Gemlibra®
Ukraina
Spis treści
ULOTKA DOTYCZĄCA STOSOWANIA LEKU HEMLIBRA® (HEMLIBRA®)
Skład:
substancja czynna: emicizumab;
1 fiolka zawiera 12 mg/0,4 ml lub 30 mg/1 ml, lub 60 mg/0,4 ml, lub 105 mg/0,7 ml, lub 150 mg/1 ml emicizumabu;
substancje pomocnicze: L-histydyna, kwas L-asparginowy, L-arginina, poloksymer 188, woda do wstrzykiwań.
Postać leku. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: sterylne, przejrzyste ciecz o barwie od bezbarwnej do lekko żółtawej, bez konserwantów.
Grupa farmakoterapeutyczna.
Leki wpływające na układ krwionośny i krwiotwórczy. Środki przeciwkrwotoczne. Witamina K i inne środki hemostatyczne. Inne środki hemostatyczne do stosowania ogólnego. Emicizumab.
Kod ATC B02B X06.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Preparat Gemlibra® wiąże aktywowany czynnik IX i czynnik X, przywracając niedostateczną funkcję czynnika VIII aktywowanego, który jest niezbędny do skutecznego hemostazy.
Farmakokinetyka.
Emicizumab wykazał kinetykę proporcjonalną do dawki w zakresie dawek od 0,3 mg/kg (0,1 części zalecanej zatwierdzonej dawki początkowej) do 6 mg/kg po podaniu podskórnej. Po wielokrotnych podskórnych wstrzyknięciach emicizumabu w dawce załadunkowej 3 mg/kg raz w tygodniu przez pierwsze 4 tygodnie, średnia (± SD) minimalna stężenie emicizumabu w osoczu wyniosła 52,6 ± 13,6 μg/ml i została osiągnięta w 5. tygodniu. Informacje dotyczące stałych średnich (± SD) stężeń emicizumabu w osoczu w stanie stacjonarnym przy stosowaniu zalecanych dawek utrzymujących przedstawiono w tabeli 1.
Tabela 1
Średnie (± SD) stężenia w stanie stacjonarnym po podaniu dawki załadunkowej emicizumabu w zależności od trybu leczenia utrzymującego
| Parametry |
Dawka ujemna |
||
| 1,5 mg/kg masy ciała raz w tygodniu |
3 mg/kg masy ciała raz na 2 tygodnie |
6 mg/kg masy ciała raz na 4 tygodnie |
|
| Cmax, ss (μg/ml) |
55,1 ± 15,9 |
58,3 ± 16,4 |
67 ± 17,7 |
| AUCss,τ (μg/ml*dobę) |
376 ± 109 |
752 ± 218 |
1503 ± 437 |
| Cmin, ss (μg/ml) |
51,2 ± 15,2 |
46,9 ± 14,8 |
38,5 ± 14,2 |
| Stosunek Cmax/Cmin (μg/ml) |
1,08 ± 0,03 |
1,26 ± 0,12 |
1,85 ± 0,47 |
AUCss,τ – pole pod krzywą „stężenie w osoczu – czas” w stanie stacjonarnym w ciągu przedziału dawkowania (τ = 1, 2 lub 4 tygodnie); Cmax, ss – maksymalne stężenie leku w osoczu w stanie stacjonarnym; Cmin, ss – minimalne stężenie leku w osoczu w stanie stacjonarnym.
Absorpcja
Po podaniu podskórnym średnie (± SD) wartości okresu półprzemiany absorpcji wynosiły 1,6 ± 1 dzień. Biologiczna dostępność po podaniu podskórnym w dawce 1 mg/kg wahała się od 80,4% do 93,1%. Analogiczne profile farmakokinetyczne obserwowano po podaniu podskórnym w okolicy brzucha, ramienia oraz uda (patrz sekcja „Sposób stosowania i dawki”).
Rozkład
Średni pozorny objętościowy rozkładu (% współczynnik zmienności [%CV]) wynosił 10,4 l (26%).
Eliminacja
Średni pozorny klirens (%CV) wynosił 0,27 l/dobę (28,4%), a średni pozorny okres półtrwania eliminacji (± SD) – 26,9 ± 9,1 dnia.
Specjalne grupy populacji
Na farmakokinetykę emicizumabu nie wpływa wiek pacjenta (od 1 do 77 lat), rasa (europejska – 62,7%, mongolska – 22,9% i czarna – 8%), status inhibitorów (obecność inhibitorów u 50%), łagodne zaburzenia funkcji wątroby (określone jako poziom bilirubiny ogólnej od 1–1,5 razy wyższy niż górna granica normy (GGN), przy dowolnym poziomie aminotransferazy asparaginianowej (AST)), umiarkowane zaburzenia funkcji wątroby (określone jako poziom bilirubiny ogólnej od 1,5–3 razy wyższy niż GGN i przy dowolnym poziomie AST), jak również łagodne zaburzenia funkcji nerek (określone jako klirens kreatyniny 60–89 ml/min) i umiarkowane zaburzenia funkcji nerek (określone jako klirens kreatyniny 30–59 ml/min). Nie badano stosowania emicizumabu u pacjentów z ciężkimi zaburzeniami funkcji wątroby lub nerek.
U dzieci poniżej 6 miesiąca życia przewidywane stężenia emicizumabu były o 19–33% niższe niż u pacjentów starszych, szczególnie przy stosowaniu dawki utrzymującej 3 mg/kg masy ciała raz na 2 tygodnie lub 6 mg/kg masy ciała raz na 4 tygodnie.
Dzieci
Skuteczność i bezpieczeństwo stosowania leku Gemlibra® u dzieci zostały potwierdzone. Zastosowanie leku Gemlibra® u dzieci z hemofilią typu A wspierają wyniki dwóch randomizowanych badań (HAVEN 1 i HAVEN 3) oraz dwóch badań nieporównawczych (HAVEN 2 i HAVEN 4). Do wszystkich badań klinicznych włączono dzieci z jednej grupy wiekowej – 47 nastolatków w wieku od 12 do 18 lat. Tylko do badania HAVEN 2 włączono dzieci z różnych grup wiekowych – 55 dzieci w wieku od 2 do 12 lat oraz 5 dzieci w wieku od 1 miesiąca do 2 lat. Nie zaobserwowano różnic w skuteczności leku u dzieci z różnych grup wiekowych.
Minimalne stężenia emicizumabu w osoczu w stanie stacjonarnym były porównywalne u dorosłych i dzieci powyżej 6 miesiąca życia przy stosowaniu dawek równoważnych według masy ciała. U dzieci poniżej 6 miesiąca życia przewiduje się niższe stężenia emicizumabu.
Masa ciała. Pozorny klirens i objętość rozkładu emicizumabu wzrastały wraz ze zwiększającą się masą ciała (od 9 do 156 kg). Dawkowanie w miligramach na kilogram (mg/kg) zapewnia podobne ekspozycje na emicizumab w całym zakresie wartości masy ciała.
Immunogenność
Wykrywanie powstawania przeciwciał zależy w dużym stopniu od czułości i specyficzności testu. Różnice w metodach analiz utrudniają znaczące porównanie częstości powstawania przeciwciał do leku w opisanych poniżej badaniach z częstością w innych badaniach, w tym z badaniami leku Gemlibra® lub innych leków zawierających emicizumab.
Na podstawie danych z badań klinicznych leku Gemlibra® u 5,1% pacjentów (34/668) wykryto przeciwciała skierowane przeciwko emicizumabowi. Uczestnicy otrzymywali emicizumab przez średni zakres międzykwartylowy (IQR) wynoszący 103,1 (82,4–148,1) tygodnia. Próbki do testowania na obecność przeciwciał pobierano na początku leczenia oraz okresowo w trakcie całego badania.
Wpływ przeciwciał skierowanych przeciwko lekowi na farmakokinetykę
Próbki pozytywne na przeciwciała poddano dalszej analizie pod kątem obecności przeciwciał neutralizujących emicizumab za pomocą zmodyfikowanego testu chromogennego FVIII. Ogółem 668 pacjentów poddano badaniu pod kątem obecności przeciwciał skierowanych przeciwko emicizumabowi. U 5,1% pacjentów (34/668) wykryto przeciwciała przeciwko emicizumabowi, a u 2,7% pacjentów (18/668) pojawiły się przeciwciała przeciwko emicizumabowi, które były neutralizujące in vitro. Spośród tych 2,7% pacjentów (18/668) przeciwciała neutralizujące emicizumab nie wywierały klinicznie istotnego wpływu na farmakokinetykę leku Gemlibra® u 2,1% pacjentów (14/668), natomiast obniżenie stężenia emicizumabu w osoczu zaobserwowano u 0,6% pacjentów (4/668).
Wpływ przeciwciał skierowanych przeciwko lekowi na farmakodynamikę
U jednego pacjenta (1/668; 0,1%) pojawiły się przeciwciała neutralizujące emicizumab, co spowodowało obniżenie stężenia emicizumabu w osoczu i dalszą utratę skuteczności leku (objawiającą się jako krwawienie przebłonowe) po 5 tygodniach leczenia. Leczenie tego pacjenta lekiem Gemlibra® zostało wcześnie przerwane (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Note: The translation preserves all original formatting, including Markdown and HTML tags, and correctly renders "ГЕМЛІБРА®" as "Gemlibra®".
Właściwości kliniczne.
Wskazania.
Profilaktyka rutynowa mająca na celu zapobieganie lub zmniejszenie częstości epizodów krwawień u dorosłych oraz dzieci od momentu urodzenia, chorych na hemofilię typu A (wrodzony niedobór czynnika VIII krzepnięcia krwi) z tworzeniem się lub bez tworzenia się inhibitorów czynnika VIII.
Przeciwwskazania.
Podwyższona wrażliwość na emicizumab lub którykolwiek z substancji pomocniczych leku.
Interakcje z innymi lekami i inne rodzaje interakcji.
Badania interakcji z innymi lekami
Nie przeprowadzono żadnych badań interakcji leku Gemlibra® z innymi lekami.
Hiperkoagulacja przy jednoczesnym stosowaniu z AKPP
Doświadczenie kliniczne wskazuje na możliwość wystąpienia interakcji między lekiem Gemlibra® a aktywowanym kompleksem protrombinowym (AKPP) (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Wpływ leku na wyniki badań laboratoryjnych
Lek Gemlibra® przywraca aktywność kofaktora tenazy aktywnego czynnika VIII (aFVIII). Badania laboratoryjne oceniające wewnętrzny szlak aktywacji kaskady krzepnięcia (np. czas tromboplastyny częściowej aktywowanej – APTT) mierzą ogólny czas krzepnięcia, w tym czas potrzebny do aktywacji trombiną czynnika VIII do aFVIII, i wykazują nadmierne skrócenie czasu krzepnięcia podczas stosowania leku Gemlibra® bez konieczności aktywacji trombiną. Nadmierne skrócenie czasu krzepnięcia wewnętrznego będzie nadal wpływać na wyniki wszystkich analiz jednoczynnikowych opartych na oznaczeniu APTT, w tym jednoetapowego oznaczenia aktywności FVIII; natomiast analizy jednoczynnikowe wykorzystujące metodę chromatyczną lub immunologiczną nie są wpływowane przez lek Gemlibra® i mogą być stosowane do monitorowania wskaźników krzepnięcia podczas leczenia, z ważnymi uwagami dotyczącymi chromatycznego oznaczenia aktywności FVIII, jak opisano poniżej.
Testy chromatycznego oznaczenia aktywności FVIII mogą być wykonywane z wykorzystaniem ludzkich lub bydlęcych białek krzepnięcia. Testy zawierające ludzkie czynniki krzepnięcia są wrażliwe na lek Gemlibra®, ale mogą zawyżać ocenę klinicznego potencjału hemostatycznego leku Gemlibra®. Z kolei testy oparte na bydlęcych czynnikach krzepnięcia są niewrażliwe na lek Gemlibra® (aktywność nie jest oznaczana) i mogą być stosowane do monitorowania aktywności endogennej lub podanego dożylnie FVIII, a także do wykrywania inhibitorów przeciwko FVIII.
Lek Gemlibra® nie traci aktywności w obecności inhibitorów FVIII, co może prowadzić do fałszywie ujemnego wyniku testu Bethesda na funkcjonalne hamowanie FVIII metodą clotting. W zamian może być stosowany chromatyczny test Bethesda z wykorzystaniem bydlęcego FVIII, który jest niewrażliwy na lek Gemlibra®.
Dzięki długiemu okresowi półtrwania lek Gemlibra® może wpływać na parametry krzepnięcia krwi nawet do 6 miesięcy po podaniu ostatniej dawki (patrz sekcja „Właściwości farmakologiczne”).
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania.
| OSTRZEŻENIE: TROMBOTYCZNA MIKROANGIOPATIA I ZAKRZEPICA W badaniach klinicznych opisano przypadki trombotycznej mikroangiopatii i zdarzeń zakrzepowo-zatorowych u pacjentów otrzymujących dawki profilaktyczne leku Gemlibra®, gdy stosowano stężony kompleks protrombinowy aktywowany (PCCa) przez 24 godziny lub dłużej przy średniej skumulowanej dawce > 100 IU/kg/24 godz. Należy monitorować wystąpienie objawów trombotycznej mikroangiopatii i zdarzeń zakrzepowo-zatorowych podczas stosowania PCCa. Należy przerwać podawanie PCCa i odłożyć podanie kolejnej dawki leku Gemlibra® w przypadku wystąpienia objawów. |
Trombotyczna mikroangiopatia związana z leczeniem Gemlibra® i AKPCC
Zgłoszono przypadki trombotycznej mikroangiopatii (TMA) u pacjentów otrzymujących dawki profilaktyczne Gemlibra®, gdy stężenie aktywowanego kompleksu protrombinowego (AKPCC) stosowano przez 24 godziny lub dłużej w średniej skumulowanej dawce >100 IU/kg/24 h. W badaniach klinicznych o rozwoju trombotycznej mikroangiopatii zgłaszano u 0,8% pacjentów (3 na 391) oraz u 8,1% pacjentów (3 na 37), którzy otrzymali co najmniej jedną dawkę AKPCC. W badaniach ADAMTS13 u pacjentów obserwowano trombocytopenię, mikroangiopatyczną anemię hemolityczną oraz ostre uszkodzenie nerek bez wyraźnych objawów niedoczynności.
Objawy poprawy stanu obserwowano w ciągu jednego tygodnia od momentu przerwania stosowania AKPCC. U jednego pacjenta kontynuowano leczenie Gemlibra® po ustąpieniu objawów TMA.
U pacjentów otrzymujących leczenie profilaktyczne Gemlibra® należy dokładnie ocenić wszystkie korzyści i ryzyko, jeśli konieczne jest zastosowanie AKPCC. Ze względu na długi okres półtrwania Gemlibra® ryzyko interakcji z AKPCC może utrzymywać się przez okres do 6 miesięcy po podaniu ostatniej dawki. Podczas stosowania AKPCC należy monitorować rozwój TMA. Należy natychmiast przerwać leczenie AKPCC i wstrzymać profilaktykę Gemlibra® w przypadku pojawienia się objawów klinicznych i/lub laboratoryjnych wskazujących na rozwój TMA oraz rozpocząć leczenie zgodnie z wskazaniami klinicznymi. Należy rozważyć możliwe korzyści i ryzyko kontynuacji profilaktyki Gemlibra® po ustąpieniu TMA w każdym indywidualnym przypadku.
Tromboembolia związana z leczeniem Gemlibra® i AKPCC
Zgłoszono przypadki zakrzepicy u pacjentów otrzymujących dawki profilaktyczne Gemlibra®, gdy stężenie aktywowanego kompleksu protrombinowego (AKPCC) stosowano przez 24 godziny lub dłużej w średniej skumulowanej dawce >100 IU/kg/24 h. W badaniach klinicznych o przypadkach zakrzepicy zgłaszano u 0,5% pacjentów (2 na 391) oraz u 5,4% pacjentów (2 na 37), którzy otrzymali co najmniej jedną dawkę AKPCC.
Żaden przypadek zakrzepicy nie wymagał leczenia antykoagulantami. Objawy poprawy stanu lub wyzdrowienia obserwowano w ciągu jednego tygodnia od momentu przerwania stosowania AKPCC. U jednego pacjenta kontynuowano leczenie Gemlibra® po ustąpieniu objawów zakrzepicy.
U pacjentów otrzymujących leczenie profilaktyczne Gemlibra® należy dokładnie ocenić wszystkie korzyści i ryzyko, jeśli konieczne jest zastosowanie AKPCC. Ze względu na długi okres półtrwania Gemlibra® ryzyko interakcji z AKPCC może utrzymywać się przez okres do 6 miesięcy po podaniu ostatniej dawki. Podczas stosowania AKPCC należy monitorować rozwój tromboembolii. Należy natychmiast przerwać leczenie AKPCC i wstrzymać profilaktykę Gemlibra® w przypadku pojawienia się objawów klinicznych, wyników badań obrazowych lub oznak laboratoryjnych wskazujących na rozwój tromboembolii oraz rozpocząć leczenie zgodnie z wskazaniami klinicznymi. Należy rozważyć możliwe korzyści i ryzyko kontynuacji profilaktyki Gemlibra® po zakończeniu zdarzenia zakrzepowego w każdym indywidualnym przypadku.
Immunogenność
Leczenie Gemlibra® może wywoływać powstawanie przeciwciał przeciw lekowi. O przeciwciałach przeciw emicyzumabowi zgłaszano u 5,1% pacjentów (34/668), którzy otrzymywali leczenie Gemlibra® w badaniach klinicznych. U większości pacjentów z przeciwciałami przeciw emicyzumabowi nie obserwowano zmian stężenia Gemlibra® we krwi ani zwiększenia liczby krwawień; jednak w rzadkich przypadkach (częstość <1%) obecność przeciwciał neutralizujących obniżających stężenie we krwi może wiązać się z utratą skuteczności (patrz sekcja „Farmakokinetyka”).
Należy obserwować pacjentów pod kątem pojawienia się objawów klinicznych utraty skuteczności leczenia (czyli zwiększenia liczby krwawień przebijających), a w przypadku ich wystąpienia należy natychmiast ocenić przyczynę i rozważyć zmianę terapii w przypadku podejrzenia przeciwciał neutralizujących przeciw emicyzumabowi.
Wpływ na parametry koagulogramu
Gemlibra® wpływa na wyniki badań laboratoryjnych dotyczących wewnętrznego szlaku aktywacji kaskady krzepnięcia, w tym czas krzepnięcia (APTT), częściowy czas tromboplastynowy aktywowany (APTT) oraz wszystkie testy oparte na oznaczaniu APTT, w tym jednoetapowe oznaczanie aktywności czynnika VIII (FVIII) (tabela 2). Z tego powodu nie należy stosować badań laboratoryjnych wewnętrznego szlaku aktywacji kaskady krzepnięcia metodą clottingową u pacjentów leczonych Gemlibra® do monitorowania jej aktywności, określania dawki terapii zastępczej ani w celu antykoagulacji lub oznaczania miana inhibitorów FVIII (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”). Badania laboratoryjne, na które wpływa lub nie wpływa Gemlibra®, przedstawiono w tabeli 2.
Tabela 2
Wyniki testów koagulacyjnych, na które wpływa lub nie wpływa Gemlibra®
| Wyniki, na które wpływa lek Gemlibra® |
Wyniki, na które nie wpływa lek Gemlibra® |
| Aktywowany częściowy czas tromboplastynowy (APTT) Test Bethesda (metodą koagulacyjną) do oznaczania miana inhibitorów FVIII Jednostopniowy, jednofaktorowy test oparty na APTT Oporna na aktywowaną białek C (APC-R) oparta na APTT Czas krzepnięcia aktywowany (ACT) |
Test Bethesda (z wołowym substratem chromogennym) do oznaczania miana inhibitorów FVIII Czas trombinowy (TT) Jednostopniowy, jednofaktorowy test oparty na czasie protrombinowym (PT) Jednofaktorowy test chromogenny innych czynników oprócz FVIII* Badania immunologiczne (np. ELISA, metody turbdynometryczne) Testy genetyczne określające czynniki krzepnięcia (np. czynnik V Leiden, protrombina 20210) |
* Ważne informacje dotyczące oznaczania aktywności FVIII metodą chromogeniczną znajdują się w sekcji „Interakcje z innymi lekami i inne rodzaje interakcji”.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
Brak dostępnych danych dotyczących ryzyka ciężkich wad wrodzonych lub poronień związanych ze stosowaniem leku Gemlibra® u ciężarnych kobiet. Badania funkcji rozrodczej na zwierzętach z zastosowaniem emicizumabu nie były prowadzone. Nie wiadomo, czy lek Gemlibra® może powodować szkodę płodowi lub wpływać na płodność u kobiet w ciąży. Lek Gemlibra® można stosować w czasie ciąży wyłącznie wtedy, gdy potencjalna korzyść dla kobiety przewyższa potencjalne ryzyko dla płodu.
W każdej ciąży istnieje tło ryzyka wad wrodzonych, utraty płodu lub innych niekorzystnych skutków. Szacowane tło ryzyka ciężkich wad wrodzonych lub poronień w docelowych populacjach jest nieznane. W ogólnej populacji Stanów Zjednoczonych szacowane ryzyko ciężkich wad wrodzonych lub poronień w czasie potwierdzonych klinicznie ciąży wynosi odpowiednio 2–4% i 15–20%.
Karmienie piersią
Brak informacji dotyczących wydzielania emicizumabu w mleku matki, możliwego wpływu na niemowlę karmione piersią ani wpływu na wydzielanie mleka. Wiadomo, że ludzki IgG występuje w ludzkim mleku matki. Korzyści karmienia piersią dla rozwoju i zdrowia należy porównać z kliniczną potrzebą matki stosowania leku Gemlibra®, możliwym ryzykiem wystąpienia działań niepożądanych u niemowlęcia karmionego piersią oraz ogólnym stanem zdrowia matki.
Antykoncepcja
Kobietom w wieku rozrodczym należy stosować środki antykoncepcyjne w czasie leczenia lekiem Gemlibra®.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Brak danych sugerujących, że leczenie lekiem Gemlibra® zwiększa ryzyko działań niepożądanych wpływających na zdolność prowadzenia pojazdów lub obsługiwanie maszyn.
Sposób stosowania i dawki.
Wyłącznie do wstrzykiwania podskórnie.
Zalecana dawka ładunkowa wynosi 3 mg/kg masy ciała, podawana podskórnie raz w tygodniu przez pierwsze 4 tygodnie, a następnie lek stosuje się w dawce utrzymaniu:
- 1,5 mg/kg masy ciała raz w tygodniu lub
- 3 mg/kg masy ciała raz na dwa tygodnie lub
- 6 mg/kg masy ciała raz na cztery tygodnie.
Wybór dawki utrzymania powinien opierać się na preferencjach lekarza klinicznego, biorąc pod uwagę tryby dawkowania, które mogą sprzyjać poprawie przestrzegania reżimu leczenia przez pacjenta.
Profilaktyczne stosowanie leków działających obejściowo należy przerwać jeden dzień przed rozpoczęciem stosowania leku Gemlibra® w celu profilaktyki.
Profilaktyczne stosowanie czynnika VIII (FVIII) może być kontynuowane przez pierwszy tydzień stosowania leku Gemlibra® w celu profilaktyki.
Pominięta dawka
Jeśli pominięto dawkę leku Gemlibra®, należy ją podać tak szybko, jak to możliwe, a następnie kontynuować normalny harmonogram dawkowania. Nie należy podawać dwóch dawek w tym samym dniu w celu nadrobienia pominiętej dawki.
Zalecenia specjalne dotyczące dawkowania
Dzieci
Dostosowania dawki u dzieci nie zaleca się. Brak danych z badań przeprowadzonych u dzieci poniżej 1 roku życia.
Pacjenci w wieku podeszłym
Badania kliniczne leku Gemlibra® obejmowały niewystarczającą liczbę pacjentów w wieku 65 lat i starszych, aby można było ustalić, czy odpowiadają oni na leczenie inaczej niż młodsze osoby. Inne doniesienia dotyczące doświadczeń klinicznych nie wykazały różnic w odpowiedzi na leczenie między pacjentami starszymi i młodszych.
Przygotowanie i podawanie
Lek Gemlibra® przeznaczony jest do stosowania wyłącznie pod nadzorem lekarza. Po odpowiednim przeszkoleniu w technice wykonywania wstrzykiwań podskórnych pacjent może samodzielnie wykonać zastrzyk lub może go wykonać osoba opiekująca się pacjentem, jeśli lekarz uzna to za możliwe. Samodzielne podawanie nie jest zalecane dzieciom poniżej 7. roku życia.
Poniżej przedstawiono szczegółowe instrukcje dotyczące przygotowania i stosowania leku Gemlibra® (patrz sekcja „Wskazówki dotyczące stosowania”).
Przed zastosowaniem należy wizualnie sprawdzić lek Gemlibra® pod kątem obecności cząstek stałych lub zmiany koloru. Gemlibra® do wstrzykiwania podskórnie to roztwór od bezbarwnego do lekko żółtawego. Nie należy stosować leku, jeśli widoczne są cząstki stałe lub zmiana koloru.
Do pobrania leku Gemlibra® z fiolki i jego podania podskórnie potrzebne są strzykawka, igła transferowa z filtrem oraz igła do wstrzykiwań.
Zobacz poniżej sekcję „Wskazówki dotyczące stosowania” w celu uzyskania instrukcji dotyczących postępowania z lekiem Gemlibra® w przypadku stosowania kilku fiolki w połączeniu.
Nie należy mieszać w jednym zastrzyku leku Gemlibra® z fiolki zawierających ten lek w różnych stężeniach (tj. 30 mg/ml i 150 mg/ml).
Zobacz poniżej kryteria wyboru zalecanego wariantu produktu medycznego. Dawki leku Gemlibra® do 1 ml należy podawać za pomocą strzykawki 1 ml. Można użyć strzykawki 1 ml spełniającej następujące kryteria: przezroczysta strzykawka z polipropylenu lub poliwęglanu z końcówką Luer-Lock (jeśli brak, można użyć strzykawki z końcówką Luer Slip), podziałka co 0,01 ml, sterylna, przeznaczona wyłącznie do wstrzykiwań, jednorazowego użytku, bez lateksu, apirogenne.
Zastrzyki leku Gemlibra® o objętości od 1 ml do 2 ml należy wykonywać za pomocą strzykawek 2 ml lub 3 ml spełniających następujące kryteria: przezroczysta strzykawka z polipropylenu lub poliwęglanu z końcówką Luer-Lock (jeśli brak, można użyć strzykawki z końcówką Luer Slip), podziałka co 0,1 ml, sterylna, przeznaczona wyłącznie do wstrzykiwań, jednorazowego użytku, bez lateksu, apirogenne.
Należy użyć igły transferowej z filtrem spełniającej następujące kryteria: igła ze stali nierdzewnej z połączeniem typu Luer-Lock (jeśli brak, można użyć igły z połączeniem typu Luer Slip), sterylna, rozmiar 18G, długość od 1 do 1½ cala, zakończona skośnym jednostronnym ostrzem lub tępym (lub półtłustym) końcem, jednorazowego użytku, bez lateksu, z filtrem 5 mikrometrów, apirogenne.
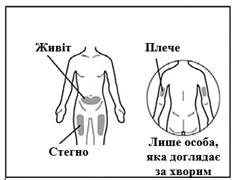
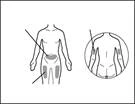
Można użyć igły do wstrzykiwań spełniającej następujące kryteria: igła ze stali nierdzewnej z połączeniem typu Luer-Lock (jeśli brak, można użyć igły z połączeniem typu Luer Slip), sterylna, rozmiar 26G (dopuszczalny zakres: 25–27G), długość głównie 3/8 cala lub maksymalnie ½ cala, jednorazowego użytku, bez lateksu, apirogenne, wyposażona w zabezpieczenie. Każdą nową dawkę leku należy podawać w inne miejsce anatomiczne (zewnętrzna powierzchnia ramienia, uda lub dowolny kwadrant brzucha), różne niż miejsce poprzedniego zastrzyku. Nie należy wykonywać zastrzyku w znamiona, blizny ani w obszary wrażliwe, z sinicami, z oznakami zaczerwienienia, grube lub uszkodzone obszary skóry. Wstrzykiwanie leku Gemlibra® w zewnętrzną powierzchnię ramienia powinno być wykonywane wyłącznie przez osobę opiekującą się pacjentem lub przez personel medyczny.
Należy zutylizować każdą pozostałą ilość niewykorzystanego leku Gemlibra® pozostawioną w jednorazowej fiolce.
Przechowywanie leku Gemlibra®:
- Przechowywać lek Gemlibra® w temperaturze od 2 do 8 °C w oryginalnym opakowaniu w celu ochrony przed światłem. Nie zamrażać. Nie wstrząsać.
- Przed zastrzykiem zamknięte fiolki z lekiem Gemlibra® można wyjąć z lodówki, a następnie ponownie odstawić do lodówki. Temperatura i całkowity czas przechowywania leku Gemlibra® poza lodówką nie powinny przekraczać 30 °C i 7 dni (przy temperaturze do 30 °C).
- Po pobraniu leku Gemlibra® z fiolki pozostałą zawartość należy zutylizować, jeśli lek Gemlibra® nie został natychmiast wykorzystany.
Wskazówki dotyczące stosowania
Sprawdzenie leku Gemlibra® i komponentów zestawu dostawy
- Zebrać wszystkie poniżej wymienione komponenty do przygotowania i wykonania zastrzyku.
- Sprawdzić datę ważności na pudełku kartonowym, etykiecie fiolki i zestawie dostawy, jak wskazano poniżej. Nie należy stosować leku ani wymienionych poniżej komponentów, jeśli ta data już upłynęła.
- Nie stosować leku, jeśli:
-
lek jest mętny, nieprzezroczysty lub ma intensywny kolor;
-
lek zawiera cząstki;
-
brakuje nakrętki nad przeciwną stroną.
- Sprawdzić komponenty zestawu dostawy pod kątem uszkodzeń. Nie należy ich używać, jeśli są uszkodzone lub upadły na podłogę.
- Umieścić komponenty zestawu dostawy na czystej, dobrze oświetlonej, płaskiej powierzchni roboczej.
| Skład opakowania obejmuje : |
||
|
|
|
|
|
|
| Skład opakowania nie obejmuje: |
||
Uwaga: jeśli do podania przepisanej dawki konieczne jest użycie zawartości więcej niż jednej fiolki, należy użyć nowego wacika alkoholowego do każdej fiolki.
|
|
|
|
||
Uwaga: jeśli do podania przepisanej dawki
|
||
|
||
|
|
|
| Przygotowanie fiolki i miejsca wstrzyknięcia
|
|
|
|
||





Przygotowanie strzykawki do wstrzyknięcia
- Lek Gemlibra® nie należy przechowywać w strzykawce.
- Lek Gemlibra® w strzykawce należy natychmiast podać podskórnie.
Ważne informacje dotyczące postępowania po wstrzyknięciu:
- Nie należy masować miejsca wstrzyknięcia po jego wykonaniu.
- W przypadku pojawienia się kropli krwi w miejscu wstrzyknięcia, można przycisnąć je sterylną watą lub gazą przez co najmniej 10 sekund, aż do ustania krwawienia.
- Jeśli pojawi się siniak (niewielki obszar krwawienia pod skórą), można również nałożyć na miejsce wstrzyknięcia worek z lodem, delikatnie przyciskając. Jeśli krwawienie nie ustaje, należy skontaktować się z lekarzem.
Unieszkodliwianie używanych fiolki Gemlibra®, igieł i strzykawek
Ważne: zawsze należy przechowywać pojemnik na przedmioty ostry w miejscu niedostępnym dla dzieci.
- Natychmiast po użyciu umieścić używane igły i strzykawki w pojemniku na przedmioty ostry. Nie wyrzucać żadnych odsłoniętych igieł i strzykawek wraz z odpadami komunalnymi.
- Jeśli nie posiada się pojemnika na przedmioty ostry, można użyć pojemnika do użytku domowego, który:
- wykonany jest z wytrzymałego tworzywa sztucznego;
- można szczelnie zamknąć przekłucioodporną pokrywą, aby zapobiec przypadkowemu wydostaniu się ostrych przedmiotów na zewnątrz;
- jest stabilny i może stać podczas użytkowania;
- nie wycieka;
- posiada odpowiednie oznakowanie wskazujące na obecność niebezpiecznych odpadów wewnątrz pojemnika.
- Gdy pojemnik na przedmioty ostry jest wypełniony w przybliżeniu do pełna, należy postępować zgodnie z lokalnymi wytycznymi dotyczącymi właściwego sposobu unieszkodliwiania pojemnika na przedmioty ostry.
- Nie wyrzucać używanego pojemnika na przedmioty ostry wraz z odpadami komunalnymi, chyba że lokalne wytyczne zezwalają na takie działanie. Nie należy ponownie używać pojemnika na przedmioty ostry.
Instrukcja krok po kroku wykonania wstrzyknięcia leku Gemlibra®
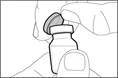
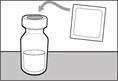
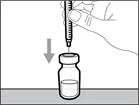
| Krok 1. Ściągnąć pokrywkę z fiolki i oczyścić górną część korka |
||
|
|
|
|
|
|
|
|
|
|
|
|
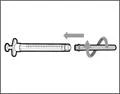
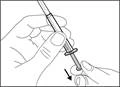
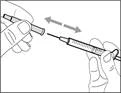
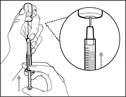
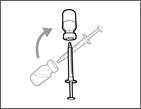
- Trzymaj strzykawkę za cylinder tak, aby igła przelewowa była skierowana do góry. * Ostrożnie odepnij nakrywkę igły przelewowej od siebie (patrz rys. 16). Nie wyrzucaj nakrywki. Połóż nakrywkę igły przelewowej na czystą, płaską powierzchnię. Po pobraniu roztworu leku należy ponownie założyć ją na igłę przelewową. * Nie dotykaj końcówki igły i nie kładź jej na powierzchnię po zdjęciu nakrywki. || Krok 4. Wprowadzić powietrze do fiolki | | |
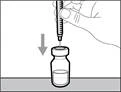
| * Trzymaj fiolkę na płaskiej, stabilnej powierzchni i wsuń igłę przelewową ze strzykawką w środek przekładki fiolki w kierunku w dół (patrz rys. 17). |
|
|
|
||||||
| Krok 5. Przenieść lek Gemlibra® do strzykawki
|
|||||||
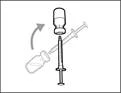
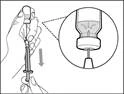
Ważne: jeśli przepisana dawka przekracza ilość leku Gemlibra® w fiolce, należy nabrać cały lek Gemlibra® i przejść do sekcji „Łączenie fiolki”.
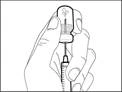
| Krok 6. Usunięcie pęcherzyków powietrza |
|
|
|
Uwaga: przed przejściem do następnego kroku należy upewnić się, że w strzykawce znajduje się wystarczająca ilość leku Gemlibra® na pełną dawkę. Jeśli nie uda się założyć całego leku Gemlibra®, należy umieścić fiolkę w pozycji pionowej, aby pobrać resztę. |
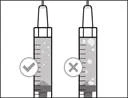
| Nie używaj igły transferowej do wstrzykiwania leku Gemlibra®, ponieważ może to spowodować uraz, taki jak ból i krwawienie. |
||||
|
|
|
|||
| Krok 8. Oczyść miejsce wstrzyknięcia |
||||
|
|
|
|||
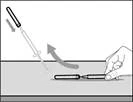
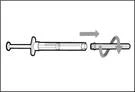
| Krok 10. Przyłącz igłę do strzykawki do wstrzykiwań |
||||
|
|
|
|||
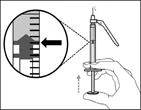
| Krok 13. Ustaw tłoczek na oznaczeniu odpowiadającym przepisanej dawce |
||||
|
|
|
|||
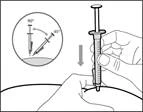
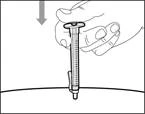
| Krok 14. Wstrzyknięcie podskórne (pod skórę) |
||||
|
|
|
|||
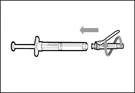
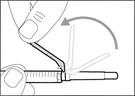
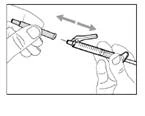
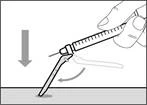
- Przesuń ochronną membranę do przodu o 90º w kierunku od cylindra strzykawki. * Trzymając strzykawkę jedną ręką, naciśnij membranę ochronną w dół na szeroką powierzchnię szybkim, pewnym ruchem, aż do usłyszenia kliknięcia (patrz rys. 32). ||
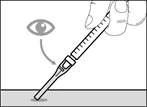
| * Jeśli nie usłyszałeś(aś) kliknięcia, sprawdź, czy igła jest całkowicie przykryta membraną ochronną (patrz rys. 33). * Cały czas trzymaj palce z tyłu membrany ochronnej i z dala od igły. * Nie odłączaj igły iniekcyjnej od strzykawki. |
Krok 17. Zutylizuj (wyrzuć) używaną strzykawkę i igłę (patrz rys. 34)
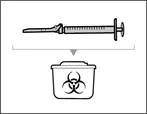
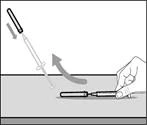
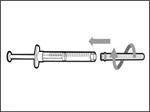
- Usuń strzykawkę i igłę do przetłaczania z pierwszego fiolki. * Jedną ręką włóż igłę do przetłaczania do kapturka i skieruj ją do góry, aby kaptur zakrył igłę (patrz rys. 35). * Gdy tylko kaptur zakryje igłę do przetłaczania, jedną ręką przesuń go w kierunku strzykawki, aby szczelnie ją zamknąć i zapobiec przypadkowemu ukłuciu igłą. | || Krok B. Zdejmij igłę do przetłaczania | | | |
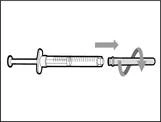
| | * Trzymaj nową fiolkę na płaskiej powierzchni roboczej i włóż nową igłę do przetłaczania ze strzykawką centralnie w korka fiolki w kierunku w dół (patrz rys. 39). | | |
| | | * Trzymaj igłę do przetłaczania w fiolce i odwróć ją do góry nogami (patrz rys. 40). | | |
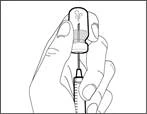
| | | * Skieruj igłę do góry i wprowadź powietrze ze strzykawki do fiolki nad roztworem leku. * Kontynuuj naciskanie palcem na tłoczek strzykawki (patrz rys. 41). * Nie wprowadzaj powietrza do roztworu leku, ponieważ może to prowadzić do powstawania pęcherzyków powietrza lub piany w leku. | | | Krok F. Nabierz lek Gemlibra® do strzykawki | | | | | |
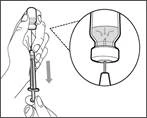
| | * Przesuń koniec igły w dół, tak aby znalazł się wewnątrz roztworu. * Powoli pociągnij tłoczek do tyłu, unikając powstawania pęcherzyków powietrza lub piany. Napełnij strzykawkę lekiem w ilości większej niż wymagana dawka (patrz rys. 42). * Bądź ostrożny: nie wyciągaj tłoczka ze strzykawki. Uwaga: przed wykonaniem kolejnych kroków należy upewnić się, że w strzykawce znajduje się wystarczająca ilość leku Gemlibra® na pełną dawkę. Jeśli nie uda się nabrać całego leku Gemlibra®, należy obrócić fiolkę do pozycji pionowej, aby pobrać resztę. | | | | Nie należy stosować igły do przetłaczania do wstrzyknięcia leku Gemlibra®, ponieważ może to spowodować urazy, takie jak ból i krwawienie. | | | | | | | Powtórz kroki A–F dla każdej dodatkowej fiolki, aż uzyskasz ilość leku Gemlibra® większą niż wymagana dawka. Po ich zakończeniu pozostaw igłę do przetłaczania w fiolce i wróć do kroku 6. Kontynuuj wykonywanie pozostałych kroków (kroki 7–17). | | | | | | | | | |
Dzieci.
Lek stosuje się dzieciom zgodnie z instrukcjami podanymi w sekcji „Sposób stosowania i dawki”.
Przedawkowanie.
Doświadczenie z przedawkowaniem leku Gemlibra® jest ograniczone. Przypadkowe przedawkowanie może prowadzić do hiperkoagulacji.
W przypadku przypadkowego przedawkowania należy natychmiast skontaktować się z lekarzem i pozostawać pod opieką medyczną.
Niepożądane działania
Poniższe poważne niepożądane działania opisano w innych sekcjach instrukcji:
- Mikroangiopatia zakrzepowa związana z zastosowaniem leku Gemlibra® i CAPK (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
- Zakrzepica i zatorowość związane z zastosowaniem leku Gemlibra® i CAPK (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”).
Dane z badań klinicznych
Ponieważ badania kliniczne są prowadzone w różnych warunkach, częstość niepożądanych działań obserwowanych podczas badań klinicznych stosowania leku nie może być bezpośrednio porównywana z częstością niepożądanych działań innego leku ustalonych w trakcie innych badań klinicznych i może nie odzwierciedlać częstości występowania tych działań w praktyce klinicznej.
Ogólnie niepożądane działania u dzieci otrzymujących leczenie lekiem Gemlibra® były podobne pod względem rodzaju do tych obserwowanych u dorosłych pacjentów z hemofilią typu A.
Poniższe niepożądane działania określono na podstawie połączonych danych dwóch randomizowanych badań z udziałem dorosłych i młodzieży (HAVEN 1 i HAVEN 3), jednego badania nieporównawczego z udziałem dorosłych i młodzieży (HAVEN 4), jednego badania nieporównawczego z udziałem dzieci (HAVEN 2) oraz badania doboru dawki, w których łącznie 391 mężczyzn z hemofilią typu A otrzymało co najmniej jedną dawkę leku Gemlibra® w celu profilaktycznego leczenia. Wśród nich 281 pacjentów (72%) to osoby dorosłe w wieku od 18 lat, 50 (13%) to młodzież w wieku od 12 do 18 lat, 55 (14%) to dzieci w wieku od 2 do 12 lat oraz 5 pacjentów (1%) to dzieci w wieku od 1 miesiąca do 2 lat. Mediana czasu trwania leczenia we wszystkich badaniach wynosiła 34,1 tygodnia (0,1–224,4 tygodnia).
Najczęściej zgłaszanymi niepożądanymi działaniami obserwowanymi u ≥10% pacjentów otrzymujących leczenie lekiem Gemlibra®, były reakcje w miejscu wstrzyknięcia, ból głowy i artrodynia.
Czterech pacjentów (1%) z badań klinicznych, którzy otrzymywali profilaktyczne leczenie lekiem Gemlibra®, przerwało terapię z powodu niepożądanych działań, w tym mikroangiopatii zakrzepowej, martwicy skóry, zakrzepowego zapalenia żył powierzchownych, bólu głowy i reakcji w miejscu wstrzyknięcia.
Tabela 3
Niepożądane działania zgłaszane u ≥5% pacjentów według połączonych danych badań klinicznych leku Gemlibra®
| Układ organizmu |
Reakcja niepożądana |
Liczba pacjentów n (%) (N = 391) |
| Zaburzenia ogólne oraz miejsca wstrzyknięcia |
Reakcje w miejscu wstrzyknięcia * |
85 (22 %) |
| Pirogieksja |
23 (6 %) |
|
| Zaburzenia ze strony układu nerwowego |
Ból głowy |
57 (15 %) |
| Zaburzenia ze strony układu pokarmowego |
Biegunka |
22 (6 %) |
| Zaburzenia ze strony układu mięśniowo-szkieletowego i tkanki łącznej |
Artrodynia |
59 (15 %) |
* Obejmuje siniaki w miejscu wstrzyknięcia, dyskomfort w miejscu wstrzyknięcia, zaczerwienienie w miejscu wstrzyknięcia, krwiaka w miejscu wstrzyknięcia, zgrubienie (indurację) w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia, świąd w miejscu wstrzyknięcia, wysypkę w miejscu wstrzyknięcia, reakcje w miejscu wstrzyknięcia, obrzęk w miejscu wstrzyknięcia, pokrzywkę w miejscu wstrzyknięcia oraz uczucie ciepła w miejscu wstrzyknięcia.
Charakterystyka leczenia CAPK we wszystkich badaniach klinicznych
W trakcie leczenia CAPK zaobserwowano 130 przypadków u 36 pacjentów, z których w 37 przypadkach (10 %) CAPK stosowano w średniej dawce skumulowanej > 100 IU/kg/24 h przez 24 godziny lub dłużej; dwa z 13 przypadków wiązały się z wystąpieniem zdarzeń zakrzepowych, a trzy z 13 przypadków wiązały się z wystąpieniem TMА (tabela 5). W pozostałych przypadkach podczas leczenia CAPK nie zaobserwowano zdarzeń zakrzepowych ani TMА.
Tabela 4
Charakterystyka leczenia* CAPK we wszystkich badaniach klinicznych
| Trwania leczenia AKPK |
Średnia skumulowana dawka KAPK po 24 godzinach (jedn./kg/24 godz.) |
||
| < 50 |
50–100 |
> 100 |
|
| < 24 godziny |
11 |
76 |
18 |
| 24–48 godziny |
0 |
6 |
3a |
| > 48 godziny |
1 |
5 |
10a,b,b,b |
* Przypadek leczenia FVIII definiuje się jako wszystkie dawki FVIII przyjęte przez pacjenta z dowolnego powodu i przed 36-godzinną przerwą w leczeniu.
a Zdarzenie zakrzepowe.
b Mikroangiopatia zakrzepowa.
Opis poszczególnych działań niepożądanych
Reakcje w miejscu wstrzyknięcia
O reakcjach w miejscu wstrzyknięcia (RMW) zgłoszono łącznie u 85 pacjentów (22%). Wszystkie RMW obserwowane podczas badań klinicznych leku Gemlibra® miały łagodny lub umiarkowany nasilenie i w 93% przypadków ustąpiły bez leczenia. Najczęściej zgłaszane RMW to zaczerwienienie w miejscu wstrzyknięcia (11%), świąd w miejscu wstrzyknięcia (4%) i ból w miejscu wstrzyknięcia (4%).
Inne rzadsze reakcje (< 1%)
Rhabdomioliza
O rhabdomiolizie zgłoszono u dwóch dorosłych pacjentów z bezobjawowym wzrostem stężenia kinazy kreatynowej w surowicy bez objawów ze strony nerek i układu mięśniowo-szkieletowego. W obu przypadkach rhabdomioliza wystąpiła po zwiększeniu aktywności fizycznej.
Doświadczenie z okresu po rejestracji
Podczas stosowania leku Gemlibra® po rejestracji zidentyfikowano poniżej wymienione działania niepożądane. Ze względu na dobrowolny charakter zgłaszania tych działań z populacji nieokreślonej liczby pacjentów, nie zawsze możliwe jest wiarygodne oszacowanie ich częstości ani ustalenie związku przyczynowego z narażeniem na lek.
Zaburzenia ze strony skóry i tkanki podskórnej: wysypka, pokrzywka, obrzęk naczynioruchowy.
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich ustawowe przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku za pośrednictwem zautomatyzowanego systemu informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua».
Okres ważności.
24 miesiące.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8 °C w oryginalnym opakowaniu w celu ochrony przed światłem. Nie zamarzać. Nie wstrząsać. Przechowywać w miejscu niedostępnym dla dzieci.
Niekompatybilność.
Nie zaobserwowano niekompatybilności leku Gemlibra® z zalecanymi strzykawkami i igłami.
Opakowanie.
12 mg/0,4 ml lub 30 mg/1 ml, lub 60 mg/0,4 ml, lub 105 mg/0,7 ml, lub 150 mg/1 ml w fiolce.
1 fiolka w pudełku kartonowym.
Kategoria wydawania.
Na receptę.
Producent.
F. Hoffmann-La Roche Ltd
Adres siedziby producenta i miejsca wykonywania działalności.
Wurmisweg, 4303 Kaiseraugst, Szwajcaria