Femoston®
Ukraina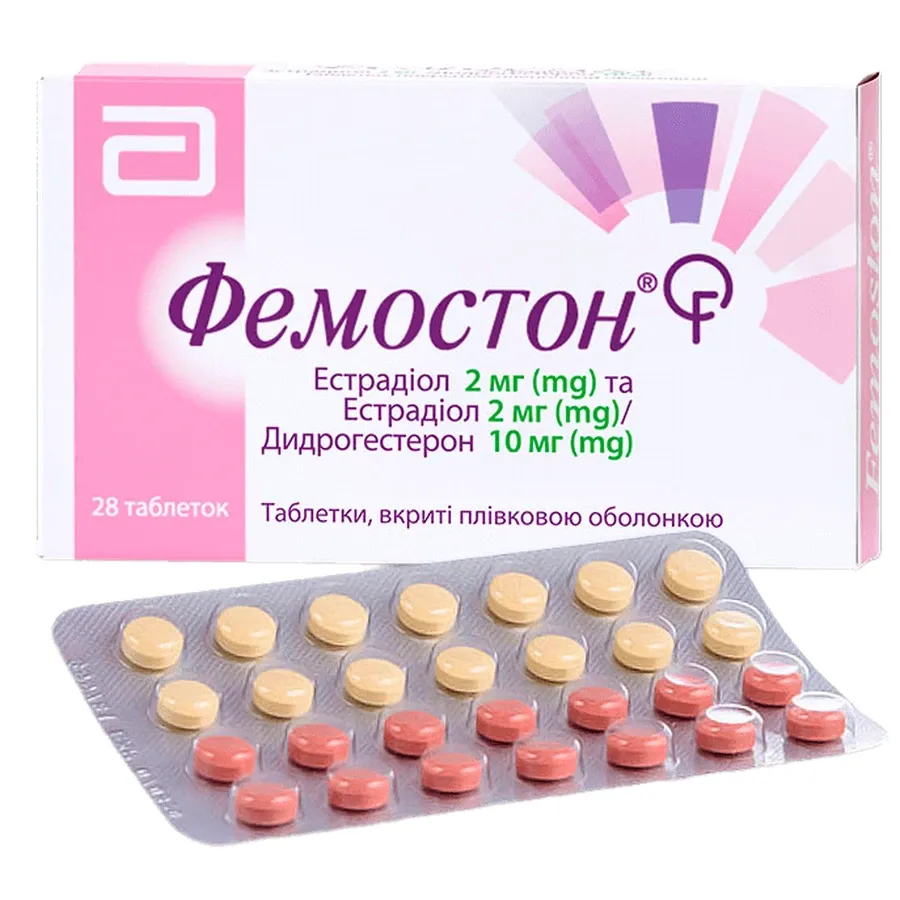
Spis treści
INSTRUKCJA dot. stosowania leku Femoston®
Skład:
tabletki estradiolu
substancja czynna: estradiol;
1 tabletka zawiera hemihydrat estradiolu, zmikronizowany, odpowiadający 1 mg lub 2 mg estradiolu;
substancje pomocnicze: laktoza, monohydrat; hipromeloza (HPMC 2910), skrobia kukurydziana, dwutlenek krzemu koloidalny bezwodny, stearynian magnezu;
powłoka filmowa: mieszana powłoka filmowa White I (hipromeloza (HPMC 2910), glikol polietylenowy 400, dwutlenek tytanu (E 171)) – dla tabletek zawierających 1 mg estradiolu;
mieszana powłoka filmowa Pink I (hipromeloza (HPMC 2910), glikol polietylenowy 400, talk, tlenek żelaza czerwony (E 172), tlenek żelaza czarny (E 172), tlenek żelaza żółty (E 172), dwutlenek tytanu (E 171)) – dla tabletek zawierających 2 mg estradiolu;
tabletki estradiolu i dydrogesteronu
substancje czynne: estradiol; dydrogesteron;
1 tabletka zawiera hemihydrat estradiolu, zmikronizowany, odpowiadający 1 mg lub 2 mg estradiolu; 10 mg dydrogesteronu, zmikronizowanego;
substancje pomocnicze: laktoza, monohydrat; hipromeloza (HPMC 2910), skrobia kukurydziana, dwutlenek krzemu koloidalny bezwodny, stearynian magnezu;
powłoka filmowa: mieszana powłoka filmowa Grey I (glikol polietylenowy 3350, talk (E 553b), alkohol polowinylowy, tlenek żelaza czarny (E 172), dwutlenek tytanu (E 171)) – dla tabletek zawierających 1 mg estradiolu i 10 mg dydrogesteronu;
mieszana powłoka filmowa Yellow II (hipromeloza (HPMC 2910), glikol polietylenowy 400, talk, tlenek żelaza żółty (E 172), dwutlenek tytanu (E 171)) – dla tabletek zawierających 2 mg estradiolu i 10 mg dydrogesteronu.
Postać farmaceutyczna. Tabletki powlekane.
Główne właściwości fizykochemiczne:
tabletka zawierająca 1 mg estradiolu: okrągła, dwuwypukła, powlekana powłoką filmową, biała tabletka z oznaczeniem „379” po jednej stronie; średnica 7 mm, masa tabletki około 144 mg;
tabletka zawierająca 2 mg estradiolu: okrągła, dwuwypukła, powlekana powłoką filmową, ceglasto-czerwona tabletka z oznaczeniem „379” po jednej stronie; średnica 7 mm, masa tabletki około 144 mg;
tabletka zawierająca 1 mg estradiolu i 10 mg dydrogesteronu: okrągła, dwuwypukła, powlekana powłoką filmową, szara tabletka z oznaczeniem „379” po jednej stronie; średnica 7 mm, masa tabletki około 144 mg;
tabletka zawierająca 2 mg estradiolu i 10 mg dydrogesteronu: okrągła, dwuwypukła, powlekana powłoką filmową, żółta tabletka z oznaczeniem „379” po jednej stronie; średnica 7 mm, masa tabletki około 144 mg.
Grupa farmakoterapeutyczna. Leki stosowane w chorobach układu moczowo-płciowego oraz hormony płciowe. Preparaty połączone zawierające progestageny i estrogeny do stosowania sekwencyjnego. Kod ATC G03F B08.
Właściwości farmakologiczne.
Farmakodynamika.
Estrydiol
Aktywny składnik 17β-estradiol jest chemicznie i biologicznie podobny do naturalnego żeńskiego hormonu płciowego – estradiolu. Zastępuje on spadek produkcji własnych estrogenów u kobiet w okresie menopauzy i łagodzi objawy menopauzy.
Estrogeny zapobiegają utracie masy tkanki kostnej po menopauzie lub ovariectomii.
Dydrogesteron
Dydrogesteron jest doustnie aktywnym progestagenem, którego działanie jest porównywalne z działaniem dożylnie podawanego progesteronu.
Ponieważ estrogeny stymulują wzrost endometrium, jeśli nie stosuje się progestagenu, zwiększają one ryzyko hiperplazji endometrium i raka. Dodanie progestagenu do terapii znacząco zmniejsza indukowane estrogenami ryzyko hiperplazji endometrium u kobiet z zachowanym macicą.
Dane z badań klinicznych
Umiarkowanie objawów niedoboru estrogenów i poprawa profilu krwawień.
Umiarkowanie objawów menopauzy obserwowano już w pierwszych tygodniach leczenia.
Regularne krwawienia odstawienie o średniej długości trwania 5 dni przy stosowaniu leku Femoston®, estrydiolu 2 mg + estrydiolu/dydrogesteronu 2 mg/10 mg, obserwowano u 89% kobiet. Krwawienie odstawienie zaczynało się zazwyczaj w 28. dniu cyklu. Krwawienia przebijające lub wydzieliny krwiste notowano u 22% kobiet w pierwszych 3 miesiącach leczenia oraz u 19% kobiet w okresie 10–12 miesięcy leczenia. Amenoreę (brak krwawień lub wydzieliny krwistej) obserwowano w 12% cykli w pierwszym roku leczenia.
Regularne krwawienia odstawienie o średniej długości trwania 5 dni przy stosowaniu leku Femoston®, estrydiolu 1 mg + estrydiolu/dydrogesteronu 1 mg/10 mg, obserwowano u 76% kobiet. Krwawienie odstawienie zaczynało się zazwyczaj w ostatnim dniu fazy progestagenowej (średnio w 28. dniu cyklu). Krwawienia przebijające lub wydzieliny krwiste notowano u około 23% kobiet w pierwszych 3 miesiącach leczenia oraz u 15% kobiet w okresie 10–12 miesięcy leczenia. Amenoreę (brak krwawień lub wydzieliny krwistej) obserwowano w 21% cykli w pierwszym roku leczenia.
Profilaktyka osteoporozy
Niedobór estrogenów po menopauzie wiąże się ze zwiększonym uogólnieniem tkanki kostnej i zmniejszeniem masy kostnej. Wpływ estrogenów na gęstość tkanki kostnej jest zależny od dawki. Ochronne działanie utrzymuje się tylko podczas stosowania leku. Po przerwaniu terapii hormonalnej zastępczej (HRT) szybkość utraty masy kostnej jest taka sama, jak u kobiet, które nie otrzymywały tej terapii.
Dane z badania WHI (Women Health Initiative) oraz metaanaliza innych badań wskazują, że stosowanie HRT głównie u zdrowych kobiet w postaci monoterapii estrogenem lub w połączeniu z progestagenem zmniejsza ryzyko złamania kości udowej, kręgów oraz innych złamań spowodowanych osteoporozą. HRT może również zapobiegać złomaniom u kobiet z niską gęstością tkanki kostnej i/lub rozpoznaną osteoporozą, ale dane na ten temat są ograniczone.
Po dwóch latach leczenia lekiem Femoston®, estrydiolu 2 mg + estrydiolu/dydrogesteronu 2 mg/10 mg, gęstość tkanki kostnej (GKK) w odcinku lędźwiowym kręgosłupa zwiększyła się o 6,7 ± 3,9% (średnia wartość ± odchylenie standardowe). Gęstość tkanki kostnej w odcinku lędźwiowym kręgosłupa zwiększyła się lub pozostała niezmieniona u 94,5% kobiet.
U kobiet przyjmujących lek Femoston®, estrydiolu 1 mg + estrydiolu/dydrogesteronu 1 mg/10 mg, gęstość tkanki kostnej w odcinku lędźwiowym kręgosłupa zwiększyła się o 5,2 + 3,8% (średnia wartość ± odchylenie standardowe). Gęstość tkanki kostnej w odcinku lędźwiowym kręgosłupa zwiększyła się lub pozostała niezmieniona u 93,0% kobiet.
Femoston® wpływa również na GKK kości udowej.
Po dwóch latach leczenia lekiem Femoston®, estrydiolu 2 mg + estrydiolu/dydrogesteronu 2 mg/10 mg, GKK zwiększyła się o 2,6 ± 5,0% (średnia wartość ± odchylenie standardowe) w szyjce kości udowej, o 3,5 ± 5,0% (średnia wartość ± odchylenie standardowe) w strefie trochanteru oraz o 4,1 ± 7,4% (średnia wartość ± odchylenie standardowe) w trójkącie Warda. GKK w trzech strefach kości udowej zwiększyła się lub pozostała niezmieniona po leczeniu lekiem Femoston®, estrydiolu 2 mg + estrydiolu/dydrogesteronu 2 mg/10 mg, u 71–88% kobiet.
Po dwóch latach terapii lekiem Femoston®, estrydiolu 1 mg + estrydiolu/dydrogesteronu 1 mg/10 mg, GKK w szyjce kości udowej zwiększyła się o 2,7 ± 4,2% (średnia wartość ± odchylenie standardowe), o 3,5 ± 5,0% (średnia wartość ± odchylenie standardowe) – w strefie trochanteru i o 2,7 ± 6,7% (średnia wartość ± odchylenie standardowe) – w trójkącie Warda. GKK w trzech strefach kości udowej zwiększyła się lub pozostała niezmieniona po leczeniu lekiem Femoston®, estrydiolu 1 mg + estrydiolu/dydrogesteronu 1 mg/10 mg, u 67–78% kobiet.
Farmakokinetyka.
Estrydiol
Wchłanianie
Wchłanianie estradiolu zależy od rozmiaru cząstek: mikronizowany estradiol szybko wchłania się z przewodu pokarmowego.
W |następnej| tabeli 1 przedstawiono|uwydatniać| średnie stałe parametry farmakokinetyczne estradiolu (E2), estronu (E1) oraz estronu siarczanu (E1S) dla każdej dawki mikronizowanego estradiolu|potem||wiele.
Dane przedstawiono jako średnie (SD).
Tabela 1
Estrydiol 1 mg
| Parametry |
E2 |
E1 |
Parametry |
E1S |
| Cmax (pg/ml) |
71 (36) |
310 (99) |
Cmax (ng/ml) |
9,3 (3,9) |
| Cmin (pg/ml) |
18,6 (9,4) |
114 (50) |
Cmin (ng/ml) |
2,099 (1,340) |
| Cav (pg/ml) |
30,1 (11,0) |
194 (72) |
Cav (ng/ml) |
4,695 (2,350) |
| AUC0-24 (pg*godz/ml) |
725 (270) |
4767 (1857) |
AUC0-24 (ng*godz/ml) |
112,7 (55,1) |
Estradiol 2 mg
| Parametry |
E2 |
E1 |
Parametry |
E1S |
| Cmax (pg/ml) |
103,7 (48,2) |
622,2 (263,6) |
Cmax (ng/ml) |
25,9 (16,4) |
| Cmin (pg/ml) |
48 (30) |
270 (138) |
Cmin (ng/ml) |
5,7 (5,9) |
| Cav (pg/ml) |
68 (31) |
429 (191) |
Cav (ng/ml) |
13,1 (9,4) |
| AUC0-24 (pg*godz/ml) |
1619 (733) |
10209 (4561) |
AUC0-24 (ng*godz/ml) |
307,3 (224,1) |
Rozkład
Estrogeny występują w postaci niezwiązanej lub związanej. Około 98–99 % dawki estradiolu wiąże się z białkami osocza krwi, z czego 30–52 % z albuminą oraz około 46–69 % z globuliną wiążącą hormony płciowe (SHBG).
Biotransformacja
Po podaniu doustnym estradiol ulega intensywnemu metabolizmowi. Głównymi metabolitami niekonjuguowanymi i konjuguowanymi są estron i siarczan estronu. Metabolity te mogą bezpośrednio wykazywać działanie estrogenowe lub po przekształceniu w estradiol. Siarczan estronu może podlegać obiegowi enterohepaticznemu.
Wydalanie
W moczu głównymi związkami są glukuronidy estronu i estradiolu. Okres półtrwania wydalania wynosi od 10 do 16 godzin. Estrogeny są wydzielane z mlekiem matki.
Zależność od dawki i czasu
Przy codziennym doustnym stosowaniu leku Femoston® stężenie estradiolu osiąga stan równowagi po około pięciu dniach. W większości przypadków stężenie stanu równowagi osiągane jest w okresie od 8 do 11 dnia stosowania.
Dydrogesteron
Wchłanianie
Po podaniu doustnym dydrogesteron jest szybko wchłaniany, z Tmax wynoszącym 0,5–1,5 godz.
W |następnej tabeli 2 przedstawiono|wyobrażać| średnie ustalone parametry farmakokinetyczne dydrogesteronu| (D) oraz dihydrodydrogesteronu| (DHGD)|potem|||.
Dane przedstawiono jako średnie (SD).
Tabela 2
Dydrogesteron 10 mg
| Parametry |
D |
DGJ |
| Cmax (ng/ml) |
2,54 (1,80) |
62,50 (33,10) |
| Cmin (ng/ml) |
0,13 (0,07) |
3,70 (1,67) |
| Cav (ng/ml) |
0,42 (0,25) |
13,04 (4,77) |
| AUCτ (ng*god/ml) |
10,17 (5,96) |
312,90 (114,54) |
Po podaniu dawki jednorazowej jedzenie opóźnia osiągnięcie szczytowego stężenia dydrogesteronu we krwi o około 1 godzinę, co prowadzi do obniżenia szczytowych stężeń dydrogesteronu we krwi o około 20%, bez wpływu na stopień ekspozycji na dydrogesteron i DHG.
Rozkład
Po doustnym podaniu dydrogesteronu widoczny objętość rozkładu jest znaczna i wynosi około 22000 l. Dydrogesteron i DHG wiążą się z białkami osocza krwi w ponad 90%.
Biotransformacja
Po doustnym podaniu dydrogesteron szybko ulega metabolizmowi z powstaniem DHG. Stężenia głównego aktywnego metabolitu, 20α-dyhydrodydrogesteronu (DHG), osiągają szczyt w tym samym czasie, co dydrogesteron. Stężenia DHG we krwi są znacznie wyższe w porównaniu z substancją wyjściową. Stosunek AUC i Cmax DHG do dydrogesteronu wynosi odpowiednio około 25 i 20. Średni okres półtrwania eliminacji dydrogesteronu i DHG wynosi około 15 godzin. Ogólną cechą wszystkich metabolitów jest zachowanie konfiguracji 4,6-dien-3-on substancji wyjściowej oraz brak hydroksylacji w pozycji 17α. Wyjaśnia to brak efektów estrogenowych i androgenowych dydrogesteronu.
Wydalanie
Po doustnym podaniu znakowanego dydrogesteronu średnio 63% dawki wydala się z moczem. Widoczny całkowity klirens osoczowy dydrogesteronu jest wysoki i wynosi około 20 l/min. Pełne wydalanie odbywa się w ciągu 72 godzin. DHG występuje w moczu głównie w formie koniugatu z kwasem glukuronkowym.
Zależność od dawki i czasu
Farmakokinetyka po dawkowaniu jednorazowym i wielokrotnym ma charakter liniowy w zakresie dawek doustnych od 2,5 do 20 mg. Porównanie kinetyki dawki jednorazowej i wielokrotnej wskazuje, że farmakokinetyka dydrogesteronu i DHG nie zmienia się przy powtarzalnym stosowaniu. Stan równowagi osiągany jest zazwyczaj po 3 dniach leczenia.
Charakterystyki kliniczne.
Wskazania.
Zastępcza terapia hormonalna (ZTH) w celu złagodzenia objawów spowodowanych niedoborem estrogenów u kobiet w okresie menopauzalnym, nie wcześniej niż 6 miesięcy po ostatniej menstruacji.
Profilaktyka osteoporozy u kobiet w okresie postmenopauzalnym z wysokim ryzykiem złamania. Femoston® należy stosować jedynie u pacjentek, u których występuje nietolerancja lub istnieją przeciwwskazania do stosowania innych leków w celu zapobiegania osteoporozie (patrz rozdział „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Doświadczenie w leczeniu kobiet powyżej 65. roku życia jest ograniczone.
Przeciwwskazania.
- Rozpoznany wcześniej, obecny lub podejrzewany nowotwór złośliwy piersi;
- ustalone lub podejrzewane nowotwory wrażliwe na estrogeny (np. raka endometrium);
- ustalone lub podejrzewane nowotwory wrażliwe na progestageny;
- meningioma lub meningioma w wywiadzie;
- nieprawidłowe krwawienia z dróg rodnych o nieustalonej przyczynie;
- nieleczona hiperplazja endometrium;
- aktualnie występujące lub w wywiadzie zakrzepica żył głębokich (ZŻG), zatorowość płucna (ZP) lub inne zakrzepcze choroby żylnych;
- obecność zaburzeń trombofilii (np. niedobór białka C, białka S lub antytrombiny, patrz rozdział „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”);
- ostre lub niedawno przebyte choroby zakrzepcze tętnicze (np. choroba wieńcowa, zawał mięśnia sercowego);
- ostre lub w wywiadzie choroby wątroby, jeśli wartości funkcji wątroby nie zostały znormalizowane;
- znana nadwrażliwość na substancje czynne lub na którąkolwiek z substancji pomocniczych leku;
- porfiria.
Interakcje z innymi lekami oraz inne rodzaje oddziaływań.
Badania interakcji leków nie były przeprowadzane.
Skuteczność estrogenów i progestagenów może być zaburzona
- Metabolizm estrogenów (oraz progestagenów) może być zwiększony podczas jednoczesnego stosowania substancji, które mogą indukować enzymy biorące udział w metabolizmie leków, w szczególności enzymy CYP450 2B6, 3A4, 3A5, 3A7. Do takich substancji należą leki przeciwpadaczkowe (fenobarbital, karbamazepina, fenytoina) oraz leki przeciwbakteryjne/przeciwwirusowe (np. ryfampicyna, ryfabutyna, nevirapyna, efawiorenc).
- Mimo że rytonawir i nelfinawir są znane jako silne inhibitory CYP450 3A4, A5, A7, w rzeczywistości wykazują działanie indukcyjne przy jednoczesnym stosowaniu ze steroidowymi hormonami.
- Preparaty roślinne zawierające ziele św. Jana (Hypericum perforatum) mogą również zwiększać metabolizm estrogenów (oraz progestagenów) poprzez CYP450 3A4.
- Klinicznie zwiększone tempo metabolizmu estrogenów i progestagenów może objawiać się zmniejszeniem skuteczności oraz zmianami w profilu krwawień.
Wpływ ZTH z estrogenami na inne leki
Wykazano, że doustne środki antykoncepcyjne zawierające estrogeny, stosowane jednocześnie z lamotrydżyną, znacząco obniżają jej stężenia w osoczu w wyniku stymulacji glukuronidacji lamotrydżyny. Może to prowadzić do utraty kontroli nad napadami padaczkowymi. Choć potencjalna interakcja między terapią zastępczą a lamotrydżyną nie była badana, należy się spodziewać podobnego efektu, który może prowadzić do zmniejszenia kontroli nad napadami u kobiet stosujących oba leki jednocześnie.
Interakcje farmakodynamiczne
Podczas badań klinicznych leczenia wirusa zapalenia wątroby typu C (WZW C) kombinacją ombitaswir/paritaprewir/rytonawir i dasabuwer z lub bez rybawiryny, znacznie częściej obserwowano przekroczenie górnej granicy normy stężenia alaninotransaminazy (ALT) ponad 5-krotnie u kobiet stosujących leki zawierające etynylestradiol, takie jak połączone środki antykoncepcyjne (PSA). Ponadto, podczas leczenia glekaprewirem/pibrantaszwirem lub sofosbuwirem/welptaswirem/woksilaprewirem, podwyższenie stężenia ALT obserwowano również u kobiet stosujących leki zawierające etynylestradiol, takie jak PSA. U kobiet stosujących leki zawierające inne estrogeny niż etynylestradiol, np. estradiol, w połączeniu z ombitaswirem/paritaprewirem/rytonawirem i dasabuwerm z lub bez rybawiryny, stopień podwyższenia stężenia ALT był podobny do kobiet niebiorących estrogenów; jednakże, ze względu na ograniczoną liczbę kobiet stosujących inne estrogeny, należy zachować ostrożność przy jednoczesnym stosowaniu tego leku z następującą terapią skojarzoną: ombitaswirem/paritaprewirem/rytonawirem i dasabuwerm z lub bez rybawiryny, glekaprewirem/pibrantaszwirem lub sofosbuwirem/welptaswirem/woksilaprewirem (patrz rozdział „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Estrogeny mogą zakłócać metabolizm innych leków
Estrogeny mogą hamować enzymy CYP450 biorące udział w metabolizmie leków poprzez inhibicję konkurencyjną. Należy szczególnie wziąć to pod uwagę w odniesieniu do leków o wąskim indeksie terapeutycznym, takich jak:
- takrolimus i cyklosporyna A (CYP450 3A4, 3A3);
- fentanil (CYP450 3A4);
- teofilina (CYP450 1A2).
Klinicznie może to prowadzić do wzrostu stężenia tych substancji we krwi do poziomów toksycznych. W związku z tym może być konieczna staranna kontrola stężenia tych leków przez dłuższy okres, a także zmniejszenie dawki takrolimusu, fentanilu, cyklosporyny A oraz teofiliny.
Szczególne ostrzeżenia dotyczące stosowania.
W celu leczenia objawów niedoboru estrogenów u kobiet w okresie menopauzy, terapię hormonalną (HRT) należy rozpoczynać tylko wtedy, gdy objawy te znacząco obniżają jakość życia. Skuteczność i ryzyko związane z HRT należy regularnie oceniać, co najmniej raz w roku, a leczenie należy kontynuować tylko wtedy, gdy korzyści przewyższają ryzyko.
Dane dotyczące ryzyka związanego z HRT w leczeniu przedwczesnej menopauzy są ograniczone. Jednak ze względu na niski poziom absolutnego ryzyka u młodszych kobiet, stosunek korzyści do ryzyka u tych kobiet jest bardziej korzystny niż u kobiet starszych.
Badanie lekarskie / dalsza obserwacja
Przed rozpoczęciem HRT lub w przypadku konieczności wznowienia HRT po przerwie należy dokładnie ustalić pełen wywiad medyczny (w tym wywiad rodzinny). Badanie fizykalne (w tym badanie ginekologiczne i badanie piersi) należy wykonać, biorąc pod uwagę wywiad, przeciwwskazania i ostrzeżenia dotyczące stosowania. Podczas leczenia zaleca się regularne kontrole, których częstotliwość i zakres określa się indywidualnie. Należy poinformować kobiety, o jakich zmianach w piersiach należy powiadomić lekarza lub pielęgniarkę (patrz sekcja poniżej „Rak piersi”). Badania, w tym odpowiednie metody wizualizacji, np. mammografię, należy przeprowadzać zgodnie z obowiązującą praktyką przesiewową, dostosowaną do indywidualnych potrzeb klinicznych.
Choroby, w których należy obserwować stan pacjentek
Pacjentki z obecnymi lub wcześniejszymi schorzeniami wymienionymi poniżej, lub ich pogorszeniem podczas ciąży lub poprzedniej terapii hormonalnej, powinny być poddawane dokładnej obserwacji. Należy pamiętać, że te stany mogą nawracać lub ich przebieg może się pogarszać podczas leczenia lekiem Femoston®. Obejmują one:
- mięsak macicy (fibromy macicy) lub endometriozę;
- czynniki ryzyka chorób zakrzepowo-zatorowych (patrz poniżej);
- czynniki ryzyka nowotworów wrażliwych na estrogeny, np. pierwszego stopnia predyspozycji genetycznej do raka piersi;
- nadciśnienie tętnicze;
- choroby wątroby (gruczolak wątroby);
- cukrzycę z objawami naczyniowymi lub bez nich;
- kamice pęcherza żółciowego;
- migrenę lub (silny) ból głowy;
- toczeń rumieniowaty układowy;
- hiperplazję endometrium w wywiadzie (patrz poniżej);
- padaczkę;
- astmę oskrzelową;
- otosklerozę.
Meningioma
Zgłaszano przypadki wystąpienia meningiom (pojedynczych i wielokrotnych) podczas stosowania leku Femoston®. Pacjenci powinni być poddawani nadzorowi w celu wykrycia objawów i oznak meningiom zgodnie z praktyką kliniczną. Jeśli u pacjenta zdiagnozowano meningiomę, wszelkie leczenie lekiem Femoston® należy natychmiast przerwać (patrz sekcja „Przeciwwskazania”). Po przerwaniu leczenia obserwowano zmniejszenie guza.
Przyczyny natychmiastowego przerwania terapii
Terapię hormonalną należy natychmiast przerwać w przypadku stwierdzenia przeciwwskazania, a także w następujących sytuacjach:
- wystąpienie żółtaczki lub zaburzeń funkcji wątroby;
- istotny wzrost ciśnienia tętniczego;
- pojawienie się po raz pierwszy bólu głowy typu migrenowego;
- ciąża.
Hiperplazja i rak endometrium
U kobiet z zachowaną macicą ryzyko wystąpienia hiperplazji endometrium i raka wzrasta przy długotrwałym stosowaniu HRT tylko z estrogenami. Obserwowany wzrost ryzyka raka endometrium u kobiet przyjmujących wyłącznie leki estrogenowe waha się od 2 do 12 razy w porównaniu z tymi, które ich nie przyjmują, w zależności od długości leczenia i dawki estrogenów (patrz sekcja „Działania niepożądane”). Po przerwaniu leczenia ryzyko może pozostać podwyższone przez co najmniej 10 lat.
Cykliczne łączenie leku estrogenowego z progestagenem przez co najmniej 12 dni na miesiąc / cykl 28-dniowy lub bezprzerwne skojarzone leczenie estrogenem i progestagenem u kobiet z zachowaną macicą zapobiega nadmiernemu ryzyku związanemu z samodzielnym stosowaniem leków estrogenowych.
W pierwszych miesiącach leczenia mogą występować nieregularne krwawienia maciczne lub wydzieliny krwiste. Jeśli pojawiają się one po pewnym czasie od rozpoczęcia leczenia lub trwają po jego przerwaniu, należy ustalić ich przyczynę, w tym poprzez wykonanie biopsji endometrium, aby wykluczyć nowotwory złośliwe.
Rak piersi
Wszystkie dostępne dane wskazują na zwiększone ryzyko raka piersi u kobiet przyjmujących skojarzoną estrogenowo-progestagenową HRT lub samodzielnie estrogenową HRT. Ryzyko to zależy od długości stosowania.
Skojarzone leczenie estrogenem i progestagenem
Badanie randomizowane, kontrolowane placebo „Women’s Health Initiative Study” (WHI), jak i metaanaliza badań epidemiologicznych prospektywnych, wykazały zgodnie zwiększone ryzyko wystąpienia raka piersi u kobiet stosujących skojarzoną estrogenowo-progestagenową HRT. Zwiększony ryzyko staje się widoczne po około 3 (1–4) latach (patrz sekcja „Działania niepożądane”).
Monoterapia estrogenem
Badanie WHI nie wykazało zwiększonego ryzyka wystąpienia raka piersi u kobiet po histerectomii, które przyjmują HRT tylko z lekami estrogenowymi. W badaniach eksperymentalnych głównie raportowano niewielki wzrost ryzyka rozpoznania raka piersi, który jest znacznie niższy niż u pacjentek przyjmujących kombinacje estrogenów i progestagenów (patrz sekcja „Działania niepożądane”).
Wyniki dużego metaanalizy wykazały, że po przerwaniu leczenia zwiększone ryzyko z czasem maleje, a czas potrzebny do powrotu do poziomu wyjściowego zależy od długości poprzedniego stosowania HRT. W przypadkach, gdy HRT stosowano dłużej niż 5 lat, ryzyko może utrzymywać się przez 10 lat lub dłużej. Terapia hormonalna zastępcza, szczególnie skojarzona estrogenowo-progestagenowa, zwiększa gęstość obrazów mammograficznych, co może negatywnie wpłynąć na wykrywanie raka piersi metodami radiologicznymi.
Rak jajników
Rak jajników występuje znacznie rzadziej niż rak piersi. Dane epidemiologiczne z dużego metaanalizy wykazały nieco zwiększone ryzyko u kobiet, którym stosowano monoterapię estrogenem lub kombinację estrogenów z progestagenem jako terapię hormonalną zastępczą; ryzyko to pojawia się w ciągu 5 lat stosowania i maleje z czasem po przerwaniu terapii. Niektóre inne badania, w tym badanie WHI, wskazują, że stosowanie skojarzonej HRT może być związane z takim samym lub nieco niższym ryzykiem (patrz sekcja „Działania niepożądane”).
Zakrzepica żylna (VTE)
HRT wiąże się z 1,3–3-krotnym wzrostem ryzyka wystąpienia zakrzepicy żylnej (VTE), tj. zakrzepicy żył głębokich lub zatoru tętnicy płucnej. Takie schorzenie jest najbardziej prawdopodobne w pierwszym roku stosowania HRT (patrz sekcja „Działania niepożądane”).
Pacjentki z znanymi powikłaniami trombofilii mają zwiększone ryzyko VTE, a HRT może dodatkowo zwiększać to ryzyko. Dlatego terapia hormonalna zastępcza jest przeciwwskazana dla tej grupy pacjentek (patrz sekcja „Przeciwwskazania”).
Ogólnie uznawanymi czynnikami ryzyka rozwoju VTE są: stosowanie estrogenów, starszy wiek, duże zabiegi chirurgiczne, długotrwała immobilizacja, otyłość (BMI > 30 kg/m²), ciąża / okres poporodowy, toczeń rumieniowaty układowy (SLE) i rak. Nie ma jednomyślnej opinii na temat roli żylaków w rozwoju VTE.
Tak jak u wszystkich pacjentów pooperacyjnych, należy podjąć środki zapobiegawcze w celu zapobiegania VTE po zabiegu chirurgicznym. Jeśli po planowanym zabiegu przewiduje się długotrwałą immobilizację, zaleca się tymczasowe przerwanie HRT 4–6 tygodni przed zabiegiem. Leczenie można wznowić dopiero wtedy, gdy kobieta w pełni odzyska ruchomość.
Kobietom bez VTE w wywiadzie, ale z rodziną pierwszego stopnia z krwawieniem w młodym wieku, można zaproponować badania przesiewowe po dokładnym omówieniu ich ograniczeń (badania przesiewowe wykrywają tylko część zaburzeń trombofilii).
Jeśli zidentyfikowano wrodzone zaburzenie trombofilii towarzyszące krwawieniu w rodzinie lub jeśli zaburzenie jest ciężkie (np. niedobór antytrombiny, białka S lub białka C lub kombinacja zaburzeń), HRT jest przeciwwskazana.
U kobiet, które już przyjmują stałą terapię przeciwkrzepliwą, należy dokładnie rozważyć korzyści i ryzyko stosowania HRT.
Jeśli VTE rozwinie się po rozpoczęciu terapii, lek należy natychmiast odstawić. Pacjentki należy uprzedzić o konieczności natychmiastowego zgłoszenia się do lekarza w przypadku wystąpienia potencjalnych objawów zatoru krwi (np. bolesnego obrzęku nogi, nagłego bólu w klatce piersiowej, duszności).
Choroba wieńcowa serca (CHD)
Nie ma dowodów z randomizowanych badań kontrolowanych na temat ochrony przed zawałem mięśnia sercowego u kobiet z lub bez CHD, które przyjmują skojarzoną estrogenowo-progestagenową HRT lub HRT tylko z estrogenem.
Skojarzone leczenie estrogenem i progestagenem
Względne ryzyko wystąpienia CHD podczas stosowania skojarzonej estrogenowo-progestagenowej HRT jest nieco zwiększone. Ponieważ absolutne ryzyko podstawowe CHD zależy w dużej mierze od wieku, liczba dodatkowych przypadków CHD spowodowanych przez stosowanie estrogenów i progestagenów jest bardzo mała u zdrowych kobiet w wieku bliskim menopauzie, ale będzie wzrastać z wiekiem.
Monoterapia estrogenem
Dane z randomizowanych badań kontrolowanych nie wykazały zwiększonego ryzyka CHD u kobiet po histerectomii, które przyjmują monoterapię estrogenem.
Udar niedokrwienny
Skojarzone leczenie estrogenem i progestagenem oraz monoterapia estrogenem wiążą się ze wzrostem ryzyka udaru niedokrwiennego do 1,5 razy. Względne ryzyko nie zmienia się z wiekiem ani czasem, który upłynął od wystąpienia menopauzy. Jednak ponieważ absolutne ryzyko podstawowe udaru zależy w dużej mierze od wieku, całkowite ryzyko udaru u kobiet przyjmujących HRT będzie wzrastać z wiekiem (patrz sekcja „Działania niepożądane”).
Inne stany
-
Estrogeny mogą powodować zatrzymanie płynów, dlatego należy uważnie obserwować pacjentów z zaburzeniami czynności serca lub nerek.
-
Kobiety z wcześniejszą hipertriglicerydemią powinny być poddawane dokładnej obserwacji podczas terapii zastępczej estrogenami lub hormonalnej terapii zastępczej, ponieważ rzadko u kobiet z tą patologią poziom triglicerydów w osoczu krwi znacznie wzrastał podczas leczenia estrogenami, co prowadziło do zapalenia trzustki.
-
Egzogenne estrogeny mogą wywoływać lub nasilać objawy dziedzicznej lub nabytej angioedemy.
-
Estrogeny zwiększają poziom globuliny wiążącej tyroksynę (TBG), co prowadzi do zwiększenia krążących hormonów tarczycy, mierzonych jako poziom jodu związanego z białkiem (PBI), poziomów T4 (w analizie z użyciem kolumn lub radioimmunologicznej) lub poziomów T3 (za pomocą radioimmunologicznej analizy). Wiązanie trijodotyroniny (T3) jest zmniejszone w wyniku podwyższonych poziomów TBG. Stężenia wolnego trijodotyroniny (T3) i tyroksyny (T4) nie zmieniają się. Poziomy innych białek wiążących w surowicy, globuliny wiążącej kortykosteroidy (CBG) i globuliny wiążącej hormony płciowe (SHBG), mogą wzrastać, co prowadzi do zwiększenia stężenia kortykosteroidów i hormonów płciowych we krwi. Stężenia wolnych i/lub biologicznie aktywnych hormonów nie zmieniają się. Mogą wzrastać stężenia innych białek osocza (substratu angiotensyny-reniny, alfa-1-antytrypsyny, cerulooplasminy).
-
HRT nie poprawia funkcji poznawczych. Uzyskano pewne dane wskazujące na zwiększone ryzyko rozwoju możliwej demencji u kobiet rozpoczynających długotrwałe skojarzone lub wyłącznie estrogenowe HRT po 65. roku życia.
-
Pacjentki z rzadkimi chorobami genetycznymi, takimi jak nietolerancja galaktozy, całkowity niedobór laktozy lub zespół złego wchłaniania glukozy-galaktozy, nie powinny przyjmować tego leku.
-
Femoston® nie należy do środków antykoncepcyjnych.
Zwiększenie poziomu ALAT
Podczas badań klinicznych z udziałem pacjentów leczonych zakażenia wirusem zapalenia wątroby C (HCV) kombinacją ombitaswir/paritaprewir/rytonawir i dasabuwirow z lub bez rybawiryny, przekroczenie górnej granicy normy poziomu ALAT ponad 5 razy częściej obserwowano u kobiet przyjmujących leki zawierające etynylestradiol, takie jak skojarzone hormonalne środki antykoncepcyjne (COC). Ponadto, również wśród pacjentów przyjmujących glekaprewir/pibrentaswir lub sofosbuwir/velpataswir/woksalaprewir, zwiększenie poziomu ALAT obserwowano u kobiet przyjmujących leki zawierające etynylestradiol, takie jak COC. U kobiet przyjmujących leki zawierające inne estrogeny niż etynylestradiol, takie jak estradiol, w połączeniu z ombitaswir/paritaprewir/rytonawir i dasabuwir z lub bez rybawiryny, stopień zwiększenia poziomu ALAT był podobny do kobiet nie przyjmujących żadnych estrogenów; jednak ze względu na ograniczoną liczbę kobiet przyjmujących inne estrogeny, należy zachować ostrożność przy jednoczesnym stosowaniu tego leku z następującą terapią skojarzoną: ombitaswir/paritaprewir/rytonawir i dasabuwir z lub bez rybawiryny, glekaprewir/pibrentaswir lub sofosbuwir/velpataswir/woksalaprewir (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”).
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
Femoston® nie jest wskazany do stosowania w czasie ciąży. Jeśli ciąża wystąpiła podczas leczenia lekiem Femoston®, należy natychmiast przerwać przyjmowanie leku.
Brakuje wystarczających danych dotyczących stosowania estradiolu/dydrogesteronu u kobiet w czasie ciąży.
W literaturze naukowej donoszono, że stosowanie niektórych progestagenów wiązano ze zwiększonym ryzykiem rozwoju hipospadii. Jednak z powodu mieszanych czynników podczas ciąży nie można jednoznacznie określić wpływu progestagenów na rozwój hipospadii.
Obecnie wyniki większości badań epidemiologicznych dotyczących przypadkowego wpływu na płód kombinacji estrogenów i progestagenów wskazują na brak ryzyka teratogennego lub toksycznego dla płodu.
Karmienie piersią
Femoston® nie jest wskazany do stosowania w czasie karmienia piersią.
Wpływ na płodność.
Femoston® nie jest wskazany do stosowania u kobiet w wieku rozrodczym.
Wpływ na zdolność do prowadzenia pojazdów lub obsługi mechanizmów.
Femoston® nie wpływa lub ma nieznaczny wpływ na zdolność do prowadzenia pojazdów lub obsługi mechanizmów.
Sposób stosowania i dawki.
Femoston® stosuje się doustnie codziennie zgodnie z trybem ciągłym sekwencyjnym, jak opisano poniżej.
Leczenie rozpoczyna się od przyjmowania jednej tabletki zawierającej 1 mg lub 2 mg estradiolu, 1 raz na dobę, codziennie przez pierwsze 14 dni cyklu 28-dniowego.
Następnie przez kolejne 14 dni przyjmuje się 1 tabletę dziennie zawierającą 1 mg lub 2 mg estradiolu oraz 10 mg dydrogesteronu, 1 raz na dobę, zgodnie z oznaczeniem na 28-dniowej opakowaniu kalendarzowym.
Po zakończeniu cyklu 28-dniowego, od dnia 29, należy natychmiast rozpocząć nowy cykl 28-dniowy.
Cykle leczenia następują jeden po drugim i są ciągłe.
W leczeniu niedoboru estrogenów u kobiet w okresie menopauzy dawkę początkową i utrzymawczą należy dobrać jako najniższą skuteczną dawkę, a czas trwania leczenia powinien być jak najkrótszy (patrz również punkt „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Ogólnie sekwencyjną terapię skojarzoną należy rozpoczynać od leku Femoston® zawierającego estradiol 1 mg + estradiol/dydrogesteron 1 mg/10 mg.
Dawkę należy dobrać indywidualnie, w zależności od odpowiedzi klinicznej.
U kobiet, które nie stosują hormonalnej terapii zastępczej, lub u kobiet przechodzących z ciągłej terapii skojarzonej hormonalnej terapii zastępczej, leczenie można rozpocząć w dowolny dogodny dzień. U kobiet przechodzących z cyklicznej lub ciągłej sekwencyjnej hormonalnej terapii zastępczej, leczenie należy rozpocząć bezpośrednio od następnego dnia po zakończeniu poprzedniego cyklu.
W przypadku pominięcia przyjęcia tabletki zaleca się kontynuowanie przyjmowania kolejnej tabletki bez uzupełniania pominiętej dawki. W przypadku pominięcia przyjęcia tabletki może zwiększyć się ryzyko wystąpienia krwawień przebijających lub wydzieliny krwistej.
Femoston® można przyjmować niezależnie od posiłku.
Dzieci.
Nie wskazuje się stosowania leku Femoston® u tej kategorii pacjentów.
Przedawkowanie.
Tak estradiol, jak i dydrogesteron są substancjami o niskiej toksyczności. W przypadku przedawkowania mogą wystąpić takie objawy jak nudności, wymioty, uczulenie piersi, zawroty głowy, ból brzucha, senność/wykończalność oraz krwawienie odstawienie. Mało prawdopodobne jest, że w przypadku przedawkowania będzie konieczne jakiekolwiek leczenie specyficzne lub objawowe.
Informacje powyżej dotyczą również przedawkowania u dzieci.
Niepożądane działania.
Najczęstsze niepożądane działania u pacjentek, którym podawano terapię estradiolu/dydrogesteronem w trakcie badań klinicznych, to ból głowy, ból brzucha, ból/wrażliwość piersi oraz ból w lędźwiach.
Podczas badań klinicznych (n = 5108) obserwowano niepożądane działania wymienione w tabeli 3, z podaną poniżej częstością:
Tabela 3
| Klasy układów narządów zgodnie z MedDRA |
Bardzo często (≥ 1/10) |
Często (≥ 1/100, <1/10) |
Nieczęsto (≥ 1/1000, < 1/100) |
Rzadko (≥ 1/10000 do < 1/1000) |
| Infekcje i choroby pasożytnicze |
Kandydoza pochwy |
Zespół przypominający zapalenie pęcherza |
||
| Nowotwory łagodne, złośliwe i niejednoznaczne |
Wzrost miomy |
|||
| Układ krwiotwórczy i chłonny |
Anemia hemolityczna* |
|||
| Układ odpornościowy |
Nadwrażliwość |
|||
| Układ nerwowy |
Ból głowy |
Migrena, zawroty głowy |
Meningioma* |
|
| Układ psychiczny |
Depresja, pobudzenie |
Wpływ na libido |
||
| Układ wzroku |
Zwiększona krzywizna rogówki*. Nietolerancja soczewek kontaktowych* |
|||
| Serce |
Przykrwienie mięśnia sercowego |
|||
| Zaburzenia naczyniowe |
Zakrzepica żylna**, nadciśnienie tętnicze, choroba naczyń obwodowych, żylaki |
Udar* |
||
| Układ pokarmowy |
Ból brzucha |
Nudności, wymioty, wzdęcia |
Trudności trawienne |
|
| Układ wątrobowo-żółciowy |
Zaburzenia funkcji wątroby (w niektórych przypadkach z żółtaczką, osłabieniem lub niedyspozycją oraz bólem brzucha), choroba pęcherza żółciowego |
|||
| Układ skóry i tkanki podskórnej |
Reakcje alergiczne skóry (np. wysypka, pokrzywka, swędzenie) |
Świerzbienie naczynioruchowe*, rumień węzłowaty*, purpura naczyniowa; chloaza lub melaza, które mogą utrzymywać się po odstawieniu leczenia* |
||
| Układ mięśniowo-szkieletowy i tkanki łącznej |
Ból pleców |
Skurcze kończyn dolnych* |
||
| Układ rozrodczy i gruczoły mlekowe |
Ból/wrażliwość gruczołów mlekowych |
Zaburzenia cyklu menstruacyjnego (w tym krwawienia pomenopauzalne, metroragia, menorrhagia, oligo-/amenorhea, nieregularne miesiączkowanie, dysmenorea), ból w okolicy miednicy, wydzielanie z szyjki macicy |
Wzrost gruczołów mlekowych, zespół przedmiesiączkowy (PMS) |
|
| Stan ogólny i reakcje w miejscu podania |
Stany asteniczne (astenia, zmęczenie, złe samopoczucie), obrzęk obwodowy |
|||
| Odstępstwa od normy wykryte podczas badań |
Wzrost masy ciała |
Spadek masy ciała |
*Niepożądane reakcje zgłaszane w oparciu o doniesienia spontaniczne, które nie występowały podczas badań klinicznych, dodano do częstości „rzadko”.
** Szczegóły poniżej.
Ryzyko raka piersi
Zgłaszano podwojenie ryzyka rozpoznania raka piersi u kobiet przyjmujących kombinowaną terapię estrogenową i progestagenową przez ponad 5 lat.
Zwiększony ryzyko u kobiet przyjmujących monoterapię estrogenową jest niższe niż u kobiet przyjmujących kombinowaną terapię estrogenową i progestagenową.
Poziom ryzyka zależy od długości stosowania (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Poniżej przedstawiono oszacowanie ryzyka absolutnego na podstawie największego randomizowanego, placebo-kontrolowanego badania Women’s Health Initiative (WHI) oraz największego metaanalizy badań epidemiologicznych prospektywnych.
Tabela 4
Największa metaanaliza prospektywnych badań epidemiologicznych
Szacowane dodatkowe ryzyko raka piersi po 5 latach stosowania u kobiet o wskaźniku masy ciała 27 (kg/m²)
| Wiek na początku HTZ (lata) |
Liczba przypadków na 1000 kobiet, które nigdy nie stosowały HTZ, w ciągu 5-letniego okresu (50–54 lata)1 |
Stosunek ryzyka |
Liczba dodatkowych przypadków na 1000 kobiet stosujących HTZ po 5 latach |
| HTZ z zastosowaniem wyłącznie estrogenów |
|||
| 50 |
13,3 |
1,2 |
2,7 |
| HTZ z zastosowaniem kombinacji estrogenów i progestagenów |
|||
| 50 |
13,3 |
1,6 |
8,0 |
| Uwaga. Ponieważ zachorowalność na raka piersi różni się w poszczególnych krajach UE, liczba dodatkowych przypadków raka piersi również będzie się różnić proporcjonalnie. 1 Przyjęte na podstawie podstawowych wskaźników zachorowalności w Anglii w 2015 roku u kobiet o wskaźniku masy ciała 27 (kg/m2). |
|||
Tabela 5
Obliczone dodatkowe ryzyko raka piersi po 10 latach stosowania u kobiet z indeksem masy ciała 27 (kg/m²)
| Wiek na początku HTZ (lata) |
Liczba przypadków na 1000 kobiet, które nigdy nie stosowały HTZ, w ciągu 10 lat (50–59 lat)1 |
Stosunek ryzyka |
Liczba dodatkowych przypadków na 1000 kobiet stosujących HTZ, po 10 latach |
| HTZ z zastosowaniem wyłącznie estrogenów |
|||
| 50 |
26,6 |
1,3 |
7,1 |
| HTZ z zastosowaniem kombinacji estrogenów i progestagenów |
|||
| 50 |
26,6 |
1,8 |
20,8 |
| 1 Przyjęte na podstawie podstawowych wskaźników zachorowań w Anglii w 2015 roku u kobiet o wskaźniku masy ciała 27 (kg/m2). Uwaga: ponieważ wskaźniki zachorowań na raka piersi różnią się w poszczególnych krajach UE, liczba dodatkowych przypadków raka piersi również będzie się proporcjonalnie zmieniać. |
|||
Tabela 6
Badania WHI w USA: dodatkowe ryzyko raka piersi po 5 latach stosowania
| Zakres wiekowy (lata) |
Liczba przypadków na 1000 kobiet w grupie placebo w ciągu 5 lat |
Stosunek ryzyka i 95% przedział ufności (PU) |
Liczba dodatkowych przypadków na 1000 kobiet stosujących HRT przez 5 lat (95% PU) |
| Monoterapia estrogenowa koniugowanymi estrogenami kanińskimi (CEE) |
|||
| 50–79 |
21 |
0,8 (0,7–1,0) |
|
| CEE + MPA skojarzona terapia estrogenowo-progesteronowa HRT ‡ |
|||
| 50–79 |
17 |
1,2 (1,0–1,5) |
+4 (0–9) |
| ‡ W analizie ograniczonej do kobiet nie stosujących HRT przed rozpoczęciem badania nie stwierdzono wyraźnego ryzyka w pierwszych 5 latach leczenia; po 5 latach ryzyko było wyższe niż u kobiet nie stosujących HRT. 2 Badanie WHI z udziałem kobiet po hysterectomii, które nie wykazały zwiększonego ryzyka raka piersi. CCE – koniugowane estrogeny kanińskie, MPA – acetat medroksyprogesteronu |
|||
Ryzyko raka endometrium
Kobiety w okresie pomenopauzalnym z zachowanym macicą
Ryzyko raka endometrium wynosi około 5 przypadków na 1000 kobiet z zachowanym macicą, które nie stosują HTZ.
HTZ z zastosowaniem wyłącznie estrogenów nie jest zalecane kobietom z zachowanym macicą, ponieważ zwiększa ono ryzyko raka endometrium (patrz sekcja „Szczególne środki ostrożności podczas stosowania”). W zależności od długości trwania monoterapii estrogenami oraz dawki estrogenów, w badaniach epidemiologicznych wzrost ryzyka raka endometrium wahał się od 5 do 55 dodatkowych przypadków rozpoznanych u każdej 1000 kobiet w wieku od 50 do 65 lat.
Dodanie progestagenu do monoterapii estrogenami przez co najmniej 12 dni w cyklu może zapobiegać temu zwiększeniu ryzyka. W badaniu Million Women Study stosowanie przez pięć lat skojarzonej HTZ (kolejnej lub ciągłej) nie zwiększało ryzyka raka endometrium (stosunek ryzyka 1,0 (0,8–1,2)).
Rak jajników
Stosowanie HTZ wyłącznie z estrogenami lub skojarzonej terapii estrogenowo-progestagenowej HTZ wiązało się z nieco zwiększonym ryzykiem raka jajników (patrz sekcja „Szczególne środki ostrożności podczas stosowania”).
W danych metaanalizy uzyskanych z 52 badań epidemiologicznych, donoszono o zwiększonego ryzyka rozwoju raka jajników u kobiet stosujących HTZ w porównaniu z kobietami, które nigdy HTZ nie stosowały (stosunek ryzyka 1,43, 95 %; przedział ufności (PU) 1,31–1,56). U kobiet w wieku od 50 do 54 lat, które stosowały HTZ przez 5 lat, prowadziło to do 1 dodatkowego przypadku na 2000 kobiet. U kobiet w wieku od 50 do 54 lat, które nie stosowały HTZ, rak jajników rozpoznaje się u około 2 na 2000 kobiet w ciągu 5 lat.
Ryzyko zatorowości żylnej
HTZ wiąże się z 1,3–3-krotnym wzrostem względnego ryzyka wystąpienia zatorowości żylnej (ZŻ), tj. zakrzepicy żył głębokich lub zatoru tętnicy płucnej. Wystąpienie takiego zjawiska jest bardziej prawdopodobne w pierwszym roku stosowania HTZ (patrz sekcja „Szczególne środki ostrożności podczas stosowania”). Poniżej przedstawiono wyniki badań WHI.
Tabela 7
Badanie WHI: dodatkowe ryzyko ZŻ w ciągu 5 lat stosowania
| Zakres wiekowy (lata) |
Liczba przypadków na 1000 kobiet w grupie placebo przez 5 lat |
Stosunek ryzyka i 95 % przedział ufności |
Liczba dodatkowych przypadków na 1000 kobiet przyjmujących ZGH przez 5 lat (95 % przedział ufności) |
| Peroralna jednoterapia hormonalna estrogenowa3 |
|||
| 50–59 |
7 |
1,2 (0,6–2,4) |
1 (–3–10) |
| Peroralne przyjmowanie kombinacji estrogenów i progestagenu |
|||
| 50–59 |
4 |
2,3 (1,2–4,3) |
5 (1–13) |
3 Badania u kobiet z usuniętym macicą.
Zagrożenie chorobą wieńcową
Zagrożenie chorobą wieńcową jest nieco zwiększone u kobiet przyjmujących skojarzoną terapię hormonalną estrogen-progesteron po 60. roku życia (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”).
Zagrożenie udarem niedokrwiennym
Stosowanie monoterapii estrogenem oraz skojarzonej terapii hormonalnej estrogen-progesteron wiąże się ze wzrostem względnego ryzyka udaru niedokrwiennego do 1,5-krotności. Ryzyko udaru krwotocznego nie wzrasta podczas stosowania TH.
Względne ryzyko nie zależy od wieku ani długości stosowania, jednak ponieważ podstawowe ryzyko w dużym stopniu zależy od wieku, całkowite ryzyko udaru u kobiet przyjmujących TH będzie wzrastać wraz z wiekiem (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”).
Tabela 8
Połączone dane badań WHI: dodatkowe ryzyko udaru niedokrwiennego\4 podczas 5-letniego stosowania
| Zakres wiekowy (lat) |
Liczba przypadków na 1000 kobiet w grupie placebo przez 5 lat |
Stosunek ryzyka i 95 % przedział ufności |
Liczba dodatkowych przypadków na 1000 kobiet przyjmujących RTG przez 5 lat (95 % przedział ufności) |
| 50–59 |
8 |
1,3 (1,1–1,6) |
3 (1–5) |
4 Różnica między udarem niedokrwiennym a udarem krwotocznym nie została stwierdzona.
Inne działania niepożądane zgłaszane w związku z terapią estrogenową/progestagenową (w tym estradiolu/dydrogesteronu):
-
nowotwory łagodne, złośliwe i o niejednoznacznej etiologii: nowotwory zależne od estrogenów, zarówno łagodne, jak i złośliwe, np. raka endometrium i jajników; zwiększenie rozmiaru nowotworów zależnych od progestagenów (np. meningiom);
-
zaburzenia układu odpornościowego: toczeń rumieniowaty układowy (SLE);
-
zaburzenia przemiany materii i odżywiania: hipertriglicerydemia;
-
zaburzenia ze strony układu nerwowego środkowego: możliwa demencja, chorea, nasilenie przebiegu padaczki;
-
zaburzenia ze strony naczyń: zakrzepica tętnicza;
-
zaburzenia ze strony przewodu pokarmowego: zapalenie trzustki (u kobiet z istniejącą hipertriglicerydemią);
-
zaburzenia ze strony skóry i tkanki podskórnej: rumień wielopostaciowy;
-
zaburzenia ze strony nerek i dróg moczowych: nietrzymanie moczu;
-
zaburzenia ze strony układu rozrodczego i gruczołów mlekowych: zmiany włóknisto-torbielowate w gruczołach mlekowych, erozja szyjki macicy;
-
wady wrodzone i genetyczne: nasilenie przebiegu porfirii;
-
odchylenia od normy wykryte podczas badań: podwyższony ogólny poziom hormonów tarczycy.
Zgłaszanie działań niepożądanych po rejestracji leku ma duże znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich uprawnieni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 3 lata.
Warunki przechowywania.
Przechowywać w oryginalnym opakowaniu w temperaturze nie wyższej niż 30 °C. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
Opakowanie kombi: 28 tabletek w blisterze (14 tabletek o białym kolorze, pokrytych powłoką filmową, po 1 mg + 14 tabletek o szarym kolorze, pokrytych powłoką filmową, po 1 mg/10 mg w blisterze). Po 1, 2 lub 3 blisterach w pudełku.
Opakowanie kombi: 28 tabletek w blisterze (14 tabletek o czerwono-czerwonym kolorze, pokrytych powłoką filmową, po 2 mg + 14 tabletek o żółtym kolorze, pokrytych powłoką filmową, po 2 mg/10 mg w blisterze). Po 1, 2 lub 3 blisterach w pudełku.
Kategoria wydawania. Na receptę.
Producent. Abbott Biologicals B.V./ Abbott Biologicals B.V.
Miejsce położenia producenta oraz adres miejsca prowadzenia działalności.
Veerweg 12, 8121 AA Olst, Holandia/ Veerweg 12, 8121 AA Olst, The Netherlands.