Cabomectics
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku CABOMETYX®
Skład:
substancja czynna: kabozantynib (S)-maloł
1 tabletka powlekana zawiera kabozantynibu (S)-maloł odpowiadające kabozantynibowi 20 mg lub 40 mg, lub 60 mg;
substancje pomocnicze: celuloza mikrokryształowa, laktoza bezwodna, hydroksypropyloceluloza, sodowa sól kroskarboksymetlocelulozy, dwutlenek krzemu koloidalny (bezwodny), stearynian magnezu;
powłoka filmowa: Opadry Yellow (03K92254): hydroksypropyloceluloza (HPMC) 2910 (E 464), dwutlenek tytanu (E 171), triacetyna, tlenek żelaza żółty (E 172).
Postać leku.
Tabletki powlekane.
Główne właściwości fizykochemiczne.
Cabomectics, tabletki powlekane, 20 mg
Okrągłe tabletki powlekane, żółte, z oznaczeniem tłoczonym „XL” po jednej stronie i „20” po drugiej stronie tabletu.
Cabomectics, tabletki powlekane, 40 mg
Trójkątne tabletki powlekane, żółte, z oznaczeniem tłoczonym „XL” po jednej stronie i „40” po drugiej stronie tabletu.
Cabomectics, tabletki powlekane, 60 mg
Owalne tabletki powlekane, żółte, z oznaczeniem tłoczonym „XL” po jednej stronie i „60” po drugiej stronie tabletu.
Grupa farmakoterapeutyczna.
Leki przeciwnowotworowe. Inhibitory kinaz białkowych. Kabozantynib.
Kod ATC L01E X07.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Kabozantynib – niskocząsteczkowy inhibitor tyrozynokinazy, który hamuje wiele receptorowych tyrozynokinaz (RTK), uczestniczących w procesach wzrostu nowotworu i angiogenezie, patologicznych zmianach tkanki kostnej, oporności na leki oraz progresji przerzutowej nowotworu. W wyniku oceny aktywności hamowania różnych kinaz, kabozantynib został sklasyfikowany jako inhibitor receptorów MET (receptor czynnika wzrostu hepatocytów) oraz receptorów VEGF (czynnik wzrostu śródbłonka naczyń). Ponadto kabozantynib hamuje inne tyrozynokinazy, w tym receptor GAS6 (AXL), RET, ROS1, TYRO3, MER, receptor czynnika wzrostu komórek macierzystych (KIT), TRKB, podobną do fms tyrozynokinazę-3 (FLT3) oraz TIE-2.
Skutki farmakodynamiczne
Kabozantynib wykazywał dawkowo zależne hamowanie wzrostu guza, regresję guza oraz/lub ograniczenie przerzutów w szerokim zakresie modeli przedklinicznych nowotworów.
Elektrofizjologia kardiologiczna
W kontrolowanym badaniu klinicznym u chorych na raka rdzeniastego tarczycy zaobserwowano wydłużenie interwału QT skorygowanego wg Fridericia (QTcF) o 10–15 ms w dniu 29 (ale nie w dniu 1) po rozpoczęciu leczenia kabozantynibem (w dawce 140 mg na dobę). Ten efekt nie był związany ze zmianami morfologicznymi fali przedsionkowej ani wystąpieniem nowych arytmii serca. W tym badaniu żaden pacjent leczony kabozantynibem nie miał potwierdzonego QTcF > 500 ms, co nie zostało potwierdzone przy leczeniu NKK lub GCT (w dawce 60 mg).
Skuteczność i bezpieczeństwo kliniczne
Skuteczność kliniczna w przypadku nowotworu komórek nerkowych (NKK) po wcześniejszej terapii lekami wpływającymi na czynnik wzrostu śródbłonka naczyń (VEGF)
Bezpieczeństwo i skuteczność leku Cabomectics w leczeniu nowotworu komórek nerkowych po wcześniejszej terapii lekami wpływającymi na VEGF oceniano w randomizowanym, otwartym, wieloośrodkowym badaniu fazy III (METEOR). Pacjentów z rozsianym jasnokomórkowym nowotworem komórek nerkowych (NKK), którzy wcześniej otrzymali co najmniej jeden cykl leczenia inhibitorami tyrozynokinaz receptora VEGF (VEGFR TKI), podzielono losowo (1:1) na grupę przyjmującą Cabomectics (N = 330) lub ewerolimus (N = 328). Pacjenci mogli wcześniej otrzymać inne cykle terapii, w tym cytokiny lub przeciwciała skierowane przeciwko VEGF, receptorem śmierci zaprogramowanej komórek-1 (PD-1) lub ich ligandom. Dozwolone były również wcześniejsze leczone przypadki z przerzutami do mózgu. Przeżycie bez postępu choroby oceniano przez komitet przeprowadzający niezależną, ślepą ekspertyzę radiologiczną, a analizę pierwotną przeprowadzono wśród pierwszych 375 zrandomizowanych pacjentów. Punkty końcowe wtórne obejmowały odsetek pacjentów z obiektywną odpowiedzią oraz przeżycie ogólne (PO). Oceny guza przeprowadzano co 8 tygodni przez pierwsze 12 miesięcy, a następnie co 12 tygodni.
Podstawowe dane demograficzne i charakterystyka choroby były podobne w grupach przyjmujących Cabomectics i ewerolimus. Większość pacjentów to mężczyźni (75%), średni wiek wynosił 62 lata. 71% pacjentów otrzymało tylko jeden poprzedni cykl VEGFR TKI; 41% pacjentów otrzymało sunitynib jako jedyny poprzedni cykl VEGFR TKI. Według kryteriów kategorii ryzyka prognostycznego Memorial Sloan Kettering Cancer Center (MSKCC), 46% miało rokowanie korzystne (0 czynników ryzyka), 42% – pośrednie (1 czynnik ryzyka), 13% – złe (2 lub 3 czynniki ryzyka). 54% pacjentów miało przerzuty do 3 lub więcej narządów, w tym do płuc (63%), węzłów chłonnych (62%), wątroby (29%) i kości (22%). Średnia długość leczenia wynosiła 7,6 miesiąca (zakres 0,3–20,5) u pacjentów przyjmujących Cabomectics oraz 4,4 miesiąca (zakres 0,21–18,9) u pacjentów leczonych ewerolimusem.
Stwierdzono istotne statystycznie poprawienie przeżycia bez postępu choroby przy stosowaniu Cabomectics w porównaniu z ewerolimusem (rysunek 1 oraz tabela 1). Planowany analizę pośredni przeżycia ogólnego przeprowadzono podczas analizy przeżycia bez postępu choroby i nie osiągnięto granicy istotności statystycznej (202 zdarzenia, HR = 0,68 [0,51, 0,90], p = 0,006). W kolejnej, niezaplanowanej analizie pośredniej przeżycia ogólnego wykazano istotne statystycznie poprawienie u pacjentów przyjmujących Cabomectics w porównaniu z grupą ewerolimusu (320 zdarzeń, mediana 21,4 miesiąca i 16,5 miesiąca, HR = 0,66 [0,53, 0,83], p = 0,0003, rysunek 2). Porównywalne wyniki dotyczące PO zaobserwowano w dalszej (opisowej) analizie 430 zdarzeń.
Analiza badawcza przeżycia bez postępu choroby oraz przeżycia ogólnego w grupie „wszyscy zrandomizowani pacjenci, którzy otrzymali co najmniej jedną dawkę badanego leku” również wykazała wiarygodne wyniki na korzyść Cabomectics w porównaniu z ewerolimusem w różnych podgrupach według wieku (< 65 lat w porównaniu z > 65 lat), płci, grupy ryzyka Memorial Sloan Kettering Cancer Center (korzystna, pośrednia, zła), statusu ECOG (0 lub 1), czasu od postawienia diagnozy do randomizacji (< 1 roku w porównaniu z > 1 roku), statusu MET guza (wysoki w porównaniu z niskim lub nieznanym), przerzutów do kości (brak w porównaniu z obecnością), przerzutów wisceralnych (brak w porównaniu z obecnością), przerzutów do kości i przerzutów wisceralnych (brak w porównaniu z obecnością), liczby poprzednich cykli VEGFR-TKI (1 w porównaniu z > 2), długości trwania pierwszej terapii VEGF (< 6 miesięcy w porównaniu z > 6 miesięcy). Wyniki dotyczące odsetka pacjentów z obiektywną odpowiedzią przedstawiono w tabeli 2.
| Liczba pacjentów w grupie ryzyka Cabomectics Everolimus |
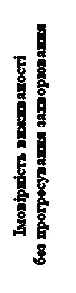
| Cabomectics Everolimus |
| Miesiące |
Rys. 1. Krzywa przeżycia wolnego od progresji choroby, określona przez niezależny komitet ekspertów radiologii, u pacjentów z nienaczyniakowym rakiem komórkowym (NKRK) po wcześniejszym leczeniu lekami wpływającymi na czynnik wzrostu śródbłonka naczyń (VEGF) (pierwszych 375 pacjentów wybranych losowo) (metoda Kaplana-Meiera, badanie METEOR)
Tabela 1
Podsumowanie wyników przeżycia wolnego od progresji choroby (PFS), określonego przez niezależny komitet ekspertów radiologii, u pacjentów z NKRK po wcześniejszym leczeniu lekami wpływającymi na czynnik wzrostu śródbłonka naczyń (VEGF) (badanie METEOR)
| Pierwotna analiza PFS w populacji |
Populacja ITT |
|||
| Punkt końcowy |
Cabomectics |
Everolimus |
Cabomectics |
Everolimus |
| N = 187 |
N = 188 |
N = 330 |
N = 328 |
|
| Mediana czasu PFS (95 % CI – przedział ufności), miesiące |
7,4 (5,6; 9,1) |
3,8 (3,7; 5,4) |
7,4 (6,6; 9,1) |
3,9 (3,7; 5,1) |
| Stosunek ryzyka (HR) (95 % CI), wartość p1 |
0,58 (0,45; 0,74), p < 0,0001 |
0,51 (0,41; 0,62), p < 0,0001 |
||
1 Ustratyzfikowany test rangowy logarytmiczny.
| Liczba pacjentów w grupie ryzyka Cabomectics Everolimus |
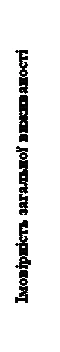
| Cabomectics Everolimus |
| Miesiące |
Rys. 2. Krzywa całkowitego przeżycia u pacjentów z RCC po wcześniejszej terapii lekami oddziałującymi na czynnik wzrostu śródbłonka naczyń (VEGF) (metoda Kaplana-Meiera, badanie METEOR)
Tabela 2
Podsumowanie danych dotyczących liczby pacjentów z obiektywną odpowiedzią zgodnie z raportem niezależnego komitetu ekspertyz radiologicznych (IRC) oraz raportem badaczy wśród chorych na RCC po wcześniejszej terapii lekami oddziałującymi na czynnik wzrostu śródbłonka naczyń (VEGF)
| Koniec punktu |
Pierwotna analiza wskaźnika obiektywnej odpowiedzi w populacji ITT, dane IRC |
Wskaźnik obiektywnej odpowiedzi w populacji ITT, dane badaczy |
||
| Cabomectics |
Everolimus |
Cabomectics |
Everolimus |
|
| N = 330 |
N = 328 |
N = 330 |
N = 328 |
|
| Wskaźnik obiektywnej odpowiedzi (tylko częściowa obiektywna odpowiedź) (95% CI) |
17 % (13 %, 22 %) |
3 % (2 %, 6 %) |
24 % (19 %, 29 %) |
4 % (2 %, 7 %) |
| Wartość p1 |
p < 0,0001 |
p < 0,0001 |
||
| Częściowa odpowiedź |
17 % |
3 % |
24 % |
4 % |
| Mediana czasu do pierwszej odpowiedzi, miesiące (95% CI) |
1,91 (1,6; 11,0) |
2,14 (1,9; 9,2) |
1,91 (1,3; 9,8) |
3,50 (1,8; 5,6) |
| Stabilizacja choroby jako najlepsza odpowiedź |
65 % |
62 % |
63 % |
63 % |
| Postęp choroby jako najlepsza odpowiedź |
12 % |
27 % |
9 % |
27 % |
1 Kryterium chi-kwadrat.
Dane kliniczne dotyczące leczenia pacjentów z komórkowym rakiem nerek, którzy wcześniej nie otrzymywali leczenia
Bezpieczeństwo i skuteczność leku CABOMECTICS u pacjentów z komórkowym rakiem nerek, którzy wcześniej nie otrzymywali leczenia, oceniano w randomizowanym, otwartym, wieloośrodkowym badaniu (CABOSUN). Pacjenci (N = 157) z wcześniej nielеченym, lokalnie zaawansowanym lub przerzutowym jasnokomórkowym rakiem nerek (NKK) zostali zrandomizowani (1:1) do otrzymywania leku CABOMECTICS (N = 79) lub sunitinibu (N = 78). Pacjenci powinni byli mieć chorobę o średnim lub wysokim ryzyku zgodnie z definicjami kategorii ryzyka według klasyfikacji Międzynarodowego Konsorcjum Baz Danych dotyczących przerzutowego NKK (IMDC). Pacjentów losowano według grupy ryzyka IMDC oraz obecności przerzutów kostnych (tak/nie). Około 75 % pacjentów poddano operacji usunięcia nerki przed rozpoczęciem leczenia.
Pacjent powinien mieć jeden lub dwa poniższe czynniki ryzyka w przypadku średniego ryzyka oraz trzy lub więcej czynników w przypadku wysokiego ryzyka: czas od rozpoznania NKK do leczenia systemowego < 1 rok, hemoglobina < NMN (dolna granica normy), skorygowany poziom wapnia > GMN (górna granica normy), wskaźnik KPS < 80 %, neutrofile > GMN, płytki > GMN.
Pierwotnym punktem końcowym była przeżycie bez postępu choroby. Wtórne punkty końcowe skuteczności określono jako wskaźnik obiektywnej odpowiedzi oraz ogólne przeżycie. Ocena guza była przeprowadzana co 12 tygodni.
Wyjściowe dane demograficzne i charakterystyka choroby były podobne w grupach stosowania leku CABOMECTICS i sunitinibu. Większość pacjentów stanowili mężczyźni (78 %); średni wiek wynosił 62 lata. Rozkład pacjentów według grup ryzyka IMDC był następujący: 81 % pacjentów w grupie średniego ryzyka (1–2 czynniki ryzyka), a 19 % – w grupie wysokiego ryzyka (≥ 3 czynników ryzyka). Większość pacjentów (87 %) miała ocenę według statusu funkcjonalnego ECOG 0 lub 1; 13 % – ocenę 2. Przerzuty kostne wykryto u 36 % pacjentów.
Statystycznie istotne poprawy przeżycia bez postępu choroby, ocenianej retrospektywnie przez niezależny komitet prowadzący ślepe radiologiczne recenzje (IRC), wykazały korzyści leku CABOMECTICS w porównaniu do sunitinibu (rysunek 3, tabela 3). Wyniki analizy przeprowadzonej przez badacza i IRC dotyczące przeżycia bez postępu choroby były zgodne między sobą.
Pacjenci z dodatnim i ujemnym statusem MET wykazali korzystny efekt leku CABOMECTICS w porównaniu do sunitinibu; przy czym większą aktywność obserwowano u pacjentów z dodatnim statusem MET w porównaniu do pacjentów z ujemnym statusem MET (HR = 0,32 (0,16; 0,63) w porównaniu do 0,67 (0,37; 1,23) odpowiednio).
Terapię lekiem CABOMECTICS wiązano z tendencją do dłuższego przeżycia w porównaniu do sunitinibu (tabela 3). Badanie to nie było wystarczająco mocyne, aby przeanalizować ogólne przeżycie, a uzyskane dane były nie dojrzałe.
Dane dotyczące wskaźnika obiektywnej odpowiedzi przedstawiono w tabeli 3.
| Liczba pacjentów w grupie ryzyka Cabomectics Everolimus |
| Cabomectics Everolimus |
| Miesiące |
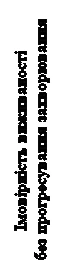
Rys. 3. Krzywa przeżycia wolnego od progresji choroby u pacjentów z RCC, którzy wcześniej nie byli leczoni (dane IRC, metoda Kaplana-Meiera)
Tabela 3
Wyniki skuteczności u pacjentów z RCC, którzy wcześniej nie byli leczeni (populacja ITT, badanie CABOSUN)
| Wskaźnik |
Cabomectics (N = 79) |
Sunitynib (N = 78) |
| Przeżycie wolne od postępu choroby według danych IRC |
||
| Mediana przeżycia wolnego od postępu (95 % CI) |
8,6 (6,2; 14,0) |
5,3 (3,0; 8,2) |
| HR (95 % CI); skategoryzowane, b |
0,48 (0,32; 0,73) |
|
| Dwustronna wartość p logarytmicznego testu rangowego: skategoryzowane |
p = 0,0005 |
|
| Przeżycie wolne od postępu choroby według danych badacza |
||
| Mediana przeżycia wolnego od postępu (95 % CI) |
8,3 (6,5; 12,4) |
5,4 (3,4; 8,2) |
| HR (95 % CI), skategoryzowane, b |
0,56 (0,37; 0,83) |
|
| Dwustronna wartość p logarytmicznego testu rangowego: skategoryzowane |
p = 0,0042 |
|
| Całkowite przeżycie |
||
| Mediana całkowitego przeżycia w miesiącach (95 % CI) |
30,3 (14,6; NE) |
21,0 (16,3; 27,0) |
| HR (95 % CI), skategoryzowane, b |
0,74 (0,47; 1,14) |
|
| Wskaźnik obiektywnej odpowiedzi, n (%) według danych IRC |
||
| Pełne odpowiedzi |
0 |
0 |
| Cząstkowe odpowiedzi |
16 (20) |
7 (9) |
| Odpowiedź obiektywna (tylko częściowe odpowiedzi) |
16 (20) |
7 (9) |
| Stabilizacja choroby |
43 (54) |
30 (38) |
| Postęp choroby |
14 (18) |
23 (29) |
| Wskaźnik obiektywnej odpowiedzi, n (%) według danych badacza |
||
| Pełne odpowiedzi |
1 (1) |
0 |
| Cząstkowe odpowiedzi |
25 (32) |
9 (12) |
| Odpowiedź obiektywna (tylko częściowe odpowiedzi) |
26 (33) |
9 (12) |
| Stabilizacja choroby |
34 (43) |
29 (37) |
| Postęp choroby |
14 (18) |
19 (24) |
a Zgodnie z przyjętymi w UE zasadami cenzurowania danych.
b Czynniki stratyfikacji zgodnie z IxRS obejmowały kategorie ryzyka IMDC (ryzyko średnie, ryzyko wysokie oraz obecność przerzutów do kości (tak/nie)).
c Obliczone na podstawie modelu proporcjonalnych ryzyk Coxa z korektą uwzględniającą czynniki stratyfikacji zgodnie z IxRS. Stosunek ryzyka < 1 wskazuje na korzyść przeżycia bez postępu choroby dla kabozantynibu.
Kliniczne dane dotyczące raka hepatocelularnego (HCC)
Bezpieczeństwo i skuteczność leku Cabomectics oceniano w randomizowanym, podwójnie ślepych, placebo-kontrolowanym badaniu fazy III (CELESTIAL). Pacjenci (N = 707) z HCC niewrażliwym na leczenie radykalne, którzy wcześniej otrzymywali sorafenib z powodu zaawansowanej postaci choroby, zostali randomizowani w stosunku 2:1 w celu otrzymania leku Cabomectics (N = 470) lub placebo (N = 237). Pacjenci mogli wcześniej otrzymać jeszcze jedną terapię systemową z powodu zaawansowanej postaci choroby oprócz sorafenibu. Randomizacja była stratyfikowana według etiologii choroby (wirus zapalenia wątroby typu B [z lub bez wirusa zapalenia wątroby typu C], wirus zapalenia wątroby typu C [bez wirusa zapalenia wątroby typu B] lub inna kategoria pacjentów), regionu geograficznego (Azja, inne regiony) oraz obecności rozsiewu choroby poza wątrobą i/lub makroskopowego zaangażowania naczyń (tak/nie).
Pierwotnym punktem końcowym oceny skuteczności była całkowita przeżywalność. Wtórne punkty końcowe skuteczności obejmowały przeżycie wolne od progresji choroby oraz wskaźnik odpowiedzi obiektywnej ocenianej przez badacza z wykorzystaniem Kryteriów Oceniania Odpowiedzi na Leczenie w Guzach Stałych (RECIST) w wersji 1.1. Oceny guzów przeprowadzano co 8 tygodni. Uczestnicy kontynuowali leczenie metodą ślepą po potwierdzeniu radiologicznym progresji choroby, dopóki odnosili korzyści kliniczne lub dopóki nie stwierdzono potrzeby dalszej terapii systemowej lub lokalnej terapii przeciwnowotworowej skierowanej na wątrobę. Przejście z placebo na kabozantynib nie było dozwolone w trakcie fazy ślepej leczenia.
Podstawowe dane demograficzne i charakterystyka choroby były podobne w grupach leczonych lekiem Cabomectics i placebo i przedstawione poniżej dla wszystkich 707 randomizowanych pacjentów:
-
mężczyźni – 82 %;
-
średni wiek pacjentów – 64 lata;
-
rasa kaukaska – 56 %, Azjaci – 34 %;
-
ocena sprawności według skali ECOG na poziomie 0 – 53 % lub na poziomie 1 – 47 %;
-
niewydolność wątroby stopnia A wg klasyfikacji Childa-Purga – 99 %, stopnia B – 1 %;
-
etiologia HCC obejmowała 38 % pacjentów z wirusem zapalenia wątroby typu B (HBV), 21 % – z wirusem zapalenia wątroby typu C (HCV), 40 % – innych (ani HBV, ani HCV);
-
obecność makroskopowego zaangażowania naczyń i/lub przerzutów poza wątrobę – 78 %;
-
poziomy alfa-fetoproteiny (AFP) ≥ 400 μg/l – 41 %;
-
lokalno-regionalna embolizacja tętnicza lub procedury chemoinfuzji – 44 %;
-
radioterapia przed leczeniem kabozantynibem – 37 %;
-
mediana czasu trwania leczenia sorafenibem – 5,32 miesiąca;
-
72 % pacjentów otrzymało jedną, a 28 % – dwie poprzednie schematy terapii systemowej w leczeniu zaawansowanej postaci choroby.
Statystycznie istotne poprawy całkowitej przeżywalności wykazano dla leku Cabomectics w porównaniu z placebo (tabela 4, rysunek 4).
Dane dotyczące przeżycia wolnego od progresji choroby oraz wskaźnik odpowiedzi obiektywnej przedstawiono w tabeli 4.
Tabela 4
Wyniki skuteczności u pacjentów z HCC (populacja ITT, badanie CELESTIAL)
| Wskaźnik |
Cabomectics (N = 470) |
Placebo (N = 237) |
| Ogólna przeżycie |
||
| Mediana ogólnego przeżycia (95 % CI), miesiące |
10,2 (9,1; 12,0) |
8,0 (6,8; 9,4) |
| HR (95 % CI)1,2 |
0,76 (0,63; 0,92) |
|
| Wartość p 1 |
p = 0,0049 |
|
| Przeżycie bez postępu choroby3 |
||
| Mediana przeżycia bez postępu choroby (95 % CI) |
5,2 (4,0; 5,5) |
1,9 (1,9; 1,9) |
| HR (95 % CI)1 |
0,44 (0,36; 0,52) |
|
| Wartość p1 |
p < 0,0001 |
|
| Ocena odsetka pacjentów bez określonych zdarzeń po 3 miesiącach leczenia (metoda Kaplana-Meiera) |
||
| % (95 % CI) |
67,0 % (62,2 %; 71,3 %) |
33,3 % (27,1 %; 39,7 %) |
| Wskaźnik obiektywnej odpowiedzi, n (%)3 |
||
| Pełne odpowiedzi (CR) |
0 |
0 |
| Cząstkowe odpowiedzi (PR) |
18 (4) |
1 (0,4) |
| Odpowiedź obiektywna (CR+PR) |
18 (4) |
1 (0,4) |
| Wartość p1,4 |
p = 0,0086 |
|
| Stabilizacja choroby |
282 (60) |
78 (33) |
| Postęp choroby |
98 (21) |
131 (55) |
1 Dwustronna, uwarstwiona, logarytmiczna ranga, w której czynnikami uwarstwienia były etiologia choroby (wirus zapalenia wątroby B [z lub bez wirusa zapalenia wątroby C], wirus zapalenia wątroby C [bez wirusa zapalenia wątroby B] lub inna kategoria pacjentów), region geograficzny (Azja, inne regiony) oraz obecność pozajelitowego rozszerzenia choroby i/lub inwazji makrososowatych (tak/nie) (na podstawie danych IVRS).
2 Obliczone na podstawie modelu proporcjonalnych ryzyk Coxa.
3 Według oceny badacza zgodnie z kryteriami RECIST 1.1.
4 Uwarstwiony test Cochrana – Mantela – Haenszla (CMH).
| Miesiące |
| Cabomectics Środek placebo |
| Liczba pacjentów w grupie ryzyka Cabomectics Placebo |

Rys. 4. Krzywa całkowitego przeżycia (metoda Kaplana-Meiera, badanie CELESTIAL)
| Miesiące |
| Liczba pacjentów w grupie ryzyka Cabomectics Placebo |
| Cabomectics Środek placebo |
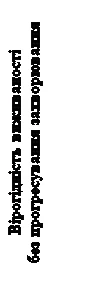
Rys. 5. Krzywa przeżycia bez postępu choroby (metoda Kaplana-Meiera, badanie CELESTIAL)
Udział systemowej terapii przeciwnowotworowej nieobjętej protokołem, niepromieniowej i lokalnie skierowanej na wątrobę (NPACT), wynosił 26% w grupie kabozantynibu i 33% w grupie placebo. Pacjentom, którzy otrzymywali taką terapię, trzeba było przerwać leczenie badanym lekiem. Analiza poszukiwawcza przeżycia całkowitego z cenzurowaniem danych dotyczących zastosowania NPACT dostarczyła danych potwierdzających wyniki analizy pierwotnej: skorygowany wskaźnik HR pod względem czynników stratyfikacji (na IxRS) wyniósł 0,66 (95% CI: 0,52, 0,84; stratyfikowana wartość p log-rank 0,0005). Średnie przeżycie całkowite oszacowane metodą Kaplana-Meiera wynosiło 11,1 miesiąca w grupie kabozantynibu w porównaniu do 6,9 miesiąca w grupie placebo, co daje obliczoną różnicę medianową na poziomie 4,2 miesiąca.
Jakość życia (QoL) oceniano niezależnie od konkretnego schorzenia za pomocą kwestionariusza EuroQoL EQ-5D-5L. Negatywny wpływ leku Cabomectics w porównaniu do placebo na wskaźnik użyteczności EQ-5D obserwowano w pierwszych tygodniach leczenia. Po tym okresie dostępne są jedynie ograniczone dane dotyczące jakości życia.
Populacja pediatryczna
Europejska Agencja Leków odwołała obowiązek przedstawienia wyników badań leku Cabomectics we wszystkich podgrupach populacji pediatrycznej w kontekście leczenia raka hepatocelularnego oraz raka nerek i miednicy nerkowej (z wyjątkiem nefroblastomy, nefroblastomatozy, mięsaka komórek jasnych, mezoblastycznej nerozy, nowotworu medularnego nerek oraz guza rhabdoidowego nerek) (patrz sekcja „Sposób stosowania i dawki” w celu uzyskania informacji dotyczącej stosowania leku u dzieci).
Farmakokinetyka.
Absorpcja
Po podaniu doustnym maksymalna stężenie kabozantynibu w osoczu osiągane jest po 3–4 godzinach. Profile stężenia we krwi w funkcji czasu wykazują drugi szczyt absorpcji około 24 godziny po podaniu, co sugeruje możliwość przeprowadzenia enterohepatalnej recyrkulacji kabozantynibu.
Powtarzanie codziennego podawania kabozantynibu w dawce 140 mg przez 19 dni doprowadziło do wzrostu średniego akumulacji kabozantynibu około 4–5 razy (na podstawie pola pod krzywą farmakokinetyczną „stężenie–czas”) w porównaniu z pojedynczą dawką; stan stacjonarny osiągany był około 15 dnia.
Wysokotłuszczowa dieta umiarkowanie zwiększyła maksymalne stężenie substancji czynnej we krwi i wartość pola pod krzywą farmakokinetyczną „stężenie–czas” (odpowiednio o 41% i 57%) w porównaniu z podaniem na czczo u zdrowych ochotników, którzy przyjmowali doustnie pojedynczą dawkę kabozantynibu 140 mg. Brak danych dotyczących wpływu posiłku spożytego godzinę po podaniu kabozantynibu.
Nie udało się wykazać bioekwiwalencji między kabozantynibem w kapsułkach a w tabletach po podaniu pojedynczej dawki 140 mg zdrowym ochotnikom. Zaobserwowano wzrost o 19% maksymalnego stężenia substancji czynnej we krwi po podaniu tabletek (Cabomectics) w porównaniu z kapsułkami (Cometriq). Różnica poniżej 10% w wartości pola pod krzywą farmakokinetyczną „stężenie–czas” obserwowano po podaniu tabletek kabozantynibu (Cabomectics) i kapsułek (Cometriq).
Rozkład
Kabozantynib charakteryzuje się wysokim stopniem wiązania z białkami osocza ludzkiego (≥ 99,7%). Na podstawie pomiarów farmakokinetycznych objętość rozkładu (Vz) wynosi około 212 l. Wiązanie z białkami osocza nie zmieniało się u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami funkcji nerek lub wątroby.
Biotransformacja
Kabozantynib metabolizuje się in vivo. Cztery metabolity występowały we krwi w stężeniach wpływowych (pole pod krzywą farmakokinetyczną „stężenie–czas”) powyżej 10% w porównaniu z pierwotnym metabolitem: XL184-N-utlenek, produkt rozszczepienia amidu XL184, monohydroksysulfat XL184 oraz sulfat produktu rozszczepienia 6-desmetyloamidu. Dwa niokonjugowane metabolity (XL184-N-utlenek i produkt rozszczepienia amidu XL184) mają < 1% docelowej aktywności hamowania kinazy w porównaniu z pierwotnym kabozantynibem, a każdy z nich stanowi < 10% całkowitego wpływu na osocze krwi związanego z lekiem.
Kabozantynib jest substratem metabolizmu CYP3A4 in vitro, ponieważ przeciwciało neutralizujące CYP3A4 hamuje powstawanie metabolitu XL184-N-utlenku o > 80% w mikrosomalnej inkubacji wątroby ludzkiej katalizowanej przez NADPH; dla porównania przeciwciała neutralizujące CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 i CYP2E1 nie wpływały na powstawanie metabolitów kabozantynibu. Przeciwciało neutralizujące CYP2C9 wykazało minimalny wpływ na powstawanie metabolitu kabozantynibu (tj. zmniejszenie o < 20%).
Wydalanie
Po doustnym podaniu kabozantynibu w dawkach od 20 mg do 140 mg średni czas półtrwania w osoczu krwi u 1883 pacjentów i 140 zdrowych ochotników wynosi około 110 godzin. Średni klirens osoczowy (klirens pozorny) wynosi prawie 2,48 l/h po osiągnięciu stanu stacjonarnego. W ciągu 48 dni po podaniu pojedynczej dawki 14C-kabozantynibu u zdrowych ochotników wykryto około 81% znakowanej izotopowo dawki, z czego niemal 54% dawki wydalono z kałem, a około 27% – z moczem.
Farmakokinetyka u szczególnych grup pacjentów
Niewydolność nerek
Wyniki badania z udziałem pacjentów z niewydolnością nerek, którym podano pojedynczą dawkę kabozantynibu 60 mg, wskazują, że średnie geometryczne stosunki obliczone metodą najmniejszych kwadratów dla kabozantynibu we krwi, maksymalnego stężenia substancji czynnej we krwi i pola pod krzywą farmakokinetyczną „stężenie–czas” od 0 do nieskończoności były wyższe o 19% i 30% u pacjentów z łagodną niewydolnością nerek (90% CI dla maksymalnego stężenia substancji czynnej we krwi: od 91,60% do 155,51%; dla pola pod krzywą „stężenie–czas” od 0 do nieskończoności: od 98,79% do 171,26%) oraz o 2% i 6–7% (90% CI dla maksymalnego stężenia substancji czynnej we krwi: od 78,64% do 133,52%; dla pola pod krzywą „stężenie–czas” od 0 do nieskończoności: od 79,61% do 140,11%) u pacjentów z umiarkowaną niewydolnością nerek w porównaniu z pacjentami z normalną funkcją nerek. Pacjentów z ostrą niewydolnością nerek nie badano.
Niewydolność wątroby
Na podstawie zintegrowanej analizy populacyjnej farmakokinetyki kabozantynibu u zdrowych ochotników i chorych na nowotwory (w tym HCC) nie zaobserwowano klinicznie istotnych różnic w średnim poziomie ekspozycji na kabozantynib we krwi u pacjentów z normalną funkcją wątroby (n = 1425) i u pacjentów z łagodną niewydolnością wątroby (n = 558). Dane dotyczące pacjentów z umiarkowaną niewydolnością wątroby (n = 15) według kryteriów NCI-ODWG (National Cancer Institute Organ Dysfunction Working Group) są ograniczone. Farmakokinetyki kabozantynibu nie oceniano u pacjentów z ciężką niewydolnością wątroby.
Rasa
Analiza populacyjnej farmakokinetyki nie wykazała klinicznie istotnych różnic w farmakokinetyce kabozantynibu w zależności od rasy pacjentów.
Dane kliniczne.
Wskazania.
Rak komórkowy nerek (RKN)
Cabomectics jest wskazany w leczeniu zaawansowanego raka komórkowego nerek (RKN):
- u dorosłych pacjentów, którzy wcześniej nie byli leczeni i u których występuje ryzyko średnie lub wysokie (patrz sekcja „Właściwości farmakodynamiczne”);
- u dorosłych pacjentów po wcześniejszej terapii lekami oddziałującymi na czynnik wzrostu śródbłonka naczyń (VEGF).
Rak komórkowy wątroby (HCC)
Cabomectics jest wskazany jako monoterapia raka komórkowego wątroby (HCC) u dorosłych pacjentów, którzy wcześniej byli leczeni sorafenibem.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych.
Interakcje z innymi lekami i inne rodzaje interakcji.
Wpływ innych leków na kabozantynib
Inhibitory i induktory CYP3A4
Wcześniejsze podawanie klotrykonazolu, silnego inhibitora CYP3A4, w dawce 400 mg raz dziennie przez 27 dni przed podaniem pojedynczej dawki kabozantynibu zwiększało stężenie kabozantynibu we krwi o 38 % (AUC) i zmniejszało klirens kabozantynibu o 29 % u zdrowych ochotników. Silne inhibitory CYP3A4 należy stosować z ostrożnością (w tym rytonawir, itrakonazol, erytromycyna, klaritromycyna, sok grejpfrutowy).
Wcześniejsze podawanie ryfampicyny, silnego induktora CYP3A4, w dawce 600 mg raz dziennie przez 31 dni przed podaniem pojedynczej dawki kabozantynibu zmniejszało stężenie kabozantynibu we krwi o 77 % (AUC) i zwiększało klirens kabozantynibu 4,3-krotnie u zdrowych ochotników. Należy unikać jednoczesnego, długotrwałego stosowania silnych induktorów CYP3A4 (w tym ryfampicyny, karbamazepiny, fenytionu, fenobarbitalu lub leków zawierających napar z zioła św. Jana (Hypericum perforatum)).
Regulatory pH przewodu pokarmowego
Wcześniejsze podawanie ezomeprazolu, inhibitora pompy protonowej (IPP), w dawce 40 mg raz dziennie przez 6 dni przed podaniem pojedynczej dawki kabozantynibu 100 mg wskazuje, że nie ma ono klinicznie istotnego wpływu na stężenie kabozantynibu we krwi (AUC) u zdrowych ochotników. Nie zaleca się dostosowywania dawki przy jednoczesnym stosowaniu kabozantynibu z lekami wpływającymi na pH żołądka (w tym IPP, antagonistami receptora H2 i lekami przeciwwysięcznymi).
Inhibitory MRP2
Dane badań in vitro wskazują, że kabozantynib jest substratem MRP2. W związku z tym podawanie inhibitorów MRP2 może prowadzić do zwiększenia stężenia kabozantynibu we krwi.
Światła kwasów żółciowych
Światła, takie jak cholestrymina i cholestagel, mogą reagować z kabozantynibem i wpływać na jego wchłanianie (lub resorpcję), co może prowadzić do obniżenia dawki i niepożądanych skutków (patrz sekcja „Farmakodynamika”). Kliniczne znaczenie tych potencjalnych oddziaływań jest nieznane.
Wpływ kabozantynibu na inne leki
Wpływ kabozantynibu na farmakokinetykę sterydowych środków antykoncepcyjnych nie był badany. Ponieważ efekt antykoncepcyjny nie może być zagwarantowany, zaleca się stosowanie dodatkowej metody antykoncepcji, w szczególności bariery.
Z uwagi na wysoki stopień wiązania kabozantynibu z białkami osocza (patrz sekcja „Farmakokinetyka”) możliwe jest oddziaływanie z warfaryną – wypieranie z białek osocza. W takim przypadku należy monitorować międzynarodowy współczynnik normalizacji (INR).
Inhibitory P-glikoproteiny
Kabozantynib jest inhibitorem (IC50 = 7,0 μM), ale nie jest substratem aktywności transportowej P-glikoproteiny (P-gp) w systemie dwukierunkowym z wykorzystaniem komórek zaprojektowanych do ekspresji P-glikoproteiny (MDCK-MDR1). Jednoczesne stosowanie inhibitorów P-gp może zwiększać stężenie kabozantynibu we krwi. Pacjentów należy ostrzec przed przyjmowaniem leków wpływających na P-gp (takich jak fexofenadyna, aliskiren, ambrysentan, dabigatranu etexilat, digoksyna, kolchicyna, maravirok, posakonazol, ranolazyna, sakagliptyna, sitagliptyna, talinolol, tolvaptan) podczas przyjmowania kabozantynibu.
Szczególne ostrzeżenia dotyczące stosowania.
Ponieważ większość działań niepożądanych może wystąpić na początku leczenia, konieczna jest staranna kontrola w pierwszych ośmiu tygodniach leczenia w celu wykrycia działań niepożądanych i zwiększonej indywidualnej wrażliwości na lek w celu możliwej modyfikacji dawki. Zwykle wcześnie występujące zdarzenia obejmują hipokalcemię, hipokaliemię, trombocytopenię, nadciśnienie tętnicze, zespół czerwonego oparzenia dłoni i stóp (ZCOPD), proteinurię oraz zaburzenia przewodu pokarmowego (ból brzucha, zapalenie błony śluzowej, zaparcia, biegunka, wymioty).
W celu leczenia podejrzanych działań niepożądanych może być konieczna tymczasowa przerwa w przyjmowaniu leku lub zmniejszenie dawki kabozantynibu (patrz sekcja „Sposób stosowania i dawki”).
W trakcie podstawowego badania klinicznego (badanie METEOR) odpowiednio 59,8 % i 70 % pacjentów z rakiem komórkowym nerek po wcześniejszej terapii lekami wpływającymi na czynnik wzrostu śródbłonka naczyń (VEGF) wymagało odpowiednio zmniejszenia dawki i przerwania terapii z powodu działań niepożądanych. U 19,3 % pacjentów konieczne było dwukrotne zmniejszenie dawki. Średni czas do pierwszego zmniejszenia dawki wynosił 55 dni, a do pierwszej przerwy w dawce – 38 dni.
U pacjentów z rakiem komórkowym nerek, którzy wcześniej nie otrzymywali leczenia, zmniejszenie dawki i przerwanie terapii zastosowano odpowiednio u 46 % i 73 % pacjentów otrzymujących kabozantynib w badaniu klinicznym (badanie CABOSUN).
W trakcie leczenia raka wątrobowokomórkowego po wcześniejszej terapii systemowej zmniejszenie dawki i przerwanie terapii miało miejsce odpowiednio u 62 % i 84 % pacjentów otrzymujących kabozantynib w badaniu klinicznym (badanie CELESTIAL). U 33 % pacjentów konieczne było dwukrotne zmniejszenie dawki. Średni czas do pierwszego zmniejszenia dawki wynosił 38 dni, a do pierwszej przerwy w dawce – 28 dni. Pacjentom z niewydolnością wątroby lekkiego lub umiarkowanego stopnia zaleca się dokładniejsze monitorowanie.
Wpływ na układ wątrobowo-żółciowy
Nieprawidłowe wyniki badań czynności wątroby (podwyższenie poziomów alaninotransferazy (ALT), asparaginianotransferazy (AST) i bilirubiny) są często obserwowane u pacjentów leczonych kabozantynibem. Przed rozpoczęciem leczenia kabozantynibem zaleca się wykonanie badań czynności wątroby (ALT, AST, bilirubina) oraz dokładne monitorowanie tych parametrów w trakcie leczenia. W przypadku pogorszenia wyników badań czynności wątroby w związku z leczeniem kabozantynibem (tj. przy braku alternatywnej przyczyny) należy postępować zgodnie z zaleceniami dotyczącymi modyfikacji dawki przedstawionymi w tabeli 5 (patrz sekcja „Sposób stosowania i dawki”).
Kabozantynib jest wydalany głównie przez wątrobę. Pacjentom z niewydolnością wątroby lekkiego lub umiarkowanego stopnia zaleca się dokładniejsze monitorowanie ogólnej bezpieczeństwa (patrz również sekcje „Szczególne ostrzeżenia dotyczące stosowania” i „Farmakokinetyka”). Encefalopatia wątrobowa podczas leczenia kabozantynibem występowała częściej u większej części pacjentów z niewydolnością wątroby umiarkowanego stopnia (klasa B wg skali Childa-Pugh). KABO\ME\TI\KS nie powinien być stosowany u pacjentów z niewydolnością wątroby ciężkiego stopnia (klasa C wg skali Childa-Pugh), ponieważ kabozantynib nie był badany w tej populacji, a jego ekspozycja może wzrosnąć u tych pacjentów.
Encefalopatia wątrobowa
W badaniu raka wątrobowokomórkowego (badanie CELESTIAL) częściej zgłaszano przypadki encefalopatii wątrobowej w grupie kabozantynibu niż w grupie placebo. W trakcie stosowania kabozantynibu występowała biegunka, wymioty, spadek apetytu i zaburzenia równowagi elektrolitowej. U pacjentów z rakiem wątrobowokomórkowym i zaburzeniem funkcji wątroby te reakcje ze strony innych układów i narządów mogą być zapowiedzią rozwoju encefalopatii wątrobowej. Zaleca się kontrolowanie stanu pacjenta pod kątem wystąpienia objawów i objawów encefalopatii wątrobowej.
Perforacje i przetoki
W trakcie leczenia kabozantynibem obserwowano poważne perforacje przewodu pokarmowego (PP) oraz przetoki, w tym zakończone śmiertelnie. Stan pacjentów z chorobą zapalną jelit (np. chorobą Crohna, wrzodziejącym zapaleniem jelita, zapaleniem otrzewnej, zapaleniem zatok, zapaleniem wyrostka robaczkowego), infiltracją guza do PP lub powikłaniami po wcześniejszej operacji PP (szczególnie jeśli wiąże się to z opóźnionym lub niepełnym gojeniem rany) należy starannie ocenić przed rozpoczęciem leczenia kabozantynibem oraz kontrolować w trakcie leczenia pod kątem wystąpienia objawów perforacji i przetok, w tym zakażeń ropnych i sepsy. Ustępująca lub okresowa biegunka w trakcie leczenia może być czynnikiem ryzyka rozwoju przetoki odbytnicy. Leczenie kabozantynibem należy całkowicie przerwać pacjentom, u których wystąpiła perforacja PP lub przetoka, której nie można odpowiednio wyleczyć.
Zaburzenia ze strony przewodu pokarmowego (PP)
Biegunka, nudności/wymioty, spadek apetytu oraz zapalenie jamy ustnej/ból w jamie ustnej są najczęściej zgłaszanymi działaniami niepożądanymi ze strony PP (patrz sekcja „Działania niepożądane”). W celu zapobiegania odwodnieniu, zaburzeniom równowagi elektrolitowej i utracie masy ciała należy szybko rozpocząć leczenie farmakologiczne, w tym leczenie wspierające przeciwwymiotne, leki przeciwbiegunkowe i środki przeciwwymiotne. W przypadku trwających lub nawracających działań niepożądanych ze strony PP należy rozważyć możliwość wstrzymania terapii lub zmniejszenia dawki lub całkowitego odstawienia kabozantynibu (patrz tabela 5).
Zdarzenia zakrzepowo-zatorowe
W trakcie leczenia kabozantynibem obserwowano przypadki zakrzepicy żylnej, w tym zatorowość płucną i zakrzepicę tętniczą, czasem zakończone śmiertelnie. Kabozantynib należy stosować z ostrożnością u pacjentów z wysokim ryzykiem wystąpienia zdarzeń zakrzepowo-zatorowych lub u pacjentów z wywiadem takich zdarzeń. W badaniu raka wątrobowokomórkowego (CELESTIAL) w trakcie leczenia kabozantynibem obserwowano zakrzepicę żyły wrotnej, w tym jeden przypadek zakończony śmiercią. Pacjenci z wcześniejszą inwazją żyły wrotnej są najprawdopodobniej bardziej narażeni na rozwój zakrzepicy żyły wrotnej. Leczenie kabozantynibem należy przerwać pacjentom z ostrym zawałem mięśnia sercowego lub z jakimkolwiek innym klinicznie istotnym powikłaniem zakrzepowo-zatorowym.
Krwawienia
Zarejestrowano przypadki ciężkich krwawień podczas przyjmowania kabozantynibu, czasem zakończone śmiertelnie. Stan pacjentów z wywiadem ciężkiego krwawienia należy starannie ocenić przed rozpoczęciem leczenia kabozantynibem.
Kabozantynib nie powinien być przepisywany pacjentom, u których występuje ciężkie krwawienie lub istnieje ryzyko jego wystąpienia.
W badaniu raka wątrobowokomórkowego (CELESTIAL) przypadki krwawień zakończone śmiercią występowały częściej przy stosowaniu kabozantynibu niż placebo. Czynniki ryzyka ciężkich krwawień u pacjentów z rozsianym rakiem wątrobowokomórkowym mogą obejmować inwazję guza do dużych naczyń krwionośnych oraz obecność marskości wątroby prowadzącej do wątrobowego przepuklinienia żył przełyku, nadciśnienia wrotnej i trombocytopenii. W badaniu CELESTIAL nie rekrutowano pacjentów leczonych jednocześnie lekami przeciwzakrzepowymi lub lekami przeciwkrzepliwymi.
Do tego badania nie włączono również pacjentów, u których nie leczono lub nie wyleczono całkowicie przepuklin z krwawieniem lub z wysokim ryzykiem krwawienia.
Aneurysmy i rozwarstwienia tętnic
Stosowanie inhibitorów szlaku VEGF u pacjentów z nadciśnieniem lub bez niego może sprzyjać powstawaniu aneurysm i/lub rozwarstwieniom tętnic. Przed rozpoczęciem przyjmowania leku należy starannie rozważyć to ryzyko u pacjentów z takimi czynnikami ryzyka, jak nadciśnienie tętnicze lub wywiad aneurysmy.
Trombocytopenia
W badaniu raka wątrobowokomórkowego (CELESTIAL) zgłaszano przypadki trombocytopenii i obniżenia poziomu płytek krwi. W trakcie leczenia kabozantynibem należy kontrolować poziom płytek krwi i dostosować dawkowanie zgodnie z nasileniem trombocytopenii (patrz tabela 5).
Powikłania w gojeniu ran
Zarejestrowano przypadki powikłań w procesie gojenia ran podczas przyjmowania kabozantynibu. Zaleca się przerwanie leczenia kabozantynibem 28 dni przed planowaną operacją, w tym stomatologiczną lub inwazyjne zabiegi stomatologiczne, jeśli to możliwe. Decyzję o wznowieniu leczenia kabozantynibem po zabiegach chirurgicznych należy podejmować po ocenie klinicznej procesu gojenia rany. Leczenie kabozantynibem należy przerwać pacjentom z powikłaniami w procesie gojenia ran wymagającymi interwencji medycznej.
Nadciśnienie tętnicze
Stosowanie kabozantynibu wiązało się ze wzrostem ciśnienia tętniczego i rozwojem kryzysu nadciśnieniowego. Przed rozpoczęciem leczenia kabozantynibem należy dokładnie ustabilizować ciśnienie tętnicze. Po rozpoczęciu stosowania kabozantynibu u wszystkich pacjentów należy regularnie monitorować ciśnienie tętnicze i w razie potrzeby leczyć standardową terapią przeciwnadciśnieniową. W przypadku ciężkiego lub trwało utrzymującego się nadciśnienia tętniczego, które utrzymuje się mimo stosowania leków przeciwnadciśnieniowych, lekarz musi podjąć decyzję o przerwaniu leczenia kabozantynibem, aż do momentu, gdy ciśnienie tętnicze zostanie skontrolowane, po czym można wznowić przyjmowanie kabozantynibu w zmniejszonej dawce.
W przypadku kryzysu nadciśnieniowego leczenie kabozantynibem należy przerwać.
Osteonekroza
Przypadki osteonekrozy żuchwy (ONŻ) obserwowano podczas stosowania kabozantynibu. Przed rozpoczęciem przyjmowania kabozantynibu oraz okresowo w trakcie terapii kabozantynibem należy przeprowadzać badanie jamy ustnej. Pacjentów należy poinformować o higienie jamy ustnej. Leczenie kabozantynibem należy przeprowadzać co najmniej 28 dni przed planowaną operacją stomatologiczną lub inwazyjnymi zabiegami stomatologicznymi, jeśli to możliwe. Należy zachować ostrożność u pacjentów przyjmujących leki związane z ONŻ, takie jak bisfosfoniany. Kabozantynib należy odstawić pacjentom, u których wystąpiła ONŻ.
Zespół czerwonego oparzenia dłoni i stóp (ZCOPD)
Przypadki wystąpienia zespołu czerwonego oparzenia dłoni i stóp (ZCOPD) zarejestrowano u pacjentów otrzymujących kabozantynib. W przypadku ciężkiego ZCOPD należy rozważyć możliwość przerwania leczenia kabozantynibem. W przypadkach wystąpienia zespołu czerwonego oparzenia dłoni i stóp należy przerwać przyjmowanie kabozantynibu do ustąpienia objawów lub ich zmniejszenia do stopnia 1.
Proteinuria
W trakcie leczenia kabozantynibem zgłaszano przypadki wystąpienia proteinurii. W trakcie leczenia kabozantynibem należy regularnie kontrolować zawartość białka w moczu. Kabozantynib należy odstawić, jeśli u pacjenta rozwija się zespół nerczycowy.
Zespół odwracalnej encefalopatii tylnej (ZOT)
Zespół odwracalnej encefalopatii tylnej (ZOT) został zarejestrowany u pacjentów otrzymujących kabozantynib. ZOT może towarzyszyć drgawki, zaburzenia wzroku, ból głowy, dezorientacja i zaburzenia neurologiczne. Pacjentom z ZOT zaleca się przerwanie leczenia kabozantynibem.
Wydlużenie odcinka QT
Kabozantynib należy stosować z ostrożnością u pacjentów z wywiadem wydłużenia odcinka QT, przyjmujących leki sprzyjające wydłużeniu odcinka QT, oraz u pacjentów z odpowiednimi chorobami serca, bradykardią lub zaburzeniami równowagi elektrolitowej. W trakcie stosowania kabozantynibu konieczne jest okresowe monitorowanie za pomocą EKG oraz oznaczanie stężenia potasu, wapnia i magnezu w surowicy.
Zaburzenia funkcji tarczycy
Wszystkim pacjentom zaleca się kontrolę laboratoryjną tarczycy przed rozpoczęciem stosowania leku. Pacjenci z istniejącym hipotyreozą lub nadczynnością tarczycy powinni być leczeni zgodnie ze standardową praktyką medyczną przed rozpoczęciem terapii kabozantynibem. W trakcie leczenia kabozantynibem należy dokładnie obserwować wszystkich pacjentów pod kątem wystąpienia objawów i objawów dysfunkcji tarczycy oraz okresowo kontrolować funkcję tarczycy. Pacjenci, u których rozwija się dysfunkcja tarczycy, powinni być leczeni zgodnie ze standardową praktyką medyczną w zależności od typu wykrytej dysfunkcji.
Nieprawidłowe wyniki badań biochemicznych krwi
Stosowanie kabozantynibu wiąże się z zwiększoną częstością zaburzeń równowagi elektrolitowej (w tym hipokaliemia, hiperkaliemia, hipomagnezemia, hipokalcemia, hiponatremia). Zaleca się kontrolowanie parametrów badań biochemicznych krwi w trakcie leczenia kabozantynibem, a w razie potrzeby stosować odpowiednie leczenie zastępcze zgodnie ze standardami praktyki klinicznej. Przypadki encefalopatii wątrobowej u pacjentów z rakiem wątrobowokomórkowym mogą być wyjaśnione zaburzeniami równowagi elektrolitowej pojawiającymi się w trakcie leczenia. Jeśli poważne odchylenia parametrów laboratoryjnych od normy utrzymują się lub powtarzają okresowo, należy rozważyć możliwość wstrzymania terapii, zmniejszenia dawki lub całkowitego odstawienia kabozantynibu (patrz tabela 5).
Inhibitory i induktory CYP3A4.
Kabozantynib jest substretem CYP3A4. Jednoczesne stosowanie kabozantynibu i inhibitora CYP3A4 ketokonazolu może prowadzić do wyraźnego zwiększenia ekspozycji na kabozantynib. Szczególna ostrożność jest wymagana podczas jednoczesnego stosowania kabozantynibu z silnymi inhibitorami CYP3A4. Jednoczesne stosowanie kabozantynibu z silnymi induktorami CYP3A4, np. ryfampicyną, może zmniejszać ekspozycję i rozkład kabozantynibu. Dlatego należy unikać jednoczesnego podawania leków będących silnymi induktorami CYP3A4 i kabozantynibu (patrz sekcje „Sposób stosowania i dawki” i „Interakcje z innymi lekami i inne rodzaje interakcji”).
Substrat glikoproteiny P
Kabozantynib jest inhibitorem (IC50 = 7,0 μM), ale nie substratem aktywności transportowej glikoproteiny P (P-gp) w dwukierunkowym systemie z wykorzystaniem komórek zaprojektowanych do ekspresji glikoproteiny P (MDCK–MDR1). Jednoczesne stosowanie inhibitorów P-gp może zwiększać stężenie kabozantynibu w osoczu krwi. Pacjentów należy uprzedzić o przyjmowaniu leków wpływających na P-gp (takich jak fexofenadyna, aliskiren, ambrisentan, dabigatran eteksylat, digoksyna, kolchicyna, maravirok, pozaokonazol, ranolazyna, sakagliptyna, sitagliptyna, talinolol, tolvaptan) podczas przyjmowania kabozantynibu (patrz sekcja „Interakcje z lekami i inne rodzaje interakcji”).
Inhibitory MRP2
Podawanie inhibitorów MRP2 może zwiększać stężenie kabozantynibu w osoczu krwi. Inhibitory MRP2 należy stosować ostrożnie (w tym cyklosporyna, efawirenz, emtrycytabina) (patrz sekcja „Interakcje z lekami i inne rodzaje interakcji”).
Ostrzeżenia dotyczące substancji pomocniczych
Pacjenci z rzadką dziedziczną nietolerancją galaktozy, pełnym niedoborem laktozy lub zespolem malabsorpcji glukozy-galaktozy nie powinni przyjmować tego leku.
1 tabletka powlekana powłoką filmową o mocy 20 mg zawiera 15,54 mg laktozy.
1 tabletka powlekana powłoką filmową o mocy 40 mg zawiera 31,07 mg laktozy.
1 tabletka powlekana powłoką filmową o mocy 60 mg zawiera 46,61 mg laktozy.
Związki sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na tabletkę, czyli jest praktycznie wolny od sodu.
Wszelkie pozostałości niewykorzystanego leku lub odpady należy utylizować zgodnie ze standardami praktyki szpitalnej.
Stosowanie w czasie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym/kontrasepcja dla mężczyzn i kobiet
Kobiety w wieku rozrodczym powinny unikać zajścia w ciążę w trakcie leczenia kabozantynibem. Kobiety, których partnerzy stosują kabozantynib, powinny unikać zajścia w ciążę. Kobiety w wieku rozrodczym i mężczyźni powinni stosować skuteczne metody antykoncepcji w trakcie leczenia i przez 4 miesiące po zakończeniu terapii. Ponieważ doustne środki antykoncepcyjne mogą nie być uważane za skuteczne metody antykoncepcji, w przypadku ich stosowania należy stosować dodatkowe środki zapobiegające (patrz sekcja „Interakcje z lekami i inne rodzaje interakcji”).
Ciąża
Brak danych dotyczących stosowania kabozantynibu u ciężarnych kobiet. Badania na zwierzętach wykazały działanie embrionotoksyczne i teratogenne. Potencjalne ryzyko dla człowieka jest nieznane. Kabozantynib nie powinien być stosowany w czasie ciąży, z wyjątkiem przypadków nagłej potrzeby klinicznej.
Karmienie piersią
Nie wiadomo, czy kabozantynib lub jego metabolity przenikają do mleka matki. Nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią, dlatego w trakcie leczenia kabozantynibem i co najmniej przez 4 miesiące po zakończeniu leczenia należy przerwać karmienie piersią.
Fertylność
Brak danych dotyczących wpływu kabozantynibu na płodność człowieka. Wyniki badań wskazują, że kabozantynib może negatywnie wpływać na płodność u mężczyzn i kobiet. Mężczyźni i kobiety powinni stosować skuteczne metody antykoncepcji w trakcie leczenia.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Kabozantynib ma nieznaczny wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn. W trakcie leczenia kabozantynibem niektórzy pacjenci zgłaszali działania niepożądane, takie jak zmęczenie i osłabienie. W trakcie leczenia należy zachować ostrożność podczas prowadzenia pojazdów i obsługiwanie maszyn.
Sposób stosowania i dawki
Leczenie lekiem CABOMECTICS powinien przepisać i prowadzić lekarz posiadający doświadczenie w stosowaniu leków przeciwnowotworowych.
Sposób stosowania
CABOMECTICS przeznaczony jest do doustnego stosowania. Tabletki należy połykać całe, nie rozgniatając. Pacjenci nie powinni spożywać pokarmu w ciągu 2 godzin przed i co najmniej 1 godziny po przyjęciu leku CABOMECTICS.
Dawki
Tabletki CABOMECTICS oraz kapsułki kabozantynibu nie są bioekwiwalentne i nie powinny być stosowane wymiennie (patrz dział „Farmakokinetyka”).
Jako monoterapia
Zalecana dawka leku CABOMECTICS w leczeniu NKK i GCK wynosi 60 mg raz dziennie. Leczenie lekiem należy kontynuować do momentu ustania klinicznych korzyści dla pacjenta lub wystąpienia nieznośnych objawów toksyczności.
Zalecane zmiany dawki
Wystąpienie podejrzanych działań niepożądanych może wymagać tymczasowego przerwania leczenia i/lub zmniejszenia dawki leku CABOMECTICS (patrz tabela 5). W razie potrzeby dawkę zaleca się zmniejszyć do 40 mg dziennie, a następnie do 20 mg dziennie.
W przypadku wystąpienia toksyczności stopnia 3 według klasyfikacji CTCAE (Common Terminology Criteria for Adverse Events) lub wyższego stopnia, lub nieznośnej toksyczności stopnia 2, należy tymczasowo wstrzymać leczenie. W przypadku wystąpienia poważnych lub nieznośnych objawów toksyczności zaleca się zmniejszenie dawki.
Nie należy przyjmować pominiętej dawki, jeśli do przyjęcia następnej dawki pozostało mniej niż 12 godzin.
Tabela 5
Zalecane modyfikacje dawek leku CABOMECTICS oraz niezbędne działania w przypadku wystąpienia działań niepożądanych
| Reakcja niepożądane i stopień ciężkości |
Zalecane zmiany dawki i działania |
| Reakcje niepożądane stopnia 1–2 dobrze tolerowane i łatwo leczalne |
Nie ma potrzeby dostosowywania dawki. Rozpocząć terapię wspierającą w razie potrzeby. |
| Reakcje niepożądane stopnia 2, które są nietolerowane i nie ustępują po zmniejszeniu dawki lub zastosowaniu terapii wspierającej |
Przerwać leczenie do czasu zmniejszenia toksyczności do poziomu ≤ stopnia 1. Wskazane jest rozpoczęcie terapii wspierającej w razie potrzeby. Wziąć pod uwagę możliwość wznowienia terapii w niższej dawce. |
| Reakcje niepożądane stopnia 3 (z wyjątkiem klinicznie nieistotnych odchyleń laboratoryjnych) |
Przerwać leczenie do czasu zmniejszenia toksyczności do poziomu ≤ stopnia 1. Wskazane jest rozpoczęcie terapii wspierającej w razie potrzeby. Wznowić terapię w niższej dawce. |
| Reakcje niepożądane stopnia 4 (z wyjątkiem klinicznie nieistotnych odchyleń laboratoryjnych) |
Wstrzymać leczenie. Udzielić odpowiedniej pomocy medycznej. Po zmniejszeniu toksyczności do poziomu ≤ stopnia 1 wznowić terapię w niższej dawce. Jeśli reakcja niepożądana utrzymuje się, całkowicie odstawić lek Cabomectics. |
Uwaga: Kryteria oceny nasilenia działań niepożądanych zgodnie z NCI-CTCAE v4.
Leki współlekowane
Należy ostrożnie współlekować leki, które są silnymi inhibitorami CYP3A4, oraz należy unikać stałego współlekowania leków, które są silnymi induktorami CYP3A4 (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności” oraz „Interakcje z innymi lekami i inne formy interakcji”).
Należy rozważyć wybór alternatywnego leku współlekowanego, który nie wykazuje lub wykazuje minimalną aktywność inhibitorową wobec izoenzymu CYP3A4.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku
Pacjenci w podeszłym wieku (≥ 65 lat) nie wymagają dostosowania dawki kabozantynibu.
Pochodzenie etniczne
Nie jest wymagane dostosowanie dawki w zależności od pochodzenia etnicznego pacjenta (patrz sekcja „Farmakokinetyka”).
Pacjenci z niewydolnością nerek
Kabozantynib należy stosować z ostrożnością u pacjentów z niewydolnością nerek o lekkim lub umiarkowanym nasileniu.
Kabozantynib jest przeciwwskazany u pacjentów z niewydolnością nerek o ciężkim nasileniu, ponieważ bezpieczeństwo i skuteczność kabozantynibu u tej grupy pacjentów nie zostały ustalone.
Pacjenci z niewydolnością wątroby
U pacjentów z niewydolnością wątroby o lekkim nasileniu nie jest wymagane dostosowanie dawki. Ze względu na ograniczone dane dotyczące stosowania kabozantynibu u pacjentów z niewydolnością wątroby o umiarkowanym nasileniu (klasa B wg skali Childa-Pugh) nie można podać zaleceń dotyczących dawkowania. U tych pacjentów zaleca się monitorowanie ogólnego bezpieczeństwa (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności” oraz „Farmakokinetyka”). Z uwagi na brak doświadczeń klinicznych dotyczących stosowania kabozantynibu u pacjentów z niewydolnością wątroby o ciężkim nasileniu (klasa C wg skali Childa-Pugh) kabozantynib nie jest zalecany tej grupie pacjentów (patrz sekcja „Farmakokinetyka”).
Pacjenci z niewydolnością serca
Dane dotyczące stosowania u pacjentów z niewydolnością serca są ograniczone. Nie ma konkretnych zaleceń dotyczących dawkowania dla tej grupy pacjentów.
Dzieci.
Bezpieczeństwo i skuteczność kabozantynibu u dzieci i młodzieży (< 18 lat) nie zostały ustalone. Brakuje odpowiednich danych. Nie stosować u dzieci.
Przedawkowanie.
Nie istnieje specyficzne leczenie przedawkowania kabozantynibu, a możliwe objawy przedawkowania nie zostały ustalone. W przypadku podejrzenia przedawkowania należy przerwać przyjmowanie kabozantynibu i rozpocząć leczenie wspierające. Wymagany jest kontrola laboratoryjnych parametrów biochemicznych i metabolizmu, monitorowanych co najmniej raz w tygodniu lub częściej, jeśli istnieją wskazania kliniczne. Działania niepożądane związane z przedawkowaniem leczy się objawowo.
Efekty uboczne.
Krótki opis profilu bezpieczeństwa
Najczęstsze powikłania u pacjentów z RCC (częstość ≥ 1 %) to zapalenie płuc, ból brzucha, biegunka, nudności, nadciśnienie tętnicze, zatorowość, hiponatremia, zatorowość płucna, wymioty, odwodnienie, osłabienie, astenia, zmniejszony apetyt, zakrzepica żył głębokich, zawroty głowy, hipomagnezemia oraz zespół czerwono-niebieski dłoni i stóp (ZCNDiS).
Najczęstsze efekty uboczne dowolnego stopnia (obserwowane u co najmniej 25 % pacjentów) u pacjentów z RCC to biegunka, osłabienie, nudności, zmniejszony apetyt, ZCNDiS, nadciśnienie tętnicze, spadek masy ciała, wymioty, dysgeuzja, zaparcia, podwyższenie aktywności AST. Nadciśnienie tętnicze obserwowano częściej u pacjentów z RCC, którzy wcześniej nie byli leczeni (67 %), w porównaniu z pacjentami z RCC, którzy wcześniej otrzymywali leki wpływające na VEGF (37 %).
Najczęstsze powikłania u pacjentów z HCC (częstość ≥ 1 %) to encefalopatia wątrobową, astenia, osłabienie, ZCNDiS, biegunka, hiponatremia, wymioty, ból brzucha oraz trombocytopenia.
Najczęstsze efekty uboczne dowolnego stopnia (obserwowane u co najmniej 25 % pacjentów) u pacjentów z HCC to biegunka, zmniejszony apetyt, osłabienie, nudności, ZCNDiS, nadciśnienie tętnicze oraz wymioty. Lista efektów ubocznych przedstawiona jest w tabeli 6.
W tabeli 6 efekty uboczne zaobserwowane podczas stosowania kabozantynibu w badaniach klinicznych lub w okresie pozarejestracyjnym są pogrupowane według układów narządów zgodnie z terminologią MedDRA i sklasyfikowane według częstości występowania dla wszystkich stopni ciężkości: bardzo często (≥ 1/10), często (≥ 1/100 i < 1/10), rzadko (≥ 1/1000 i < 1/100), nieznane (niemożliwe do oszacowania na podstawie dostępnych danych). W ramach każdej grupy według częstości występowania, reakcje niepożądane są uporządkowane według malejącej ciężkości.
Tabela 6
Reakcje niepożądane leku (RNŁ), zarejestrowane podczas stosowania kabozantynibu w badaniach klinicznych lub w okresie pozarejestracyjnym
| Układ narządów (według MedDRA) |
Bardzo często |
Często |
Nieczęsto |
Nieznane |
| Infekcje i inwazje |
absces, zapalenie płuc |
|||
| Układ krwionośny i chłonny |
anemia, trombocytopenia1 |
neutropenia1, limfopenia1 |
||
| Układ endokrynny |
hipotyreoz3 |
|||
| Zaburzenia metabolizmu i układu pokarmowego |
spadek apetytu hipomagnezemia2 hipokaliemia2 hipoalbuminemia2 |
odwodnienie hipofosfatemia2 hiponatremia2 hipokalcemia2, hiperkaliemia3, hiperbilirubinemia3 hiperglikemia3 hipoglikemia2 |
||
| Układ nerwowy |
dysgezja bóle głowy zawroty głowy |
neuropatia obwodowa (w tym czuciowa) |
drżenia, zaburzenia mózgowo-naczyniowe zespół odwracalnej encefalopatii tylnej |
|
| Układ słuchu i równowagi |
wycie w uszach |
|||
| Układ sercowo-naczyniowy |
zawał mięśnia sercowego |
|||
| Układ naczyniowy |
hipertensja tętnicza5 wylewy krwi* |
zakrzepica żylna |
kryz hipertensyjny zakrzepica tętnicza |
aneurysmy i rozwarstwienie tętnic |
| Układ oddechowy, płuca i opłucna |
osłabienie głosu udrudnione oddychanie kaszel |
zatorowość płucna |
otwarcie opłucnej |
|
| Układ pokarmowy |
biegunka* mdłości wymioty zapalenie jamy ustnej zaparcia ból brzucha,4 wzdęcia |
refluks żołądkowo-przełykowy ból w jamie ustnej sucha jamy ustnej trudności z połykaniem zapalenie okrężnicy zapalenie żołądka hemoroidy |
przebicie przewodu pokarmowego* fistała* zapalenie trzustki, przebicie jelita cienkiego, ból języka |
|
| Układ wątrobowo-żółciowy |
encefalopatia wątrobowo-żółciowa* |
zapalenie wątroby z cechami cholestazy |
||
| Układ skóry i tkanki podskórnej |
zespoł czerwonego dłoni i podeszwy stóp wysypka |
świerzbienie łysienie susza skóry rumień zapalenie skóry typu trądzikowatego zmiana koloru włosów hiperkeratoza |
zapalenie naczyń skóry |
|
| Układ mięśniowo-szkieletowy i tkanki łącznej |
ból kończyn |
skurcze mięśni artrologia |
martwica kości żuchwy |
|
| Układ nerek i dróg moczowych |
proteinuria |
|||
| Zaburzenia ogólne |
zmęczenie, zapalenie błon śluzowych osłabienie, obrzęk obwodowy |
|||
| Badania laboratoryjne |
spadek masy ciała wzrost stężenia alaninotransferazy (ALT) w surowicy wzrost stężenia asparaginianotransferazy (AST) w surowicy |
wzrost stężenia fosfatazy alkalicznej (FA) we krwi wzrost stężenia gamma-glutamylotransferazy (GGT) wzrost stężenia kreatyniny we krwi wzrost stężenia amylazy wzrost stężenia lipazy wzrost stężenia cholesterolu we krwi3 wzrost stężenia trójglicerydów we krwi3 |
||
| Urazy, zatrucia i powikłania związane z procedurami |
powikłania gojenia ran6 |
* Opis wybranych działań niepożądanych do dalszej charakterystyki.
Poniższe terminy zostały połączone w celu dostosowania do klasyfikacji częstości:
-
Obniżone parametry hematologiczne: limfopenia i zmniejszona liczba limfocytów; neutropenia i zmniejszona liczba neutrofili; trombocytopenia i zmniejszona liczba płytek krwi.
-
Obniżone parametry biochemiczne: hipoproteinemia i obniżony poziom albuminy we krwi; hipokalcemia i obniżony poziom wapnia we krwi; hipoglikemia i zmniejszony poziom glukozy we krwi; hipokaliemia i obniżony poziom potasu we krwi; hipomagnezemia i obniżony poziom magnezu we krwi; hiponatremia i obniżony poziom sodu we krwi; hipofosfatemia i zmniejszony poziom fosforu we krwi.
-
Podwyższone parametry biochemiczne: podwyższony poziom cholesterolu we krwi i hipercholesterolemia; hiperbilirubinemia i podwyższony poziom bilirubiny we krwi; hiperglikemia i podwyższony poziom glukozy we krwi; hipotyreozę i podwyższone stężenie hormonu tyreotropowego we krwi; hiperkaliemia i podwyższony poziom potasu we krwi; zwiększenie poziomu trójglicerydów i hipertriglicerydemia.
-
Ból brzucha, dyskomfort brzuszny, ból brzucha w części górnej i ból brzucha w części dolnej.
-
Nadciśnienie i wzrost ciśnienia tętniczego.
-
Zaburzenia gojenia i powikłania w miejscu nacięcia.
Opis pojedynczych działań niepożądanych
Dane dotyczące poniższych reakcji u pacjentów przyjmujących lek Cabomectics w dawce 60 mg na dobę (doustnie), uzyskane w badaniach podstawowych u pacjentów z rakiem komórkowym nerek (RKN) po terapii lekami wpływającymi na VEGF oraz u pacjentów z RKN, którzy wcześniej nie otrzymywali leczenia, a także u pacjentów z rakiem komórki wątrobowej (RKW) po wcześniejszej terapii systemowej (patrz sekcja „Farmakodynamika”).
Perforacja przewodu pokarmowego
W badaniu RKN po stosowaniu leków wpływających na VEGF (METEOR), perforację przewodu pokarmowego stopnia 2 lub 3 odnotowano u 0,9 % pacjentów z rakiem komórkowym nerek otrzymujących kabozatynib (3 na 331). Mediana czasu do wystąpienia perforacji wynosiła 10 tygodni.
W badaniu RKN z udziałem pacjentów, którzy wcześniej nie otrzymywali leczenia (CABOSUN), perforację przewodu pokarmowego stopnia 4 i 5 wykryto u 2,6 % (2 na 78) pacjentów otrzymujących kabozatynib.
W badaniu RKW (CELESTIAL) perforację przewodu pokarmowego stopnia 3 lub 4 wykryto u 0,9 % pacjentów leczonych kabozatynibem (4 na 467). Mediana czasu do wystąpienia perforacji wynosiła 5,9 tygodnia.
W programie badań klinicznych kabozatynibu odnotowano przypadki perforacji zakończone śmiercią.
Encefalopatia wątrobową
W badaniu RKW (CELESTIAL) encefalopatię wątrobową (encefalopatię wątrobową, encefalopatię, hiperamonemiczną encefalopatię) odnotowano u 5,6 % pacjentów otrzymujących kabozatynib (26 na 467). Reakcje stopnia 3–4 obserwowano u 2,8 %; zaobserwowano również jeden przypadek stopnia 5 (0,2 %). Mediana czasu do rozwoju encefalopatii wynosiła 5,9 tygodnia. W badaniach RKN (METEOR i CABOSUN) nie odnotowano przypadków encefalopatii wątrobowej.
Biegunka
W badaniu RKN po wcześniejszej terapii lekami wpływającymi na VEGF (METEOR), biegunkę stwierdzono u 74 % pacjentów otrzymujących kabozatynib (245 na 331). Reakcje stopnia 3–4 stanowiły 11 %. Mediana czasu do wystąpienia biegunki wynosiła 4,9 tygodnia.
W badaniu RKN z udziałem pacjentów, którzy wcześniej nie otrzymywali leczenia (CABOSUN), biegunkę obserwowano u 73 % pacjentów otrzymujących kabozatynib (57 na 78). Reakcje stopnia 3–4 odnotowano w 10 % przypadków.
W badaniu RKW (CELESTIAL) biegunkę obserwowano u 54 % pacjentów otrzymujących kabozatynib (251 na 467); reakcje stopnia 3–4 odnotowano w 9,9 %. Mediana czasu do wystąpienia tych przypadków wynosiła 4,1 tygodnia. W wyniku biegunki u 84 na 467 (18 %), 69 na 467 (15 %) oraz u 5 na 467 (1 %) pacjentów konieczne było odpowiednio dostosowanie dawki, przerwanie terapii oraz całkowite odstawienie leczenia.
Fistuły
W badaniu RKN po wcześniejszej terapii lekami wpływającymi na VEGF (METEOR), fistuły obserwowano u 1,2 % (4 na 331) pacjentów przyjmujących kabozatynib, w tym fistuły odbytu u 0,6 % (2 na 331). Jeden przypadek sklasyfikowano jako działanie niepożądane stopnia 3, pozostałe jako reakcje stopnia 2. Mediana czasu do wystąpienia fistuły wynosiła 30,3 tygodnia.
W badaniu RKN z udziałem pacjentów, którzy wcześniej nie otrzymywali leczenia (CABOSUN), nie wykryto fistul.
W badaniu RKW (CELESTIAL) fistuły obserwowano u 1,5 % (7 na 467) pacjentów z RKW. Mediana czasu do wystąpienia fistuły wynosiła 14 tygodni.
W programie klinicznego rozwoju kabozatynibu zgłaszano przypadki fistul zakończone śmiercią.
Krwawienie
W badaniu RKN po wcześniejszej terapii lekami wpływającymi na VEGF (METEOR), częstość występowania ciężkich zdarzeń krwotocznych (stopień ≥ 3) wynosiła 2,1 % u pacjentów z RKN otrzymujących kabozatynib (7 na 331). Mediana czasu do wystąpienia krwawienia wynosiła 20,9 tygodnia.
W badaniu RKN z udziałem pacjentów, którzy wcześniej nie otrzymywali leczenia (CABOSUN), częstość występowania ciężkich zdarzeń krwotocznych (stopień ≥ 3) wynosiła 5,1 % (4/78) u pacjentów z RKN otrzymujących kabozatynib.
W badaniu RKW (CELESTIAL) częstość występowania ciężkich zdarzeń krwotocznych (stopień ≥ 3) wynosiła 7,3 % u pacjentów otrzymujących kabozatynib (34 na 467). Mediana czasu do wystąpienia krwawienia wynosiła 9,1 tygodnia.
W programie klinicznego rozwoju kabozatynibu zgłaszano przypadki krwawień zakończone śmiercią.
Zespół odwracalnej encefalopatii tylnej (ZOET)
W badaniach METEOR, CABOSUN czy CELESTIAL nie odnotowano przypadków ZOET, jednak w innych badaniach klinicznych rzadko zgłaszano przypadki ZOET (2 na 4872 pacjentów; 0,04 %).
Hipotyreozę
W badaniu RKN po stosowaniu leków wpływających na VEGF (METEOR), przypadki hipotyreozę odnotowano u 21 % pacjentów (68 na 331).
W badaniu RKN z udziałem pacjentów, którzy wcześniej nie otrzymywali leczenia (CABOSUN), przypadki hipotyreozę odnotowano u 23 % pacjentów (18 na 78), którzy otrzymywali kabozatynib.
W badaniu RKW (CELESTIAL) przypadki hipotyreozę odnotowano u 8,1 % pacjentów (38 na 467), którzy otrzymywali kabozatynib, a reakcje stopnia 3 stanowiły 0,4 % (2 na 467).
Zgłaszanie działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w trakcie stosowania tego leku. Personel medyczny i farmaceutyczny, a także pacjenci lub ich prawni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua
Termin ważności.
4 lata.
Warunki przechowywania.
Lek nie wymaga specjalnych warunków przechowywania. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
20 mg: po 7 tabletek powlekanych, w blistrze; 4 blisterki w tece kartonowej lub po 30 tabletek powlekanych w butelce z polietylenu o wysokiej gęstości (HDPE) z polipropylenową pokrywką trudną do otwarcia dla dzieci, z trzema pojemnikami z żelem krzemionkowym osuszającym po 1 g i włóknem poliestrowym; 1 butelka w tece kartonowej;
40 mg: po 7 tabletek powlekanych, w blistrze; 4 blisterki w tece kartonowej lub po 30 tabletek powlekanych w butelce z polietylenu o wysokiej gęstości (HDPE) z polipropylenową pokrywką trudną do otwarcia dla dzieci, z trzema pojemnikami z żelem krzemionkowym osuszającym po 1 g i włóknem poliestrowym; 1 butelka w tece kartonowej;
60 mg: po 7 tabletek powlekanych, w blistrze; 4 blisterki w tece kartonowej lub po 30 tabletek powlekanych w butelce z polietylenu o wysokiej gęstości (HDPE) z polipropylenową pokrywką trudną do otwarcia dla dzieci, z trzema pojemnikami z żelem krzemionkowym osuszającym po 1 g i włóknem poliestrowym; 1 butelka w tece kartonowej.
Kategoria wydania.
Na receptę.
Producenci.
Rottendorf Pharma GmbH, Niemcy.
Pfizer France.
Tjoapak Netherlands B.V.
Adres producentów i miejsce prowadzenia działalności.
Ostenfelder Strasse 51-61, Ennigerloh, Nadrenia-Westfalia, 59320, Niemcy.
40 Boulevard de Champert, Bourgoin-Jallieu, 38300, Francja.
Nieuwe Donk 9, Etten-Leur, 4879AC, Holandia.
Wnioskodawca.
IPSEN PHARMA.
Adres wnioskodawcy.
70, rue de la Ballade, 75015 Paryż, Francja