Idelvion 3.500 IU proszek i rozpuszczalnik do roztworu do wstrzykiwań
HiszpaniaSpis treści
Ulotka: Informacja dla użytkownika
Wprowadzenie
Ulotka: informacja dla użytkownika
IDELVION 250 J, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
IDELVION 500 J, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
IDELVION 1000 J, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
IDELVION 2000 J, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
IDELVION 3500 J, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
albutrepenonakog alfa (rekombinowany czynnik krzepnięcia krwi IX)
Przed zastosowaniem tego leku należy dokładnie przeczytać całą ulotkę, ponieważ zawiera ona ważne informacje dla Ciebie.
-
Zachowaj ulotkę, ponieważ możesz mieć potrzebę ponownego zapoznania się z jej treścią.
-
W przypadku wątpliwości skonsultuj się z lekarzem, farmaceutą lub pielęgniarką.
-
Ten lek został Ci przepisany wyłącznie Tobie i nie powinieneś go przekazywać innym osobom, nawet jeśli mają takie same objawy co Ty, ponieważ może im zaszkodzić.
-
W przypadku wystąpienia działań niepożądanych skontaktuj się z lekarzem, farmaceutą lub pielęgniarką, nawet jeśli chodzi o możliwe działania niepożądane, których nie ma w niniejszej ulotce. Zobacz sekcję 4.
Zawartość ulotki:
-
Co to jest IDELVION i do czego służy
-
Co należy wiedzieć przed rozpoczęciem stosowania IDELVION
-
Jak stosować IDELVION
-
Możliwe działania niepożądane
-
Jak przechowywać IDELVION
-
Zawartość opakowania i informacje dodatkowe
1. Co to jest IDELVION i do czego służy
Co to jest IDELVION?
IDELVION to lek stosowany w leczeniu hemofilii, który zastępuje naturalny krzepnący czynnik IX. Czynna substancja IDELVION to albutrepenonakog alfa (rekombinowana białkowa proteina fuzji łącząca czynnik krzepnięcia IX z albuminą [rIX-FP]).
Czynnik IX odgrywa rolę w krzepnięciu krwi. Pacjenci z hemofilią B nie posiadają tego czynnika, co oznacza, że krew nie krzepnie tak szybko, jak powinna, co zwiększa skłonność do krwawień. IDELVION działa, zastępując czynnik IX u pacjentów z hemofilią B, umożliwiając krzepnięcie ich krwi.
Do czego stosuje się IDELVION?
IDELVION stosuje się w celu zapobiegania lub zatrzymywania krwawień spowodowanych niedostateczną ilością czynnika IX u pacjentów w każdym wieku z hemofilią B (nazywaną również wrodzoną niedoborem czynnika IX lub chorobą Christmasa).
2. Co należy wiedzieć przed zastosowaniem IDELVION
Nie stosuj IDELVION
- jeśli jesteś uczulony na substancję czynną (albutrepenonakog alfa) lub na którykolwiek z innych składników tego leku (wymienionych w sekcji 6),
- jeśli jesteś uczulony na białka chomika.
Ostrzeżenia i środki ostrożności
Zaleca się, aby za każdym razem, gdy stosujesz IDELVION, zapisywać nazwę i numer serii produktu w celu śledzenia produktów i partii, których użyto.
Śledzenie produktu
W celu poprawy śledzenia leków biologicznych nazwa i numer serii podanego leku powinny być wyraźnie zarejestrowane.
Skonsultuj się z lekarzem, farmaceutą lub pielęgniarką przed rozpoczęciem stosowania IDELVION.
-
Może dojść do reakcji alergicznych (nadwrażliwości). Produkt zawiera śladowe ilości białek chomika (zobacz również „Nie stosuj IDELVION”). Jeśli wystąpią objawy reakcji alergicznych, należy natychmiast przerwać leczenie i skontaktować się z lekarzem lub ośrodkiem opieki medycznej, w którym jesteś leczony. Twój lekarz powinien poinformować Cię o wczesnych objawach reakcji nadwrażliwości. Obejmują one pokrzywkę, ogólną wysypkę skórną, uczucie ucisku w klatce piersiowej, trudności w oddychaniu, spadek ciśnienia krwi (hipotensję) oraz anafilaksję (ciężką reakcję alergiczną powodującą poważne trudności oddechowe lub zawroty głowy).
-
Ze względu na ryzyko reakcji alergicznych związanych z czynnikiem IX, pierwsze podania IDELVION powinny być wykonywane pod nadzorem lekarskim, aby zapewnić dostęp do odpowiedniej opieki medycznej w przypadku wystąpienia reakcji alergicznych.
-
Powstawanie inhibitorów (przeciwciał neutralizujących) jest znanym powikłaniem zgłaszanym podczas leczenia IDELVION. Inhibitory uniemożliwiają prawidłowe działanie leczenia. Jeśli krwawienia nie ustępują mimo stosowania IDELVION, należy natychmiast powiadomić lekarza. Należy być regularnie kontrolowanym pod kątem rozwoju inhibitorów.
-
Jeśli cierpisz na chorobę wątroby lub serca lub niedawno przeszedłeś dużą operację, poinformuj o tym lekarza, ponieważ istnieje większe ryzyko powikłań związanych z krzepnięciem krwi.
-
Jeśli do podania IDELVION wymagane jest użycie centralnego urządzenia dostępu do żyły (CVC), lekarz weźmie pod uwagę ryzyko powikłań związanych z CVC, takich jak infekcje miejscowe, obecność bakterii we krwi (bakteriemia) oraz powstawanie skrzepliny krwi w naczyniach krwionośnych (trombosis) w miejscu wprowadzenia cewnika.
Stosowanie IDELVION z innymi lekami
- Poinformuj lekarza lub farmaceutę, jeśli stosujesz, stosowałeś niedawno lub mógłbyś potrzebować innych leków.
Ciąża i laktacja
- Jeśli jesteś w ciąży lub karmisz piersią, podejrzewasz ciążę lub planujesz zajść w ciążę, skonsultuj się z lekarzem lub farmaceutą przed zastosowaniem tego leku.
- W czasie ciąży i karmienia piersią IDELVION należy stosować tylko wtedy, gdy wyraźnie jest to konieczne.
Kierowanie pojazdami i obsługa maszyn
IDELVION nie wpływa na zdolność prowadzenia pojazdów lub obsługi maszyn.
IDELVION zawiera sód
Ten lek zawiera do 8,6 mg sodu (główny składnik soli kuchennej) w każdej fiolce. Odpowiada to 0,4% maksymalnej dziennej zalecanej dawki sodu dla dorosłego.
3. Jak stosować IDELVION
Leczenie należy rozpocząć i nadzorować lekarz posiadający doświadczenie w leczeniu zaburzeń krzepnięcia krwi. Postępuj dokładnie zgodnie z zaleceniami lekarza dotyczącymi stosowania tego leku. W razie wątpliwości skonsultuj się z lekarzem.
Dawkę IDELVION wyliczy lekarz. Ilość IDELVION potrzebna do leczenia oraz jego długość zależą od:
- stopnia ciężkości choroby
- lokalizacji i nasilenia krwawienia
- stanu klinicznego i odpowiedzi klinicznej
- masy ciała
IDELVION podaje się jako wstrzyknięcie do żyły (dożylne, IV) po odtworzeniu roztworu z proszku za pomocą rozpuszczalnika dostarczonego przez lekarza lub pielęgniarkę. IDELVION może być również podawany przez Ciebie lub inną osobę jako wstrzyknięcie dożylne, ale wyłącznie po odpowiednim przeszkoleniu.
Jeśli zastosujesz zbyt dużą dawkę IDELVION
Natychmiast skontaktuj się z lekarzem, jeśli wstrzyknąłeś więcej IDELVION niż zalecono.
Jeśli przerwiesz leczenie IDELVION
Nie przerywaj stosowania IDELVION bez wcześniejszej konsultacji z lekarzem.
Odtwarzanie i podawanie
Ogólne instrukcje
- Proszek należy zmieszać z rozpuszczalnikiem (cieczą) i pobrać z fiolki, zachowując warunki sterylności (wolne od drobnoustrojów). Lekarz pokaże Ci, jak przygotować roztwór i jak poprawnie pobrać go z fiolki.
- IDELVION nie należy mieszać z innymi lekami ani rozpuszczalnikami, z wyjątkiem wymienionych w punkcie 6.
- Otrzymany roztwór powinien być przezroczysty lub lekko opalizujący, w kolorze od żółtego do bezbarwnego, tzn. może lekko świecić w świetle, ale nie powinien zawierać widocznych cząstek. Po pobraniu lub przefiltrowaniu roztworu (patrz niżej) należy go wizualnie ocenić przed użyciem. Nie należy stosować roztworu, jeśli jest mętny lub zawiera ścięgna lub widoczne cząstki.
- Usunięcie niewykorzystanego produktu oraz wszystkich pozostałych materiałów należy przeprowadzić zgodnie z lokalnymi przepisami oraz wskazówkami lekarza.
Odtworzenie roztworu
Nie otwierając żadnych fiolki, ogrzej proszek IDELVION i ciecz do temperatury pokojowej lub temperatury ciała. Można to zrobić, pozostawiając fiolki w temperaturze pokojowej przez około godzinę lub trzymając je w dłoniach przez kilka minut.
NIE narażaj fiolki na bezpośrednie działanie ciepła. Fiolki nie powinny być ogrzewane powyżej temperatury ciała (37 °C).
Delikatnie usuń osłonki ochronne z fiolki, a następnie oczyść odsłonięte części sept gumowych za pomocą chusteczki alkoholowej. Pozwól fiolkom wyschnąć przed otwarciem opakowania Mix2Vial (zawierającego przewodnik do przenoszenia z filtrem), a następnie postępuj zgodnie z poniższymi instrukcjami.
|
|
|
|
|
|
|
|
|
Usuń fiolkę z rozpuszczalnikiem z zamocowanym niebieskim adapterem Mix2Vial. |
|
|
|
|
 1
1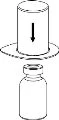 2
2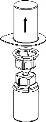 3
3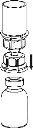 4
4 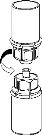 5
5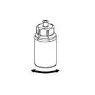 6
6 7
7Przesyłanie i podawanie
|
|
9 |
|
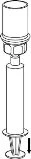 8
8Użyj zestawu do weno punkcji dostarczonego z produktem i wsuń igłę do żyły. Pozwól krwi swobodnie przepływać aż do końca rurki. Podłącz strzykawkę do nakręcanego zatyczki końcowej części zestawu do weno punkcji. Wstrzygnij powoli roztwór odtworzony (w tempie, które będzie wygodne dla Ciebie, maksymalnie do 5 ml/min) do żyły, zgodnie z instrukcjami udzielonymi przez lekarza. Staraj się, aby krew nie dostała się do strzykawki zawierającej produkt.
Sprawdź, czy nie wystąpią skutki uboczne bezpośrednio po wstrzyknięciu. Jeśli wystąpią skutki uboczne, które mogą być związane z podaniem IDELVION, wstrzyknięcie należy przerwać (zobacz również punkty 2 i 4).
Jeśli masz jakiekolwiek wątpliwości dotyczące stosowania tego leku, skonsultuj się z lekarzem, farmaceutą lub pielęgniarką.
4. Możliwe działania niepożądane
Tak jak wszystkie leki, ten lek może powodować działania niepożądane, choć nie u wszystkich osób one występują.
Skontaktuj się z lekarzem:
-
jeśli zauważysz objawy reakcji alergicznych (patrz poniżej);
-
jeśli zauważysz, że lek przestaje działać prawidłowo.
Obserwowano następujące działania niepożądane związane z lekami zawierającymi czynnik IX:
-
Możliwe są reakcje nadwrażliwościowe o charakterze alergicznym (częste), obejmujące następujące objawy: zaczerwienienie, swędzenie skóry (rozlane pokrzywki), uczucie ucisku w klatce piersiowej, trudności z oddychaniem, niskie ciśnienie krwi (hipotensja) oraz anafilaksję (ciężką reakcję powodującą silne trudności z oddychaniem i zawroty głowy). W takim przypadku należy natychmiast przerwać podawanie leku i skontaktować się z lekarzem.
-
Inhibitory: lek przestaje działać prawidłowo (trwające krwawienie). Możesz wytworzyć inhibitor (przeciwciało neutralizujące) czynnika IX (częstość nieznana), co oznacza, że czynnik IX przestanie działać prawidłowo. W takim przypadku należy natychmiast przerwać podawanie leku i skontaktować się z lekarzem.
Następujące działania niepożądane wystąpiły często przy stosowaniu IDELVION (mogą dotyczyć do 1 na 10 osób):
-
Ból głowy
-
Reakcje w miejscu wstrzyknięcia
-
Zawroty głowy
-
Wysypka
Następujące działania niepożądane wystąpiły rzadziej (mogą dotyczyć do 1 na 100 osób):
- Egzema
Działania niepożądane u dzieci i nastolatków
Oczekuje się, że działania niepożądane u dzieci będą takie same jak u dorosłych.
Zgłaszanie działań niepożądanych
Jeśli wystąpi u Ciebie jakiekolwiek działanie niepożądane, skontaktuj się z lekarzem, farmaceutą lub pielęgniarką, nawet jeśli chodzi o możliwe działania niepożądane, które nie są wymienione w niniejszym ulotce. Możesz również zgłaszać działania niepożądane bezpośrednio za pośrednictwem Hiszpańskiego Systemu Farmakowigilancji Leków stosowanych u ludzi: www.notificaRAM.es. Poprzez zgłaszanie działań niepożądanych możesz przyczynić się do dostarczenia większej ilości informacji na temat bezpieczeństwa tego leku.
5. Przechowywanie IDELVION
-
Przechowywać ten lek w miejscu niedostępnym dla dzieci.
-
Nie należy stosować tego leku po upływie daty ważności podanej na etykiecie i opakowaniu.
-
Nie przechowywać w temperaturze powyżej 25 °C.
-
Nie zamrażać.
-
Przechowywać fiolkę w opakowaniu zewnętrznym, aby chronić ją przed światłem.
-
Po odtworzeniu preparatu należy go stosować preferencyjnie natychmiast.
Jeśli odtworzony preparat nie jest podawany natychmiast, czasy i warunki przechowywania przed użyciem są odpowiedzialnością użytkownika.
6. Zawartość opakowania i informacje dodatkowe
Skład IDELVION
Substancją czynną jest:
250 JE w fiolce; po rozcieńczeniu 2,5 ml wody do sporządzania roztworów do wstrzykiwań roztwór zawiera 100 JE/ml albutrepenonakogu alfa.
500 JE w fiolce; po rozcieńczeniu 2,5 ml wody do sporządzania roztworów do wstrzykiwań roztwór zawiera 200 JE/ml albutrepenonakogu alfa.
1 000 JE w fiolce; po rozcieńczeniu 2,5 ml wody do sporządzania roztworów do wstrzykiwań roztwór zawiera 400 JE/ml albutrepenonakogu alfa.
2 000 JE w fiolce; po rozcieńczeniu 5 ml wody do sporządzania roztworów do wstrzykiwań roztwór zawiera 400 JE/ml albutrepenonakogu alfa.
3 500 JE w fiolce; po rozcieńczeniu 5 ml wody do sporządzania roztworów do wstrzykiwań roztwór zawiera 700 JE/ml albutrepenonakogu alfa.
Pozostałe składniki to:
cytrynian sodu, polisorbat 80, manitol, sacharoza i kwas chlorowodorowy (do regulacji pH).
Zobacz ostatni akapit sekcji 2.
Rozpuszczalnik: woda do sporządzania roztworów do wstrzykiwań
Wygląd zewnętrzny IDELVION i zawartość opakowania
IDELVION jest dostępne w postaci żółto-białego proszku i jest dostarczane z rozpuszczalnikiem w postaci wody do sporządzania roztworów do wstrzykiwań.
Otrzymany po rozcieńczeniu roztwór powinien być przezroczysty lub lekko mleczny, od żółtego do bezbarwnego, tzn. może wykazywać połysk w świetle, ale nie powinien zawierać widocznych cząstek.
Sposoby dozowania
Opakowanie zawierające 250, 500 lub 1 000 JE, składające się z:
1 fiolki z proszkiem
1 fiolki z 2,5 ml wody do sporządzania roztworów do wstrzykiwań
1 przewodnika z filtrem 20/20
Zestaw do podania (opakowanie wewnętrzne):
1 jednorazowa strzykawka 5 ml
1 zestaw do nakłucia żyły
2 chusteczki nasączone alkoholem
1 opatrunek niejałowy
Opakowanie zawierające 2 000 lub 3 500 JE, składające się z:
1 fiolki z proszkiem
1 fiolki z 5 ml wody do sporządzania roztworów do wstrzykiwań
1 przewodnika z filtrem 20/20
Zestaw do podania (opakowanie wewnętrzne):
1 jednorazowa strzykawka 10 ml
1 zestaw do nakłucia żyły
2 chusteczki nasączone alkoholem
1 opatrunek niejałowy
Nie wszystkie wielkości opakowań mogą być dostępne w obrocie handlowym.
Właściciel pozwolenia na dopuszczenie do obrotu i producent
CSL Behring GmbH
Emil-von-Behring-Straße 76
35041 Marburg
Niemcy
Więcej informacji na temat tego leku można uzyskać, kontaktując się z lokalnym przedstawicielem właściciela pozwolenia na dopuszczenie do obrotu:
België/Belgique/Belgien CSL Behring NV Tél/Tel: +32 15 28 89 20 | Litwa CentralPharma Communications UAB Tel: +370 5 243 0444 |
| Luksemburg CSL Behring NV Tél/Tel: +32 15 28 89 20 |
Republika Czeska CSL Behring s.r.o. Tel: + 420 702 137 233 | Węgry CSL Behring Kft. Tel.: +36 1 213 4290 |
Dania CSL Behring AB Tlf: +46 8 544 966 70 | Malta AM Mangion Ltd. Tel: +356 2397 6333 |
Niemcy CSL Behring GmbH Tel: +49 6190 75 84810 | Niderlandy CSL Behring BV Tel: + 31 85 111 96 00 |
Estonia CentralPharma Communications OÜ Tel: +3726015540 | Norwegia CSL Behring AB Tlf: +46 8 544 966 70 |
Grecja CSL Behring ΕΠΕ Τηλ: +30 210 7255 660 | Austria CSL Behring GmbH Tel: +43 1 80101 1040 |
Hiszpania CSL Behring S.A. Tel: +34 933 67 1870 | Polska CSL Behring Sp.z o.o. Tel: +48 22 213 22 65 |
Francja CSL Behring S.A. Tél: + 33 –(0)-1 53 58 54 00 | Portugalia CSL Behring Lda Tel: +351 21 782 62 30 |
Chorwacja Marti Farm d.o.o. Tel: +385 1 5588297 | Rumunia Prisum Healthcare S.R.L. Tel: +40 21 322 0171 |
Irlandia CSL Behring GmbH Tel: +49 6190 75 84700 Islandia CSL Behring AB Sími: +46 8 544 966 70 | Słowenia Emmes Biopharma Global s.r.o. podružnica v Sloveniji Tel:+ 386 41 42 0002 Słowacka Republika CSL Behring Slovakia s.r.o. Tel: +421 911 653 862 |
Włochy CSL Behring S.p.A. Tel: +39 02 34964 200 | Finlandia CSL Behring AB Puh/Tel: +46 8 544 966 70 |
Cypr CSL Behring ΕΠΕ Τηλ: +30 210 7255 660 | Szwecja CSL Behring AB Tel: +46 8 544 966 70 |
Łotwa CentralPharma Communications SIA Tel: +371 6 7450497 | |

Data ostatniej weryfikacji tego ulotnika:
Szczegółowe informacje na temat tego leku są dostępne na stronie internetowej Europejskiej Agencji Leków: http://www.ema.europa.eu oraz na stronie internetowej Hiszpańskiej Agencji Leków i Produktów Zdrowia (AEMPS) (http://www.aemps.gob.es/).
Informacja przeznaczona wyłącznie dla specjalistów medycyny:
Dawkowanie
Dawkę i długość trwania leczenia zastępczego dobiera się indywidualnie, w zależności od stopnia niedoboru czynnika IX, lokalizacji i nasilenia krwawienia oraz stanu klinicznego pacjenta.
Podawaną dawkę czynnika IX wyraża się w jednostkach międzynarodowych (j.m.) w odniesieniu do obecnego standardu WHO produktów zawierających czynnik IX. Aktywność czynnika IX w osoczu wyraża się w procentach (w porównaniu z osoczem ludzkim normalnym) lub w jednostkach międzynarodowych (w odniesieniu do międzynarodowego standardu czynnika IX osoczkowego).
Jedna jednostka międzynarodowa (j.m.) aktywności czynnika IX odpowiada ilości czynnika IX obecnej w 1 ml osocza ludzkiego normalnego.
Leczenie na żądanie
Dawkę czynnika IX oblicza się na podstawie doświadczenia klinicznego, zgodnie z którym 1 j.m. czynnika IX na kg masy ciała zwiększa aktywność czynnika IX w osoczu średnio o 1,3 j.m/dl u pacjentów w wieku ≥ 12 lat i o 1,0 j.m/dl u pacjentów w wieku < 12 lat. Wymaganą dawkę oblicza się według następującego wzoru:
Wymagana dawka (j.m.) = masa ciała (kg) × pożądane zwiększenie aktywności czynnika IX (% normalnego poziomu lub j.m/dl) × {odwrotność zaobserwowanej wartości odzysku (j.m/kg na j.m/dl)}
Przewidywane zwiększenie aktywności czynnika IX (j.m/dl lub % normalnego poziomu) = dawka (j.m.) × odzysk (j.m/dl na j.m/kg) / masa ciała (kg)
Dawkę i częstotliwość podawania należy dostosować zawsze do obserwowanej skuteczności klinicznej u każdego pacjenta.
Pacjenci w wieku < 12 lat
Przy przyrostowym odzysku wynoszącym 1 j.m/dl na 1 j.m/kg dawkę oblicza się według następującego wzoru:
Wymagana dawka (j.m.) = masa ciała (kg) × pożądane zwiększenie aktywności czynnika IX (j.m/dl) × 1 dl/kg
Przykład:
-
U pacjenta o masie ciała 20 kg z ciężką hemofilią typu B wymaga się osiągnięcia maksymalnego poziomu aktywności czynnika IX wynoszącego 50% normalnego poziomu. Odpowiednia dawka wyniesie: 20 kg × 50 j.m/dl × 1 dl/kg = 1000 j.m.
-
U pacjenta o masie ciała 25 kg po podaniu dawki 1000 j.m IDELVION oczekuje się maksymalnego wzrostu aktywności czynnika IX po iniekcji: 1000 j.m / 25 kg × 1,0 (j.m/dl na j.m/kg) = 40 j.m/dl (40% normalnego poziomu).
Pacjenci w wieku ≥ 12 lat
Przy przyrostowym odzysku wynoszącym 1,3 j.m/dl na 1 j.m/kg dawkę oblicza się według następującego wzoru:
Wymagana dawka (j.m.) = masa ciała (kg) × pożądane zwiększenie aktywności czynnika IX (j.m/dl) × 0,77 dl/kg
Przykład:
-
U pacjenta o masie ciała 80 kg z ciężką hemofilią typu B wymaga się osiągnięcia maksymalnego poziomu aktywności czynnika IX wynoszącego 50% normalnego poziomu. Odpowiednia dawka wyniesie: 80 kg × 50 j.m/dl × 0,77 dl/kg = 3080 j.m.
-
U pacjenta o masie ciała 80 kg po podaniu dawki 2000 j.m IDELVION oczekuje się maksymalnego wzrostu aktywności czynnika IX po iniekcji: 2000 j.m × 1,3 (j.m/dl na j.m/kg) / 80 kg = 32,5 j.m/dl (32,5% normalnego poziomu).
W przypadku następujących zdarzeń krwotocznych aktywność czynnika IX nie powinna być niższa niż ustalony poziom aktywności plazmatycznej (w % normalnego poziomu lub w j.m/dl) przez odpowiedni okres czasu. Poniższa tabela może służyć jako wytyczna dawkowania w przypadku epizodów krwotocznych i zabiegów chirurgicznych:
Stopień krwawienia/ rodzaj zabiegu chirurgicznego | Poziom czynnika IX wymagany (% lub UI/dl) | Częstotliwość dawkowania (godziny)/czas trwania leczenia (dni) |
Krwawienie Krwotok do stawu łagodny lub umiarkowany, krwawienie do mięśni (z wyłączeniem mięśnia udo-lędźwiowego) lub krwawienie z jamy ustnej | 30–60 | Jedna dawka powinna wystarczyć w większości przypadków krwawienia. Dawka w dalszym leczeniu powinna być podana po upływie 24–72 godzin, jeśli wystąpią dodatkowe objawy krwawienia. |
Poważne krwawienie Krwawienia potencjalnie śmiertelne, krwawienie do głębokich mięśni, w tym do mięśnia udo-lędźwiowego | 60–100 | Należy powtarzać co 24–72 godziny przez pierwszy tydzień, a następnie podawać dawkę utrzymującą raz w tygodniu, aż do ustania krwawienia i gojenia się rany. |
Mały zabieg chirurgiczny Na przykład (w tym niepowikłane usuwanie zębów) | 50–80 (przed i po zabiegu) | Jedna dawka powinna wystarczyć w większości małych zabiegów. W razie potrzeby można podać dawkę utrzymującą po upływie 24–72 godzin, aż do ustania krwawienia i gojenia się rany. |
Duży zabieg chirurgiczny | 60–100 (przed i po zabiegu) | Należy powtarzać co 24–72 godziny przez pierwszy tydzień, a następnie podawać dawkę utrzymującą 1–2 razy w tygodniu, aż do ustania krwawienia i gojenia się rany. |
Profilaktyczne leczenie
Dla długoterminowej profilaktyki w celu zapobiegania krwawieniom u pacjentów z ciężką hemofilią typu B, dawkowanie zalecane to 35–50 j.m./kg raz w tygodniu. Pacjenci dobrze kontrolowani w ramach schematu raz w tygodniu mogą być leczeni dawką do 75 j.m./kg co 10 lub 14 dni. U pacjentów powyżej 18 roku życia można rozważyć dalsze wydłużenie odstępu między dawkami.
W niektórych przypadkach, szczególnie u młodych pacjentów, może być konieczne skrócenie odstępów między dawkami lub zastosowanie wyższych dawek.
Po wystąpieniu epizodu krwawienia w trakcie profilaktyki, pacjenci powinni kontynuować swój schemat profilaktyczny w jak największym stopniu, podając 2 dawki IDELVION z co najmniej 24-godzinną różnicą, ale dłuższy odstęp może być stosowany, gdy uznaje się to za odpowiednie dla pacjenta.
Pediatryczna populacja
W długoterminowym leczeniu profilaktycznym zalecana dawka to 35–50 j.m./kg raz w tygodniu. U nastolatków w wieku 12 lat i starszych zalecenia dotyczące dawkowania są takie same jak u dorosłych (patrz wyżej).
Ostrzeżenia i szczególne środki ostrożności przy stosowaniu
Inhibitory
Po leczeniu powtarzanym produktami zawierającymi ludzki czynnik krzepnięcia IX należy monitorować pacjentów pod kątem rozwoju przeciwciał neutralizujących (inhibitorów), które należy ilościowo oznaczyć w jednostkach Bethesdy (UB) za pomocą odpowiednich testów biologicznych.
W piśmiennictwie opisano przypadki wskazujące na związek między wystąpieniem inhibitora czynnika IX a reakcjami alergicznymi. Dlatego pacjentów, u których wystąpiły reakcje alergiczne, należy ocenić pod kątem obecności inhibitora. Należy pamiętać, że pacjenci z inhibitorami czynnika IX mogą mieć zwiększone ryzyko anafilaksji po kolejnym narażeniu na czynnik IX.
Monitorowanie leczenia
W trakcie leczenia zaleca się odpowiednie kontrolowanie poziomów czynnika IX w celu ustalenia dawki do podania oraz częstotliwości przetaczania. Odpowiedzi pacjentów na czynnik IX mogą się różnić, co odzwierciedla różną połowiczny czas życia i różne wartości odbioru. Dawka oparta na masie ciała może wymagać dostosowania u pacjentów z niedowagą lub nadwagą. W szczególnym przypadku dużych zabiegów chirurgicznych, konieczne jest dokładne monitorowanie leczenia substytucyjnego za pomocą testów krzepnięcia (aktywność czynnika IX w osoczu).
W przypadku stosowania jednoetapowego testu krzepnięcia opartego na czasie częściowej tromboplastyny aktywowanej (APTT) in vitro do oznaczania aktywności czynnika IX w próbkach krwi pacjentów, wyniki aktywności czynnika IX w osoczu mogą być istotnie wpływane przez zastosowany odczynnik APTT oraz standard odniesienia używany w teście. Prawdopodobne jest, że pomiar wykonany za pomocą jednoetapowego testu krzepnięcia z użyciem odczynnika APTT opartego na kaolinie lub odczynnika APTT z Actin FS może prowadzić do zaniżenia rzeczywistego poziomu aktywności. Jest to szczególnie istotne przy zmianie laboratorium lub odczynników stosowanych w teście.