúminuria.
Durante el tratamiento, el TFGc disminuyó más lentamente en el grupo tratado con empagliflozina en comparación con el grupo placebo. La empagliflozina ralentizó la tasa anual de disminución del TFGc en comparación con placebo en 1,37 ml/min/1,73 m²/año (IC 95 %: 1,16; 1,59), basado en los resultados de un análisis previo de todas las mediciones del TFGc realizadas desde la visita del segundo mes hasta la visita final de control. En pacientes que recibieron empagliflozina, se observó una disminución inicial del TFGc que regresó al nivel basal tras la interrupción del tratamiento, como se ha demostrado en varios estudios con empagliflozina, lo que confirma que los cambios hemodinámicos desempeñan un papel en el efecto agudo de empagliflozina sobre el TFGc.
Pacientes pediátricos
Diabetes mellitus tipo 2
La eficacia clínica y seguridad del empagliflozina (10 mg con posibilidad de aumentar la dosis a 25 mg) y linagliptina (5 mg), administrados una vez al día, se evaluaron en niños y adolescentes de 10 a 17 años con diabetes mellitus tipo 2 en un estudio controlado con placebo (DINAMO) de 26 semanas, con una extensión de seguridad hasta las 52 semanas.
El tratamiento basal como complemento a la dieta y al ejercicio físico incluía metformina (51 %), combinación de metformina e insulina (40,1 %), insulina (3,2 %) o ninguno de ellos (5,7 %).
El cambio medio ajustado en el HbA1c a las 26 semanas entre empagliflozina (N=52) y placebo (N=53), que fue de -0,84 %, fue clínica y estadísticamente significativo (IC 95 %: -1,50; -0,19; p = 0,0116).
Además, el tratamiento con empagliflozina en comparación con placebo produjo un cambio medio ajustado clínicamente significativo en la glucemia en ayunas de -35,2 mg/dl (IC 95 %: -58,6; -11,7) (-1,95 mmol/l (-3,25; -0,65)).
Insuficiencia cardíaca y enfermedad renal crónica
La Agencia Europea de Medicamentos ha eximido del requisito de presentar los resultados de los estudios con el medicamento de referencia en todas las subpoblaciones pediátricas con enfermedad renal crónica e insuficiencia cardíaca (consulte la información sobre uso pediátrico en la sección «Posología y forma de administración»).
Farmacocinética
Absorción
La farmacocinética de empagliflozina se ha descrito detalladamente en voluntarios sanos y pacientes con diabetes mellitus tipo 2. Tras la administración oral, la empagliflozina se absorbe rápidamente, alcanzando una concentración máxima en plasma con un tmax medio de 1,5 horas tras la ingesta. Posteriormente, la concentración en plasma disminuyó de forma bifásica, con una fase de distribución rápida y una fase terminal relativamente lenta. Los valores medios del área bajo la curva de concentración-tiempo (AUC) y de la concentración máxima (Cmax) en estado estacionario fueron de 1870 nmol/l·h y 259 nmol/l con una dosis de 10 mg de empagliflozina, y de 4740 nmol/l·h y 687 nmol/l con una dosis de 25 mg de empagliflozina, administrada una vez al día. La exposición sistémica a empagliflozina aumentó proporcionalmente con la dosis. Los parámetros farmacocinéticos de empagliflozina en estado estacionario tras una dosis única fueron similares, lo que indica una farmacocinética lineal respecto al tiempo. No se observaron diferencias clínicamente relevantes en la farmacocinética de empagliflozina entre voluntarios sanos y pacientes con diabetes mellitus tipo 2.
La administración de 25 mg de empagliflozina tras una comida rica en calorías y en grasas provocó una reducción moderada de su exposición: la AUC disminuyó aproximadamente un 16 % y la Cmax aproximadamente un 37 % en comparación con la administración en ayunas. Este efecto de la comida sobre la farmacocinética de empagliflozina no se considera clínicamente relevante. Empagliflozina puede administrarse independientemente de la ingesta de alimentos.
Distribución
El volumen de distribución en estado estacionario es de 73,8 l. Tras la administración oral de una solución de [14C]-empagliflozina a voluntarios sanos, la distribución a los eritrocitos fue aproximadamente del 37 % y la unión a proteínas plasmáticas del 86 %.
Biotransformación
No se detectaron metabolitos principales de empagliflozina en plasma humano. Los metabolitos más abundantes fueron tres conjugados de glucurónido (2-, 3- y 6-O-glucurónido). La exposición sistémica a cada metabolito fue inferior al 10 % de la exposición total al fármaco. Los estudios in vitro indican que la glucuronidación mediada por las UDP-glucuronosiltransferasas UGT2B7, UGT1A3, UGT1A8 y UGT1A9 es la vía principal del metabolismo de empagliflozina en humanos.
Eliminación
El periodo de semivida terminal de empagliflozina es de 12,4 horas y el aclaramiento oral aparente es de 10,6 l/h. La variabilidad interindividual y residual del aclaramiento oral de empagliflozina fue del 39,1 % y 35,8 %, respectivamente. Tras la administración una vez al día, las concentraciones plasmáticas de empagliflozina en estado estacionario se alcanzaron antes de la quinta dosis. De acuerdo con el periodo de semivida, se observó un acumulación de hasta un 22 % (en relación con la AUC en plasma) en estado estacionario. Tras la administración oral de una solución de [14C]-empagliflozina a voluntarios sanos, aproximadamente el 96 % de la sustancia radiomarcada se eliminó en heces (41 %) y orina (54 %). La mayor parte de la sustancia marcada se excretó sin cambios en heces y aproximadamente la mitad de la sustancia marcada se excretó sin cambios en orina.
Pacientes con características especiales
Pacientes con disfunción renal
En pacientes con alteraciones renales leves, moderadas o graves (TFGc < 30–< 90 ml/min/1,73 m²) y en pacientes con insuficiencia renal / enfermedad renal en etapa terminal (ERET), la AUC de empagliflozina aumentó aproximadamente en un 18 %, 20 %, 66 % y 48 %, respectivamente, en comparación con sujetos con función renal normal. Los niveles plasmáticos máximos de empagliflozina fueron similares en pacientes con alteraciones renales moderadas y en pacientes con insuficiencia renal / ERET en comparación con sujetos con función renal normal. Los niveles plasmáticos máximos de empagliflozina fueron aproximadamente un 20 % más altos en pacientes con alteraciones renales leves y graves en comparación con sujetos con función renal normal. Un análisis farmacocinético poblacional mostró que el aclaramiento oral aparente de empagliflozina disminuye con la reducción del TFGc, lo que conlleva un aumento del efecto del fármaco.
Pacientes con disfunción hepática
En pacientes con disfunción hepática leve, moderada y grave según la clasificación de Child-Pugh, la AUC de empagliflozina aumentó aproximadamente en un 23 %, 47 % y 75 %, y la Cmax en aproximadamente un 4 %, 23 % y 48 %, respectivamente, en comparación con sujetos con función hepática normal.
Índice de masa corporal
El índice de masa corporal no tuvo un impacto clínicamente relevante en la farmacocinética de empagliflozina. La AUC fue un 5,82 %, 10,4 % y 17,3 % menor en pacientes con IMC de 30, 35 y 45 kg/m², respectivamente, en comparación con pacientes con IMC de 25 kg/m².
Sexo
El sexo no tuvo un impacto clínicamente relevante en la farmacocinética de empagliflozina.
Raza
La AUC fue un 13,5 % mayor en pacientes de raza mongoloide con IMC de 25 kg/m² en comparación con pacientes de otras razas con IMC de 25 kg/m².
Pacientes de edad avanzada
La edad del paciente no tuvo un impacto clínicamente relevante en la farmacocinética de empagliflozina.
Pacientes pediátricos
Se han iniciado estudios clínicos con empagliflozina en niños de 10 a 18 años con diabetes mellitus tipo 2. Los datos actuales de farmacocinética y farmacodinámica disponibles son comparables a los de adultos.
Un estudio pediátrico de Fase III evaluó la farmacocinética y farmacodinámica (cambio en HbA1c respecto al valor basal) de 10 mg de empagliflozina, con posibilidad de aumentar la dosis a 25 mg, en niños y adolescentes de 10 a 17 años con diabetes mellitus tipo 2. La relación exposición-respuesta observada fue generalmente comparable entre adultos, niños y adolescentes. La administración oral de empagliflozina produjo una exposición dentro del rango observado en pacientes adultos.
Las concentraciones mínimas geométricas medias observadas y las concentraciones medias geométricas a las 1,5 horas tras la dosis en estado estacionario fueron de 26,6 nmol/l y 308 nmol/l con 10 mg de empagliflozina una vez al día, y de 67,0 nmol/l y 525 nmol/l con 25 mg de empagliflozina una vez al día.
Características clínicas
Indicaciones
Diabetes mellitus tipo 2
Gliaf está indicado en adultos y niños a partir de 10 años para el tratamiento de la diabetes mellitus tipo 2 insuficientemente controlada, como complemento de la dieta y del ejercicio físico:
- como monoterapia, cuando el uso de metformina no sea posible debido a la intolerancia al medicamento;
- en combinación con otros medicamentos para el tratamiento de la diabetes mellitus tipo 2.
Para conocer los resultados de los estudios sobre la terapia combinada, el efecto sobre el control glucémico, los eventos cardiovasculares y renales, así como las poblaciones estudiadas, véanse las secciones «Propiedades farmacodinámicas», «Interacción con otros medicamentos y otras formas de interacción» y «Uso y dosis».
Insuficiencia cardíaca
Gliaf está indicado en adultos para el tratamiento de la insuficiencia cardíaca crónica sintomática.
Enfermedad renal crónica
Gliaf está indicado en adultos para el tratamiento de la enfermedad renal crónica.
Contraindicaciones
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Interacción con otros medicamentos y otras formas de interacción
Interacciones farmacodinámicas
Diuréticos
Empagliflozina puede potenciar el efecto diurético de los diuréticos tiazídicos y de asa, aumentando el riesgo de deshidratación e hipotensión (véase la sección «Uso y dosis»).
Insulina y fármacos estimuladores de la secreción de insulina
La insulina y los fármacos estimuladores de la secreción de insulina, como las sulfonilureas, pueden aumentar el riesgo de hipoglucemia. Cuando se administren en combinación con empagliflozina, puede recomendarse una reducción de la dosis de insulina o del estimulador de la secreción de insulina para disminuir el riesgo de hipoglucemia (véanse las secciones «Uso y dosis» y «Reacciones adversas»).
Interacciones farmacocinéticas
Efecto de otros medicamentos sobre empagliflozina
Los datos in vitro indican que la vía principal del metabolismo de empagliflozina en humanos es la glucuronidación por las UDP-glucuronosiltransferasas UGT1A3, UGT1A8, UGT1A9 y UGT2B7. Empagliflozina es sustrato de los transportadores de absorción en humanos OAT3, OATP1B1 y OATP1B3, pero no de OAT1 ni de OCT2. Empagliflozina es sustrato de la glucoproteína P (P-gp) y de la proteína de resistencia al cáncer de mama (BCRP).
La administración concomitante de empagliflozina con probenecid, un inhibidor de las enzimas UDP-glucuronosiltransferasa (UGT) y del transportador OAT3, provocó un aumento del 26 % en la concentración máxima plasmática (Cmax) de empagliflozina y un incremento del 53 % en el AUC. Estos cambios no se consideraron clínicamente relevantes.
No se ha estudiado el efecto de la inducción de UGT (incluida la inducción por rifampicina o fenitoína) sobre empagliflozina. No se recomienda el tratamiento concomitante con inductores conocidos de las enzimas UGT debido al riesgo potencial de reducción de la eficacia. Si se debe administrar simultáneamente un inductor de las enzimas UGT, es conveniente realizar un seguimiento del control glucémico para evaluar la respuesta a Gliaf.
Un estudio de interacción con gemfibrozil, un inhibidor in vitro de los transportadores OAT3 y OATP1B1/1B3, mostró que tras la administración concomitante, la Cmax de empagliflozina aumentó un 15 % y el AUC un 59 %. Estos cambios no se consideraron clínicamente relevantes.
La inhibición de los transportadores OATP1B1/1B3 durante la administración concomitante con rifampicina provocó un aumento del 75 % en la Cmax y un incremento del 35 % en el AUC de empagliflozina. Estos cambios no se consideraron clínicamente relevantes.
El efecto de empagliflozina durante la administración concomitante con verapamilo, un inhibidor de P-gp, fue similar por separado. Esto indica que la inhibición de P-gp no tiene un efecto clínicamente relevante sobre empagliflozina.
Los estudios de interacción realizados con voluntarios sanos indican que la farmacocinética de empagliflozina no se ve afectada por la administración concomitante de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, warfarina, verapamilo, ramiprilo, simvastatina, torasemida e hidroclorotiazida.
Efecto de empagliflozina sobre otros medicamentos
Empagliflozina puede aumentar la excreción renal de litio y reducir los niveles séricos de litio. Tras iniciar el tratamiento con empagliflozina o cambiar la dosis, se debe monitorizar con mayor frecuencia la concentración sérica de litio. El paciente debe ser derivado al médico que prescribe los medicamentos con litio para el control de la concentración sérica de litio.
Según datos de estudios in vitro, empagliflozina no inhibe, no inactiva ni induce las isoformas CYP450. Empagliflozina no inhibe UGT1A1, UGT1A3, UGT1A8, UGT1A9 ni UGT2B7. Se considera poco probable que se produzcan interacciones medicamentosas con las principales isoformas CYP450 o UGT entre empagliflozina y otros fármacos sustratos de estas enzimas.
Empagliflozina no inhibe P-gp en dosis terapéuticas. Según estudios in vitro, es poco probable que empagliflozina provoque interacciones con principios activos que sean sustratos de P-gp. La administración concomitante de digoxina, un sustrato de P-gp, y empagliflozina provocó un aumento del 6 % en el AUC y del 14 % en la Cmax de digoxina. Estos cambios no se consideraron clínicamente relevantes.
Empagliflozina no inhibe los transportadores de absorción en humanos, como OAT3, OATP1B1 y OATP1B3, in vitro en concentraciones clínicamente relevantes en suero, por lo que se considera poco probable una interacción medicamentosa con sustratos de estos transportadores de absorción.
Los estudios de interacción realizados con voluntarios sanos indican que empagliflozina no tiene un efecto clínicamente relevante sobre la farmacocinética de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, simvastatina, warfarina, ramiprilo, digoxina, diuréticos y anticonceptivos orales.
Pacientes pediátricos
Los estudios de interacción se han realizado únicamente en adultos.
Características de uso
Empagliflozina no debe administrarse a pacientes con diabetes mellitus tipo 1 (ver más abajo «Cetoacidosis»).
Cetoacidosis
Se han notificado casos de cetoacidosis, incluyendo casos graves y fatales, en pacientes con diabetes mellitus que recibieron inhibidores del SGLT-2 (incluyendo empagliflozina). En varios casos, la cetoacidosis se presentó de forma atípica, con solo un aumento moderado de la glucosa en sangre (menos de 14 mmol/l (250 mg/dl)). No se conoce si el aumento de la dosis de empagliflozina influye en la probabilidad de aparición de cetoacidosis. Aunque la cetoacidosis es menos probable en pacientes sin diabetes mellitus, se han registrado casos también en estos pacientes.
Debe considerarse el riesgo de cetoacidosis ante síntomas inespecíficos como náuseas, vómitos, pérdida de apetito, dolor abdominal, sed excesiva, dificultad respiratoria, confusión mental, fatiga inusual o somnolencia. Ante la aparición de estos síntomas, los pacientes deben evaluarse inmediatamente para descartar cetoacidosis, independientemente del nivel de glucosa en sangre.
Si se sospecha o se diagnostica cetoacidosis en un paciente, debe suspenderse inmediatamente el tratamiento con empagliflozina.
En caso de hospitalización por procedimientos quirúrgicos mayores o ante la aparición de enfermedades agudas graves, el tratamiento con empagliflozina debe interrumpirse. En estos pacientes se recomienda el monitoreo de cetonas. La medición de cetonas en orina tiene prioridad sobre la determinación en sangre. El tratamiento con empagliflozina puede reiniciarse cuando los niveles de cetonas se normalicen y el estado del paciente se estabilice.
Antes de iniciar el tratamiento con empagliflozina, debe evaluarse en la historia clínica del paciente factores que puedan indicar predisposición a cetoacidosis.
Con el uso de empagliflozina se han observado cetoacidosis prolongada y glucosuria prolongada.
Debido al periodo de semivida de empagliflozina, la cetoacidosis puede persistir incluso después de la suspensión del medicamento. Factores independientes del uso de empagliflozina (como el déficit de insulina) pueden prolongar los episodios de cetoacidosis.
Los pacientes con alto riesgo de cetoacidosis incluyen aquellos con baja función de las células β (por ejemplo, diabetes mellitus tipo 2 con bajo nivel de péptido C, diabetes autoinmune latente en adultos o antecedentes de pancreatitis); pacientes con estados que limitan la ingesta de alimentos o deshidratación severa; pacientes con reducción de la dosis de insulina; pacientes con necesidad aumentada de insulina debido a enfermedad aguda, intervención quirúrgica o abuso de alcohol. En estos pacientes, los inhibidores del SGLT-2 deben usarse con precaución.
No se recomienda reanudar la terapia con inhibidores del SGLT-2 en pacientes que hayan presentado previamente cetoacidosis durante el tratamiento con inhibidores del SGLT-2, a menos que se haya identificado y eliminado claramente otro factor desencadenante.
Glyafor no debe administrarse a pacientes con diabetes mellitus tipo 1. Los datos del programa de estudios clínicos en pacientes con diabetes mellitus tipo 1 mostraron un aumento en la frecuencia de cetoacidosis con el uso de empagliflozina en dosis de 10 mg y 25 mg como complemento a la insulina, en comparación con placebo.
Insuficiencia renal
Debido a la experiencia limitada, no se recomienda iniciar el tratamiento con empagliflozina en pacientes con TFG < 20 ml/min/1,73 m².
En pacientes con TFG < 60 ml/min/1,73 m², la dosis diaria recomendada de empagliflozina es de 10 mg (ver sección «Posología y forma de administración»).
La eficacia de empagliflozina en la reducción de la glucosa depende de la función renal y disminuye en pacientes con TFG < 45 ml/min/1,73 m², y probablemente es inexistente en pacientes con TFG < 30 ml/min/1,73 m² (ver secciones «Posología y forma de administración», «Farmacodinamia» y «Farmacocinética»).
Monitoreo de la función renal
Se recomienda evaluar la función renal de la siguiente manera:
- antes de iniciar empagliflozina y periódicamente durante el tratamiento, al menos una vez al año (ver secciones «Posología y forma de administración», «Reacciones adversas», «Farmacodinamia» y «Farmacocinética»);
- antes de iniciar cualquier medicamento concomitante que pueda tener un efecto adverso sobre la función renal.
Uso en pacientes con riesgo de reducción del volumen de líquido extracelular
Debido al mecanismo de acción de los inhibidores del SGLT-2, la diuresis osmótica asociada a la glucosuria puede provocar una disminución leve de la presión arterial (ver sección «Farmacodinamia»). El medicamento debe usarse con precaución en pacientes en los que una reducción de la presión arterial inducida por empagliflozina pueda representar un riesgo, por ejemplo, pacientes con enfermedades cardiovasculares, pacientes que toman medicamentos antihipertensivos o con antecedentes de hipotensión, o pacientes de 75 años o más.
En caso de desarrollar estados que puedan provocar pérdida de líquidos (como enfermedades gastrointestinales), se recomienda un monitoreo cuidadoso del grado de reducción del volumen de líquido extracelular (por ejemplo, examen físico, medición de la presión arterial, pruebas de laboratorio, incluyendo el hematocrito) y la administración de electrolitos en pacientes que reciben empagliflozina. Debe considerarse la necesidad de suspender temporalmente el tratamiento con empagliflozina hasta que se resuelva la pérdida de líquidos.
Pacientes de edad avanzada
El efecto de empagliflozina sobre la excreción urinaria de glucosa está asociado con una diuresis osmótica, lo que puede afectar el estado de hidratación. Los pacientes de 75 años o más tienen un riesgo aumentado de reducción del volumen de líquido extracelular. La mayoría de estos pacientes que recibieron empagliflozina presentaron reacciones adversas relacionadas con la reducción del volumen de líquido extracelular en comparación con los pacientes del grupo placebo (ver sección «Reacciones adversas»). Por lo tanto, se requiere especial atención al volumen de líquido extracelular cuando se administren medicamentos concomitantes que puedan provocar su reducción (por ejemplo, diuréticos, inhibidores de la ECA).
Infecciones urinarias complicadas
Se han observado casos de complicaciones por infecciones del tracto urinario, incluyendo pielonefritis y urosepsis, en pacientes que recibieron empagliflozina (ver sección «Reacciones adversas»). Debe considerarse la necesidad de suspender temporalmente el tratamiento con empagliflozina en pacientes con infecciones urinarias complicadas.
Fascitis necrotizante del periné (gangrena de Fournier)
Se han notificado casos de fascitis necrotizante del periné (también conocida como gangrena de Fournier) en hombres y mujeres con diabetes mellitus que tomaron inhibidores del SGLT-2, incluyendo empagliflozina. La gangrena de Fournier es una infección rara, pero grave y potencialmente mortal, que requiere intervención quirúrgica inmediata y tratamiento con antibióticos.
Los pacientes deben informarse de la necesidad de buscar atención médica de inmediato si presentan síntomas como dolor, sensibilidad, eritema o edema en la zona de los órganos genitales o el periné, acompañados de fiebre o malestar general. Debe tenerse en cuenta que una infección urogenital o un absceso del periné puede preceder a la fascitis necrotizante. En caso de sospecha de gangrena de Fournier, debe suspenderse inmediatamente Glyafor y comenzar rápidamente el tratamiento (incluyendo antibióticos y cirugía para la zona afectada).
Amputaciones de extremidades inferiores
En un estudio con otro inhibidor del SGLT-2 se observó un aumento en los casos de amputaciones de extremidades inferiores (principalmente del dedo del pie). No se sabe si este efecto es común a toda la clase de medicamentos. Se debe aconsejar a los pacientes con diabetes mellitus que realicen cuidados preventivos de los pies.
Afectación hepática
Durante los estudios clínicos se han notificado casos de afectación hepática con el uso de empagliflozina. No se ha establecido una relación causal entre el uso de empagliflozina y la afectación hepática.
Aumento del hematocrito
Se observa aumento del hematocrito durante el tratamiento con empagliflozina (ver sección «Reacciones adversas»). Los pacientes con aumento marcado del hematocrito deben estar bajo vigilancia y evaluarse para detectar enfermedades hematológicas subyacentes.
Enfermedad renal crónica
Los pacientes con albuminuria pueden obtener mayor beneficio del tratamiento con empagliflozina.
Enfermedad infiltrativa o miocardiopatía de takotsubo
El uso de empagliflozina en pacientes con enfermedad infiltrativa o miocardiopatía de takotsubo no ha sido estudiado específicamente. Por lo tanto, la eficacia en estos pacientes no está establecida.
Análisis de orina
En pacientes que toman Glyafor, la prueba de glucosa en orina será positiva debido al mecanismo de acción del medicamento.
Interferencia con los niveles de 1,5-anhidroglicitol (1,5-AG)
No se recomienda el monitoreo del control glucémico mediante la medición de 1,5-AG, ya que los valores de 1,5-AG no son fiables para evaluar el control glucémico en pacientes que toman inhibidores del SGLT-2. Se recomienda el uso de métodos alternativos de control glucémico.
Lactosa
Este medicamento contiene lactosa. No debe administrarse a pacientes con intolerancia hereditaria rara a la galactosa, deficiencia total de lactasa o malabsorción de glucosa-galactosa.
Sodio
Una tableta de este medicamento contiene menos de 1 mmol de sodio (23 mg), es decir, este medicamento es prácticamente libre de sodio.
Uso durante el embarazo o la lactancia
Embarazo
No existen datos sobre el uso de empagliflozina en mujeres embarazadas. Los estudios en animales indican que empagliflozina atraviesa la placenta de forma muy limitada en las últimas etapas del embarazo, y no indican efectos perjudiciales directos o indirectos sobre el desarrollo embrionario temprano. Sin embargo, los estudios en animales mostraron efectos adversos sobre el desarrollo postnatal. Como medida de precaución, se recomienda evitar el uso de Glyafor durante el embarazo.
Período de lactancia
No se sabe si empagliflozina pasa a la leche materna humana. Glyafor no debe administrarse durante la lactancia.
Función reproductiva
No se han realizado estudios sobre el efecto de Glyafor sobre la fertilidad humana.
Capacidad para conducir y operar maquinaria
Glyafor tiene un efecto insignificante sobre la capacidad para conducir vehículos o manejar maquinaria. Sin embargo, los pacientes deben informarse sobre el riesgo de hipoglucemia si Glyafor se utiliza en combinación con medicamentos de sulfonilurea y/o insulina.
Vía de administración y dosis
Dosificación
Diabetes mellitus tipo 2
La dosis recomendada inicial es de 10 mg de empagliflozina una vez al día, como monoterapia o en combinación con otros medicamentos utilizados en el tratamiento de la diabetes mellitus. En pacientes que toleran bien la empagliflozina 10 mg una vez al día, con una TFG ≥ 60 ml/min/1,73 m² y que requieran un control glucémico más estricto, la dosis puede aumentarse hasta 25 mg una vez al día. La dosis máxima diaria es de 25 mg (ver información a continuación y la sección «Propiedades farmacéuticas»).
Insuficiencia cardíaca
La dosis recomendada es de 10 mg de empagliflozina una vez al día.
Enfermedad renal crónica
La dosis recomendada es de 10 mg de empagliflozina una vez al día.
Todas las indicaciones
Cuando empagliflozina se administra en combinación con sulfonilureas o insulina, se debe considerar la posibilidad de utilizar dosis más bajas de sulfonilurea o insulina con el fin de reducir el riesgo de hipoglucemia (ver secciones «Interacción con otros medicamentos y otras formas de interacción» y «Reacciones adversas»).
Si se olvida una dosis, se debe tomar tan pronto como el paciente lo recuerde, pero no se debe tomar una dosis doble en el mismo día.
Grupos de pacientes especiales
Pacientes con insuficiencia renal
Debido a la experiencia limitada, no se recomienda iniciar el tratamiento con empagliflozina en pacientes con TFG < 20 ml/min/1,73 m².
En pacientes con TFG < 60 ml/min/1,73 m², la dosis diaria de empagliflozina es de 10 mg.
En pacientes con diabetes mellitus tipo 2, la eficacia de empagliflozina para reducir los niveles de glucosa disminuye cuando la TFG es < 45 ml/min/1,73 m² y probablemente es ausente cuando la TFG es < 30 ml/min/1,73 m². Por lo tanto, si la TFG es inferior a 45 ml/min/1,73 m², debe considerarse el uso de un tratamiento hipoglucemiante adicional (ver secciones «Propiedades farmacéuticas», «Reacciones adversas», «Farmacodinamia» y «Farmacocinética»).
Pacientes con insuficiencia hepática
No se requiere ajuste de dosis en pacientes con insuficiencia hepática. El efecto de empagliflozina puede potenciarse en pacientes con insuficiencia hepática grave. La experiencia con empagliflozina en pacientes con insuficiencia hepática grave es limitada; por lo tanto, no se recomienda su uso en esta población (ver sección «Propiedades farmacológicas»).
Pacientes de edad avanzada
No se requiere ajuste de dosis basado en la edad del paciente. En pacientes de 75 años o más, debe considerarse el riesgo aumentado de hipovolemia (ver secciones «Propiedades farmacéuticas» y «Reacciones adversas»).
Vía de administración
Los comprimidos pueden tomarse con o sin alimentos, con agua, sin masticar.
Niños
La dosis inicial recomendada es de 10 mg de empagliflozina una vez al día. En pacientes que toleran bien la empagliflozina 10 mg una vez al día y que requieran un control glucémico adicional, la dosis puede aumentarse hasta 25 mg una vez al día (ver secciones «Farmacodinamia» y «Farmacocinética»). No existen datos sobre el uso en niños con TFG < 60 ml/min/1,73 m² ni en niños menores de 10 años.
No se ha estudiado la seguridad y eficacia de empagliflozina para el tratamiento de insuficiencia cardíaca o enfermedad renal crónica en niños (menores de 18 años). El medicamento no debe utilizarse en esta población.
Sobredosis
Síntomas
Durante estudios clínicos controlados, dosis únicas de hasta 800 mg de empagliflozina en voluntarios sanos y varias dosis diarias de hasta 100 mg de empagliflozina en pacientes con diabetes mellitus tipo 2 no provocaron efectos tóxicos. Empagliflozina aumentó la excreción urinaria de glucosa, lo que condujo a un aumento del volumen urinario. El aumento observado en el volumen urinario no fue dependiente de la dosis ni clínicamente significativo. No existe experiencia en el uso de dosis superiores a 800 mg en humanos.
Tratamiento
En caso de sobredosis, se debe iniciar el tratamiento según el estado clínico del paciente. No se ha estudiado la eliminación de empagliflozina mediante hemodiálisis.
Reacciones adversas
Diabetes mellitus tipo 2
La reacción adversa más frecuente fue la hipoglucemia cuando se utilizó empagliflozina junto con sulfonilureas o insulina.
Insuficiencia cardíaca
En los estudios EMPEROR participaron pacientes con insuficiencia cardíaca y fracción de eyección del ventrículo izquierdo reducida (N = 3 726) y con fracción de eyección conservada (N = 5 985), que recibieron empagliflozina a una dosis de 10 mg o placebo. Aproximadamente la mitad de estos pacientes tenían diabetes mellitus tipo 2. La reacción adversa más frecuente, según los datos combinados de los estudios EMPEROR-Reduced y EMPEROR-Preserved, fue la hipovolemia (empagliflozina 10 mg – 11,4 %, placebo – 9,7 %).
Enfermedad renal crónica
En el estudio EMPA-KIDNEY se incluyeron pacientes con enfermedad renal crónica (N = 6 609) que recibieron 10 mg de empagliflozina o placebo. Aproximadamente el 44 % de los pacientes tenían diabetes mellitus tipo 2.
Las reacciones adversas más frecuentes en el estudio EMPA-KIDNEY fueron la gota (empagliflozina – 7,0 %, placebo – 8,0 %) y la lesión renal aguda (empagliflozina – 2,8 %, placebo – 3,5 %), que se notificaron con mayor frecuencia en pacientes que recibieron placebo.
El perfil general de seguridad del medicamento Gliaf, en general, fue coherente con todas las indicaciones estudiadas.
Las reacciones adversas se clasifican por sistemas orgánicos (según MedDRA) y frecuencia de aparición. La frecuencia se define como muy frecuente (> 1/10), frecuente (> 1/100 – < 1/10), poco frecuente (> 1/1 000 – < 1/100), rara (> 1/10 000 – < 1/1 000), muy rara (< 1/10 000) o desconocida (no puede estimarse a partir de los datos disponibles).
Reacciones adversas (según datos de estudios controlados con placebo)
Infecciones e infestaciones
Frecuentes: candidiasis vaginal, vulvovaginitis, balanitis y otras infecciones genitalesa, infecciones del tracto urinario (incluyendo pielonefritis y urosepsis)a.
Raras: fascitis necrotizante perineal (gangrena de Fournier)*.
Alteraciones del metabolismo y de la nutrición
Muy frecuentes: hipoglucemia (cuando se utiliza con sulfonilureas o insulina)a.
Frecuentes: sensación de sed.
Poco frecuentes: cetoacidosis*.
Trastornos gastrointestinales
Frecuentes: estreñimiento.
Trastornos de la piel y del tejido subcutáneo
Frecuentes: prurito (generalizado), erupción cutánea.
Poco frecuentes: urticaria, angioedema.
Trastornos vasculares
Muy frecuentes: disminución del volumen de líquido intersticiala.
Trastornos renales y del tracto urinario
Frecuentes: aumento de la miccióna.
Poco frecuentes: disuria.
Raras: nefritis tubulointersticial.
Exploraciones
Frecuentes: aumento de los lípidos en sueroa.
Poco frecuentes: aumento del nivel de creatinina en sangre / disminución de la velocidad de filtración glomerulara, aumento del hematocritoa.
a Véase información adicional en las secciones siguientes.
* Véase la sección «Instrucciones especiales de uso».
Descripción de algunas reacciones adversas
Hipoglucemia
La frecuencia de hipoglucemia dependía del tratamiento de fondo durante los estudios correspondientes y fue similar cuando se utilizó empagliflozina y placebo como monoterapia, como complemento a metformina, como complemento a pioglitazona con o sin metformina, como complemento a linagliptina y metformina, así como cuando se utilizó la combinación de empagliflozina con metformina en pacientes que recibieron tratamiento por primera vez, en comparación con pacientes que previamente habían recibido empagliflozina y metformina como componentes individuales. Se observó un aumento en la frecuencia de hipoglucemia leve cuando se añadió empagliflozina o placebo a metformina y sulfonilurea (empagliflozina 10 mg – 16,1 %, empagliflozina 25 mg – 11,5 %, placebo – 8,4 %) y como complemento a insulina basal con o sin metformina y con o sin sulfonilurea (empagliflozina 10 mg – 19,5 %, empagliflozina 25 mg – 28,4 %, placebo – 20,6 % durante las primeras 18 semanas de tratamiento, cuando la dosis de insulina no podía ajustarse; empagliflozina 10 mg – 36,1 %, empagliflozina 25 mg – 36,1 %, placebo – 35,3 % en un estudio de 78 semanas), así como cuando se añadió a insulina en inyección premezclada con o sin metformina (empagliflozina 10 mg – 39,8 %, empagliflozina 25 mg – 41,3 %, placebo – 37,2 % durante las primeras 18 semanas de tratamiento, cuando la dosis de insulina no podía ajustarse; empagliflozina 10 mg – 51,1 %, empagliflozina 25 mg – 57,7 %, placebo – 58 % en un estudio de 52 semanas).
En los estudios EMPEROR sobre insuficiencia cardíaca, se observó una frecuencia similar de hipoglucemia al añadir el medicamento a sulfonilureas o insulina (empagliflozina 10 mg – 6,5 %, placebo – 6,7 %).
Hipoglucemia grave (hipoglucemia que requiere tratamiento)
No se observó un aumento en la frecuencia de hipoglucemia grave con empagliflozina en comparación con placebo como monoterapia, como complemento a metformina, como complemento a metformina y sulfonilurea, como complemento a pioglitazona con o sin metformina, como complemento a linagliptina y metformina, ni como complemento a la terapia estándar, ni cuando se utilizó la combinación de empagliflozina con metformina en pacientes que recibieron tratamiento por primera vez, en comparación con pacientes que previamente habían recibido empagliflozina y metformina como componentes individuales. Se observó un aumento en la frecuencia de hipoglucemia grave al añadir empagliflozina o placebo a insulina basal con o sin metformina y con o sin sulfonilurea (empagliflozina 10 mg – 0 %, empagliflozina 25 mg – 1,3 %, placebo – 0 % durante las primeras 18 semanas de tratamiento, cuando la dosis de insulina no podía ajustarse; empagliflozina 10 mg – 0 %, empagliflozina 25 mg – 1,3 %, placebo – 0 % en un estudio de 78 semanas) y como complemento a insulina en inyección premezclada con o sin metformina (empagliflozina 10 mg – 0,5 %, empagliflozina 25 mg – 0,5 %, placebo – 0,5 % durante las primeras 18 semanas de tratamiento, cuando la dosis de insulina no podía ajustarse; empagliflozina 10 mg – 1,6 %, empagliflozina 25 mg – 0,5 %, placebo – 1,6 % en un estudio de 52 semanas).
En los estudios EMPEROR sobre insuficiencia cardíaca, se observó una frecuencia similar de hipoglucemia en pacientes con diabetes mellitus al recibir empagliflozina o placebo como complemento a sulfonilureas o insulina (empagliflozina 10 mg – 2,2 %, placebo – 1,9 %).
Candidiasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales
La candidiasis vaginal, vulvovaginitis, balanitis y otras infecciones genitales ocurrieron con mayor frecuencia con empagliflozina (empagliflozina 10 mg – 4,0 %, empagliflozina 25 mg – 3,9 %) en comparación con placebo (1,0 %). Estas infecciones fueron más frecuentes en mujeres que recibieron empagliflozina en comparación con el grupo placebo; la diferencia en hombres fue menos pronunciada. Las infecciones genitales fueron de intensidad leve o moderada.
En los estudios EMPEROR sobre insuficiencia cardíaca, la frecuencia de estas infecciones fue más alta en pacientes con diabetes mellitus tipo 2 (empagliflozina 10 mg – 2,3 %, placebo – 0,8 %) que en pacientes sin diabetes (empagliflozina 10 mg – 1,7 %, placebo – 0,7 %), con empagliflozina en comparación con placebo.
Se notificaron casos de fimosis/adquisición de fimosis asociados con infecciones genitales, y en algunos casos fue necesaria la circuncisión.
Aumento de la micción
El aumento de la micción (incluyendo los términos predefinidos: polaquiuria, poliuria y nicturia) se observó con mayor frecuencia en pacientes que recibieron empagliflozina (empagliflozina 10 mg – 3,5 %, empagliflozina 25 mg – 3,3 %) en comparación con el grupo placebo (1,4 %). El aumento de la micción fue principalmente de intensidad leve o moderada. La frecuencia de nicturia fue similar con placebo y empagliflozina (< 1 %).
En los estudios EMPEROR sobre insuficiencia cardíaca, la diuresis aumentada se observó con frecuencia similar en pacientes que recibieron empagliflozina o placebo (empagliflozina 10 mg – 0,9 %, placebo – 0,5 %).
Infecciones del tracto urinario
La frecuencia general de infecciones del tracto urinario notificadas como eventos adversos fue similar en pacientes que recibieron empagliflozina 25 mg y en el grupo placebo (7,0 % y 7,2 %), y más alta con empagliflozina 10 mg (8,8 %). En comparación con placebo, las infecciones del tracto urinario ocurrieron con mayor frecuencia con empagliflozina en pacientes con antecedentes de infecciones urinarias crónicas o recurrentes. La intensidad (leve, moderada, grave) de las infecciones del tracto urinario fue similar en pacientes que recibieron empagliflozina y en el grupo placebo. Las infecciones del tracto urinario fueron más frecuentes en mujeres que recibieron empagliflozina en comparación con mujeres del grupo placebo; no hubo diferencia en hombres.
Disminución del volumen de líquido intersticial
La frecuencia general de disminución del volumen de líquido intersticial (incluyendo los términos predefinidos: disminución de la presión arterial [ambulatoria], disminución de la presión arterial sistólica, deshidratación, hipotensión arterial, hipovolemia, hipotensión ortostática y síncope) fue similar en pacientes que recibieron empagliflozina (empagliflozina 10 mg – 0,6 %, empagliflozina 25 mg – 0,4 %) y en el grupo placebo (0,3 %). La frecuencia de disminución del volumen de líquido intersticial fue mayor en pacientes de 75 años o más que recibieron empagliflozina 10 mg (2,3 %) o empagliflozina 25 mg (4,3 %), en comparación con aquellos que recibieron placebo (2,1 %).
Aumento del nivel de creatinina en sangre / disminución de la velocidad de filtración glomerular
La frecuencia general de aumento del nivel de creatinina en sangre y disminución de la velocidad de filtración glomerular fue similar con empagliflozina y placebo (aumento del nivel de creatinina en sangre: empagliflozina 10 mg – 0,6 %, empagliflozina 25 mg – 0,1 %, placebo – 0,5 %; disminución de la velocidad de filtración glomerular: empagliflozina 10 mg – 0,1 %, empagliflozina 25 mg – 0 %, placebo – 0,3 %).
En general, en pacientes que recibieron empagliflozina, el aumento inicial del nivel de creatinina y la disminución de la velocidad de filtración glomerular durante el tratamiento prolongado fueron temporales o reversibles tras la interrupción del tratamiento.
Durante el estudio EMPA-REG OUTCOME, en pacientes que recibieron empagliflozina se observó una disminución inicial de la tasa de filtración glomerular estimada (eGFR) (valor medio de 3 ml/min/1,73 m²). Posteriormente, la eGFR se mantuvo estable durante el tratamiento continuo. La eGFR media regresó a los valores iniciales tras la interrupción del tratamiento, lo que sugiere que alteraciones hemodinámicas leves podrían desempeñar un papel en estos cambios de la función renal. Este fenómeno también se observó en el estudio de insuficiencia cardíaca EMPEROR y en el estudio EMPA-KIDNEY.
Aumento de los niveles de lípidos en suero
El porcentaje medio de aumento desde el valor basal con empagliflozina 10 mg y 25 mg en comparación con placebo fue: colesterol total 4,9 % y 5,7 % frente a 3,5 %; colesterol HDL 3,3 % y 3,6 % frente a 0,4 %; colesterol LDL 9,5 % y 10,0 % frente a 7,5 %; triglicéridos 9,2 % y 9,9 % frente a 10,5 %.
Aumento del hematocrito
Los cambios medios del hematocrito desde el valor basal fueron 3,4 % y 3,6 % con empagliflozina 10 mg y 25 mg, en comparación con 0,1 % con placebo. En el estudio EMPA-REG OUTCOME, los valores de hematocrito regresaron a los niveles basales a los 30 días tras la interrupción del tratamiento.
Pacientes pediátricos
En el estudio DINAMO se trataron 157 niños de 10 años o más con diabetes mellitus tipo 2, de los cuales 52 recibieron empagliflozina, 52 recibieron linagliptina y 53 recibieron placebo (véase la sección «Farmacodinámica»). Durante la fase controlada con placebo, la reacción adversa más frecuente fue la hipoglucemia, con un índice general más alto en el grupo de empagliflozina en comparación con el grupo placebo (empagliflozina 10 mg y 25 mg combinados – 23,1 %, placebo – 9,4 %). Ninguno de estos casos fue grave ni requirió tratamiento.
En general, el perfil de seguridad en niños fue similar al observado en adultos con diabetes mellitus tipo 2.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite un seguimiento continuo de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar de todos los casos sospechosos de reacciones adversas y de falta de eficacia del medicamento a través del Sistema Automatizado de Información sobre Vigilancia Farmacológica en el enlace: https://aisf.dec.gov.ua.
Período de validez. 2 años.
Condiciones de conservación
El medicamento no requiere condiciones especiales de conservación.
Conservar en un lugar fuera del alcance de los niños.
Envase. 10 comprimidos por blíster; 3 blísteres por caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. FarmaPas S.A. / PharmaPath S.A.
Dirección del fabricante y del lugar de actividad
28 Octovriu 1, Agia Varvara, 123 51, Grecia / 28is Oktovriou 1, Agia Varvara, 123 51, Greece.