Xeomin
Ucraina
Indice
ISTRUZIONI PER L'USO DEL MEDICINALE XEOMIN®
Composizione:
Principio attivo: neurotossina botulinica di tipo A di Clostridium botulinum;
un flaconcino contiene 50 unità LD50 oppure 100 unità LD50 di neurotossina botulinica di Clostridium botulinum di tipo A (150 kD), priva di proteine di complessazione [neurotossina botulinica di tipo A, purificata da colture di Clostridium botulinum (ceppo Hall)];
Eccipienti: saccarosio, albumina umana.
Forma farmaceutica. Polvere per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: sostanza solida di colore bianco o quasi bianco.
Gruppo farmacoterapeutico. Altri miorilassanti con meccanismo d'azione periferico. Tossina botulinica. Codice ATC M03AX01.
Proprietà farmacologiche.
Farmacodinamica.
La tossina botulinica di tipo A blocca il rilascio di acetilcolina inibendo la trasmissione colinergica nelle sinapsi neuromuscolari. Le terminazioni nervose nelle sinapsi neuromuscolari non rispondono più agli impulsi nervosi, interrompendo così la secrezione di neurotrasmettitori sulla placca terminale del nervo motorio (denervazione chimica). Il ripristino della trasmissione degli impulsi avviene grazie alla formazione di nuove terminazioni nervose e di nuovi collegamenti con le placche terminali dei nervi motori.
Meccanismo d'azione
Il meccanismo d'azione con cui la tossina botulinica di tipo A agisce sulle terminazioni nervose colinergiche può essere descritto come un processo sequenziale in quattro fasi:
- legame: la catena pesante della tossina botulinica di tipo A si lega con estrema selettività e affinità ai recettori presenti esclusivamente sulle terminazioni nervose colinergiche;
- internalizzazione: il restringimento della membrana della terminazione nervosa e l'assorbimento della tossina attraverso la terminazione nervosa (endocitosi);
- translocazione: il segmento amminoterminale della catena pesante della neurotossina forma un poro nella membrana vescicolare, il legame disolfuro viene scisso e la catena leggera della neurotossina passa attraverso questo poro nel citosol;
- effetto: dopo il rilascio della catena leggera, questa scinde in modo altamente selettivo la proteina bersaglio (SNAP-25), fondamentale per il rilascio dell'acetilcolina.
Il completo ripristino della funzione della placca terminale/trasmissione degli impulsi dopo iniezione intramuscolare avviene normalmente entro 3-4 mesi, periodo durante il quale le terminazioni nervose ricrescono e ristabiliscono i collegamenti con la placca del nervo motorio.
Risultati degli studi clinici
L'obiettivo di due studi clinici comparativi randomizzati in fase III con dosi singole era dimostrare l'equivalenza di efficacia del medicinale Xeomin rispetto a un altro prodotto contenente il complesso tradizionale di tossina botulinica di tipo A – onabotulinotossina A (900 kD): uno studio coinvolgeva pazienti con blefarospasmo (studio MRZ 60201-0003, n = 300), l'altro pazienti con distonia cervicale (studio MRZ 60201-0013, n = 463). I risultati degli studi indicano che Xeomin e il prodotto di confronto hanno efficacia e profilo di sicurezza equivalenti nei pazienti con blefarospasmo e nei pazienti con torcicollo spastico quando utilizzati in un rapporto di conversione del dosaggio 1:1.
Blefarospasmo
Xeomin è stato studiato in uno studio multicentrico randomizzato, in doppio cieco, controllato con placebo, di fase 3, che ha coinvolto 109 pazienti con blefarospasmo. Tutti i pazienti avevano una diagnosi clinica di blefarospasmo essenziale benigno, un punteggio di base ≥ 2 nella sottoscala di gravità della Scala di Valutazione di Yankovic (YVS) e un effetto terapeutico stabile dopo precedenti somministrazioni di onabotulinotossina A (Botox).
I pazienti sono stati randomizzati (2:1) per ricevere una singola dose di Xeomin (n = 75) o placebo (n = 34) in una dose simile (+/− 10 %) a quella utilizzata nelle ultime due procedure di trattamento con onabotulinotossina A prima dell'inizio dello studio. La dose massima consentita nello studio era di 50 unità per occhio; la dose media di Xeomin era di 32 unità per occhio.
Il parametro primario di efficacia era rappresentato dalla variazione del punteggio nella sottoscala di gravità della YVS rispetto al valore basale entro la settimana 6 dopo l'iniezione nella popolazione di tutti i pazienti trattati (popolazione ITT), con i dati mancanti sostituiti con l'ultimo valore disponibile per quel paziente (last observation carried forward). La differenza tra il gruppo Xeomin e il gruppo placebo nella variazione del punteggio della sottoscala di gravità della YVS rispetto al valore basale entro la settimana 6 nella popolazione ITT è stata di −1,0 (IC 95 % −1,4; −0,5) punti ed è risultata statisticamente significativa (p < 0,001).
Se i pazienti necessitavano di un’ulteriore iniezione, potevano partecipare al proseguimento di questo studio. I pazienti hanno ricevuto fino a cinque iniezioni di Xeomin con un intervallo minimo tra due iniezioni di almeno 6 settimane (durata totale dello studio 48-69 settimane, dose massima 50 unità per occhio). Durante l'intero studio, la mediana dell'intervallo tra le iniezioni nei pazienti trattati con NT 201 è variata tra 10,14 (primo intervallo) e 12,00 settimane (secondo e quinto intervallo).
Torcicollo spastico
Xeomin è stato studiato in uno studio multicentrico randomizzato, in doppio cieco, controllato con placebo, di fase III, che ha coinvolto 233 pazienti con distonia cervicale. Tutti i pazienti avevano una diagnosi clinica di distonia cervicale prevalentemente rotatoria e un punteggio basale totale ≥ 20 nella Scala di Valutazione del Torcicollo Spastico di Toronto Ovest (TWSTRS). I pazienti sono stati randomizzati (1:1:1) per ricevere una singola iniezione di Xeomin a una dose di 240 unità (n = 81), Xeomin a una dose di 120 unità (n = 78) o placebo (n = 74). Il numero e i siti di iniezione sono stati determinati dall'investigatore.
Il parametro primario di efficacia era rappresentato dalla variazione media quadratica del punteggio rispetto al valore basale entro la settimana 4 dopo l'iniezione nella scala TWSTRS nella popolazione di tutti i pazienti in trattamento (popolazione ITT), con i dati mancanti sostituiti dal valore basale del paziente (modello statistico completo). Le variazioni del punteggio rispetto al valore basale entro la settimana 4 dopo l'iniezione nella scala TWSTRS sono risultate significativamente maggiori nei gruppi NT 201 rispetto al gruppo placebo (p < 0,001 per tutti i modelli statistici). Inoltre, queste differenze avevano rilevanza clinica, ad esempio: −9,0 punti per il gruppo con dose di 240 unità rispetto al placebo e −7,5 punti per il gruppo con dose di 120 unità rispetto al placebo nel modello statistico completo.
Se i pazienti necessitavano di un’ulteriore iniezione, potevano partecipare al proseguimento di questo studio. I pazienti hanno ricevuto fino a cinque iniezioni di Xeomin a una dose di 120 unità o 240 unità con un intervallo minimo tra due iniezioni di almeno 6 settimane (durata totale dello studio 48-69 settimane). Durante l'intero studio, la mediana dell'intervallo tra le iniezioni nei pazienti trattati con NT 201 è variata tra 10,00 (primo intervallo) e 13,14 settimane (terzo e sesto intervallo).
Spasticità del membro superiore (negli adulti)
In uno studio di base (multicentrico, in doppio cieco, controllato con placebo) che ha coinvolto pazienti con spasticità del membro superiore dopo ictus, 148 partecipanti sono stati randomizzati per ricevere Xeomin (n = 73) o placebo (n = 75) secondo le raccomandazioni di dosaggio per il trattamento iniziale riportate nella sezione «Modalità di somministrazione e dosi». La dose cumulativa dopo fino a 6 cicli ripetuti di trattamento nello studio clinico è stata in media di 1333 unità (massimo 2395 unità) nel periodo fino a 89 settimane.
Secondo il parametro primario definito per la valutazione dell'efficacia (frequenza di risposta nei muscoli flessori del polso secondo la scala di Ashworth alla settimana 4, con risposta definita come miglioramento di almeno 1 punto su una scala di Ashworth a 5 punti), i pazienti trattati con Xeomin (frequenza di risposta – 68,5 %) avevano una probabilità 3,97 volte maggiore di sviluppare una risposta terapeutica rispetto a quelli trattati con placebo (frequenza di risposta – 37,3 %; IC 95 % – da 1,90 a 8,30; p < 0,001, popolazione ITT).
Sebbene in questo studio non fosse previsto un confronto fisso delle dosi per determinare differenze nell'effetto terapeutico tra donne e uomini, un'analisi retrospettiva ha mostrato che la frequenza di risposta era più alta nelle donne (89,3 %) rispetto agli uomini (55,6 %), ma la differenza raggiungeva la significatività statistica solo nelle donne. Tuttavia, nei pazienti maschi del gruppo trattato con Xeomin, la frequenza di risposta secondo la scala di Ashworth alla settimana 4 è risultata costantemente più alta per tutti i gruppi di muscoli trattati rispetto al gruppo placebo.
Nella fase aperta di proseguimento di questo studio di base (in cui era consentita una titolazione flessibile della dose) che ha coinvolto 145 pazienti, ai quali sono state somministrate fino a 5 iniezioni, la frequenza di risposta positiva al trattamento è stata simile tra uomini e donne; la stessa conclusione è stata raggiunta in uno studio con mascheramento dei dati per l'investigatore (numero dello studio nel registro EudraCT 2006-003036-30), in cui l'efficacia e la sicurezza di Xeomin in due diluizioni sono state valutate in 192 pazienti con spasticità del membro superiore di diversa eziologia.
In un altro studio clinico multicentrico, in doppio cieco, controllato con placebo, di fase III, hanno partecipato 317 pazienti con spasticità del membro superiore dopo ictus avvenuto almeno 3 mesi prima, che non avevano ricevuto trattamento in precedenza. Durante la fase principale dello studio, ai pazienti è stata somministrata una dose totale fissa di Xeomin (400 unità) per via intramuscolare nei principali siti predefiniti, tra cui gomito flesso, polso flesso o pugno chiuso, nonché altri gruppi muscolari interessati (n = 210). L'analisi confermativa dei parametri primari e secondari di efficacia alla settimana 4 dopo l'iniezione ha dimostrato miglioramenti statisticamente significativi nei punteggi del paziente rispondente al trattamento secondo la scala di Ashworth o nella variazione rispetto al valore basale secondo la scala di Ashworth e la scala di impressione globale dell'investigatore sul cambiamento dello stato del paziente.
296 pazienti hanno completato il ciclo di trattamento durante la fase principale e hanno partecipato al primo ciclo dello studio clinico aperto. Proseguendo nello studio, i pazienti hanno ricevuto fino a tre iniezioni. Ogni ciclo dello studio clinico aperto consisteva in una sessione di trattamento (somministrazione di Xeomin in una dose totale di 400 unità, distribuita uniformemente tra tutti i muscoli interessati), seguita da un periodo di osservazione di 12 settimane. La durata totale dello studio è stata di 48 settimane.
Il trattamento dei muscoli della spalla è stato studiato durante uno studio clinico aperto di fase III, a cui hanno partecipato 155 pazienti con necessità clinica di trattamento per spasticità combinata dei membri superiori e inferiori. Il protocollo dello studio consentiva una dose fino a 600 unità di Xeomin nel membro superiore.
Questo studio ha dimostrato una correlazione positiva tra l'aumento delle dosi di Xeomin e il miglioramento dello stato del paziente secondo la scala di Ashworth e altri parametri di efficacia, senza compromettere la sicurezza o la tollerabilità di Xeomin.
Rughe verticali tra le sopracciglia, visibili con massima contrazione (rughe glabellari interciliari)
Negli studi sull'efficacia di Xeomin per l'indicazione di rughe glabellari interciliari, hanno partecipato complessivamente 994 pazienti con rughe glabellari interciliari da moderate a marcate alla massima contrazione. Di questi, 169 pazienti (≥ 18 anni) hanno ricevuto Xeomin durante la fase principale di uno studio di base in doppio cieco, controllato con placebo, di fase III, e 236 pazienti sono stati trattati durante la fase aperta di proseguimento di questo studio. L'efficacia del trattamento delle rughe mimiche è stata definita come "assenti" o "moderate" su una scala a 4 punti, valutata dall'investigatore alla settimana 4 con massima contrazione. Lo studio ha dimostrato un'efficacia statisticamente e clinicamente significativa di 20 unità di Xeomin rispetto al placebo. Il tasso di risposta complessivo è stato del 51,5 % nel gruppo trattato con Xeomin contro lo 0 % nel gruppo placebo. Durante lo studio di base, non è stato osservato alcun peggioramento in nessun paziente trattato con Xeomin. Ciò è stato confermato da un numero maggiore di pazienti rispondenti al trattamento al giorno 30 in base alla valutazione delle rughe mimiche sul viso con massima contrazione. Sia secondo la valutazione dell'investigatore che dei pazienti, è stato osservato un numero significativamente maggiore di pazienti con risposta positiva al trattamento tra quelli che hanno ricevuto 20 unità di Xeomin rispetto al gruppo placebo.
L'analisi nei sottogruppi ha mostrato che l'efficacia nei pazienti di età pari o superiore a 50 anni è inferiore rispetto ai pazienti più giovani. Di questi, 113 pazienti avevano meno di 50 anni e 56 pazienti avevano più di 50 anni. L'efficacia negli uomini è risultata inferiore rispetto alle donne. 33 pazienti erano di sesso maschile e 136 di sesso femminile.
L'equivalenza terapeutica di Xeomin è stata dimostrata in uno studio comparativo con il prodotto di confronto Vistabel/Botox, contenente il complesso di tossina botulinica di tipo A (onabotulinotossina A, 900 kD), in due studi multicentrici randomizzati in doppio cieco (n = 631) con dosi singole (20 e 24 unità rispettivamente). I risultati dello studio hanno mostrato che Xeomin e questo prodotto di confronto hanno efficacia e profilo di sicurezza equivalenti nei pazienti con rughe glabellari interciliari (da moderate a marcate) quando utilizzati in un rapporto di conversione 1:1 (vedere la sezione «Modalità di somministrazione e dosi»).
La sicurezza con l'uso ripetuto a lungo termine (20 unità) per il trattamento delle rughe glabellari interciliari è stata dimostrata in uno studio di fase III per un periodo di trattamento fino a due anni con fino a 8 cicli consecutivi di iniezioni (MRZ 60201-0609, n = 796).
Rughe perioculari laterali, visibili con massima espressione del sorriso ("zampe di gallina")
In uno studio di fase III, 111 pazienti con rughe perioculari laterali ("zampe di gallina") da moderate a marcate con massimo sorriso sono stati trattati per 1 ciclo con 12 unità di Xeomin o placebo, somministrati in un lato (destro/sinistro), confrontando uno schema di iniezione a 3 punti e uno a 4 punti. Il successo del trattamento è stato definito come miglioramento di almeno 1 punto su una scala a 4 punti, valutato da un esperto indipendente alla settimana 4, utilizzando fotografie digitali standardizzate di entrambi gli occhi scattate con massimo sorriso, rispetto allo stato iniziale. Sia lo schema a 3 punti che quello a 4 punti hanno dimostrato un vantaggio rispetto al placebo. Con lo schema a 3 punti, la percentuale di successo è stata del 69,9 % nel gruppo Xeomin contro il 21,4 % nel gruppo placebo, e con lo schema a 4 punti – 68,7 % contro 14,3 % rispettivamente. In nessun paziente trattato con Xeomin è stato osservato un peggioramento. Ciò è stato confermato da un numero maggiore di pazienti rispondenti al trattamento al giorno 30 in base alla valutazione su una scala a 4 punti con massimo sorriso. Sia secondo la valutazione dell'investigatore che dei pazienti, è stato osservato un numero maggiore di pazienti con risposta positiva al trattamento tra quelli che hanno ricevuto 12 unità di Xeomin in un'area oculare rispetto a quelli che hanno ricevuto placebo.
Rughe mimiche superiori del viso
L'efficacia e la sicurezza dell'uso di Xeomin da 54 a 64 unità nel trattamento combinato delle rughe mimiche superiori del viso (rughe glabellari interciliari, rughe perioculari laterali e rughe orizzontali della fronte) sono state studiate in uno studio di fase III controllato con placebo, a cui hanno partecipato 156 pazienti. L'efficacia del trattamento delle rughe è stata definita come "assenti" o "moderate" con massima contrazione del muscolo frontale, valutata dall'investigatore secondo una scala estetica Merz a 5 punti. L'analisi ha dimostrato differenze statisticamente significative nel trattamento e un alto tasso di risposta a Xeomin nel trattamento delle rughe glabellari interciliari, delle rughe perioculari laterali e delle rughe orizzontali della fronte separatamente, nonché nel trattamento di tutte le aree contemporaneamente.
Complessivamente, l'82,9 % dei pazienti trattati con Xeomin ha avuto una risposta positiva al trattamento delle rughe glabellari interciliari, mentre nessun paziente ha risposto al placebo. La risposta al trattamento delle rughe perioculari laterali è stata osservata nel 63,8 % dei pazienti trattati con Xeomin, rispetto al 2,0 % dei pazienti trattati con placebo. Nel complesso, il 71,4 % dei pazienti trattati con Xeomin ha avuto una risposta positiva al trattamento delle rughe orizzontali della fronte, mentre solo un paziente (2,0 %) ha risposto al placebo. Nel trattamento delle tre aree contemporaneamente, è stata osservata una risposta positiva nella maggior parte dei pazienti nel gruppo trattato con Xeomin (54,3 %), mentre nel gruppo placebo non è stata osservata alcuna risposta (0,0 %).
La sicurezza e tollerabilità a lungo termine di 54–64 unità di Xeomin sono state dimostrate in uno studio prospettico aperto di fase III con ripetuta somministrazione della dose per un periodo di trattamento superiore a un anno con 4 cicli consecutivi di iniezioni, a cui hanno partecipato 125 pazienti con rughe mimiche superiori del viso da moderate a marcate.
Bambini
L'Agenzia europea per i medicinali ha esentato dall'obbligo di presentare i risultati degli studi di Xeomin in tutte le sottopopolazioni pediatriche per il trattamento delle rughe correlate all'attività muscolare, distonie e nei bambini di età compresa tra 0 e 24 mesi per il trattamento della spasticità muscolare (vedere la sezione «Modalità di somministrazione e dosi» per l'uso nei bambini).
Farmacocinetica.
Caratteristiche generali della sostanza attiva
Non sono stati condotti studi classici sulla farmacocinetica e distribuzione della tossina botulinica di tipo A poiché la sostanza attiva viene somministrata in quantità estremamente ridotte (picogrammi per iniezione) e si lega rapidamente e in modo irreversibile alle terminazioni nervose colinergiche.
La tossina botulinica nativa è un complesso ad alto peso molecolare che, oltre alla neurotossina (150 kD), contiene altre proteine non tossiche come emagglutinine e non emagglutinine. A differenza dei prodotti tradizionali contenenti il complesso di tossina botulinica di tipo A, Xeomin contiene la neurotossina pura (150 kD), poiché è privo delle proteine formanti complesso e quindi presenta un basso contenuto di proteine estranee. Il contenuto di proteine estranee somministrate è considerato uno dei fattori della mancata risposta secondaria al trattamento.
Come molti altri proteine, è stato dimostrato che la tossina botulinica di tipo A subisce un trasporto assonale retrogrado dopo iniezione intramuscolare. Tuttavia, non si osserva un trasporto transsinaptico retrogrado della tossina botulinica di tipo A attiva nel sistema nervoso centrale quando vengono utilizzate dosi terapeuticamente appropriate.
La tossina botulinica di tipo A legata ai recettori subisce endocitosi nelle terminazioni nervose fino a raggiungere il suo bersaglio (SNAP-25), dove viene poi degradata a livello intracellulare. Le molecole libere di tossina botulinica di tipo A che non si sono legate ai recettori presinaptici delle terminazioni nervose colinergiche vengono fagocitate e pinocitate e degradate come qualsiasi altra proteina libera circolante.
Distribuzione della sostanza attiva nell'organismo dei pazienti
Studi farmacocinetici di Xeomin nell'uomo non sono stati condotti per i motivi sopra citati.
Dati preclinici di sicurezza
I dati preclinici non hanno evidenziato particolari pericoli per l'uomo in base agli studi farmacologici standard di sicurezza cardiovascolare e intestinale.
I risultati degli studi di tossicità da somministrazione ripetuta riguardo alla tossicità sistemica di Xeomin negli animali erano principalmente correlati al suo effetto farmacodinamico, cioè atonia, paresi e atrofia dei muscoli in cui sono state effettuate le iniezioni.
Non sono state osservate manifestazioni di intolleranza locale. Gli studi sull'effetto tossico di Xeomin sulla funzione riproduttiva non hanno evidenziato effetti negativi sulla fertilità di maschi e femmine di conigli né effetti diretti sull'embrione/feto o sullo sviluppo pre- e postnatale di ratti e/o conigli. Tuttavia, la somministrazione giornaliera, settimanale o ogni due settimane di Xeomin negli studi di tossicità embrionale, a dosi che provocavano riduzione del peso corporeo della femmina, ha aumentato il numero di aborti nei conigli e leggermente ridotto il peso corporeo del feto nei ratti. La somministrazione sistemica ripetuta di Xeomin a femmine di coniglio durante (una fase sensibile sconosciuta) dell'organogenesi, come presupposto per l'insorgenza di effetti teratogeni, non era prevista come obbligatoria in questi studi.
Di conseguenza, i margini di sicurezza rispetto alla terapia clinica erano generalmente bassi in relazione alle alte dosi cliniche.
Non sono stati condotti studi di genotossicità e cancerogenicità di Xeomin.
Caratteristiche cliniche.
Indicazioni.
Xeomin è indicato per la riduzione temporanea delle rughe verticali moderate o marcate tra le sopracciglia, visibili con la massima contrazione (rughine glabellari), delle rughe periorbitali laterali visibili con il massimo sorriso ("zampe di gallina") e/o delle rughe orizzontali della fronte visibili con la massima contrazione del muscolo frontale, in adulti fino a 65 anni di età, quando la marcata evidenza di tali rughe ha un forte impatto psicologico sul paziente; per il trattamento sintomatico negli adulti di blefarospasmo, distonia cervicale prevalentemente di tipo rotatorio (torticollis spasmodico) e spasticità muscolare della mano.
Controindicazioni.
Ipersensibilità alla sostanza attiva o a uno qualsiasi degli eccipienti del medicinale, disturbi muscolari sistemici (ad esempio miastenia grave, sindrome di Lambert − Eaton), infezione o infiammazione nell'area prevista per l'iniezione.
Interazioni con altri medicinali ed altre forme di interazione.
Studi di interazione non sono stati condotti.
Teoricamente, l'effetto della neurotossina botulinica può essere potenziato dagli antibiotici aminoglicosidi o da altri medicinali che interferiscono con la trasmissione neuromuscolare, ad esempio miorilassanti di tipo tubocurarina.
Pertanto, l'uso concomitante del medicinale Xeomin con aminoglicosidi o spettinomicina richiede particolare attenzione. I miorilassanti con meccanismo d'azione periferico devono essere utilizzati con cautela, riducendo se necessario la dose iniziale del miorilassante o impiegando farmaci di durata intermedia, come vecuronio o atracurio, invece di sostanze ad azione prolungata.
Le 4-aminochinoline possono ridurre l'efficacia del medicinale Xeomin.
Caratteristiche particolari di impiego.
Informazioni generali
Prima dell’iniezione del medicinale Xeomin, il medico deve essere a conoscenza dell’anatomia del paziente e di eventuali alterazioni anatomiche dovute a precedenti interventi chirurgici.
È necessario prestare attenzione a non iniettare il medicinale Xeomin in un vaso sanguigno.
Si deve considerare che le rughe orizzontali della fronte non sono dovute soltanto all’attività mimica, ma possono anche essere il risultato della perdita di elasticità della pelle (ad esempio, legata all’invecchiamento o ai danni da esposizione al sole). In questi casi, i pazienti potrebbero non rispondere ai preparati a base di tossina botulinica.
Xeomin deve essere utilizzato con cautela nei seguenti casi:
- presenza di qualsiasi alterazione della coagulazione del sangue;
- assunzione da parte del paziente di anticoagulanti o di altre sostanze con effetto anticoagulante.
Gli effetti clinici della neurotossina botulinica di tipo A possono aumentare o diminuire dopo iniezioni ripetute. Possibili cause della variazione degli effetti clinici includono diversi metodi di ricostituzione del prodotto, intervalli tra le iniezioni, muscoli iniettati e variazioni naturali dell’attività tossica risultanti da test biologici o da una risposta secondaria assente.
Diffusione locale e sistemica dell’effetto della tossina
Effetti indesiderati possono verificarsi a seguito di iniezioni di neurotossina botulinica di tipo A in aree inadeguatamente selezionate, con possibile paralisi temporanea di gruppi muscolari adiacenti. Nel trattamento di indicazioni neurologiche, dosi elevate possono causare paralisi in muscoli distanti dal sito di iniezione.
Sono stati riportati effetti indesiderati potenzialmente correlati alla diffusione della tossina botulinica in aree distanti dal sito di iniezione (vedi sezione «Effetti indesiderati»). Alcuni di questi possono essere potenzialmente letali; sono stati inoltre riportati casi fatali, talvolta associati a disfagia, polmonite e/o grave deperimento.
Nei pazienti sottoposti a dosi terapeutiche può verificarsi una debolezza muscolare eccessiva.
I pazienti o le persone che si prendono cura di loro devono essere informati della necessità di ricorrere immediatamente all’assistenza medica in caso di comparsa di disturbi della deglutizione, del linguaggio o della respirazione.
Sono stati riportati casi di disfagia anche dopo iniezioni in aree non correlate alla muscolatura del collo.
Presenza di disturbi neuromuscolari preesistenti
Esiste un rischio di sviluppare una debolezza muscolare eccessiva nei pazienti con disturbi neuromuscolari. Il medicinale a base di tossina botulinica deve essere somministrato a questi pazienti solo sotto la supervisione di uno specialista e solo se il beneficio terapeutico supera il rischio. I pazienti con disfagia e storia di aspirazione devono essere trattati con particolare cautela.
Non è raccomandato l’uso di Xeomin per indicazioni estetiche in pazienti con disfagia e storia di aspirazione.
Xeomin deve essere usato con cautela:
- nei pazienti con sclerosi laterale amiotrofica;
- nei pazienti con altre malattie che causano disfunzione neuromuscolare periferica;
- nei pazienti in cui i muscoli bersaglio presentano debolezza o atrofia marcate.
Reazioni di ipersensibilità
Sono state osservate reazioni di ipersensibilità con l’uso del medicinale a base di neurotossina botulinica. In caso di reazioni gravi (ad esempio anafilattiche) e/o immediate di ipersensibilità, deve essere avviato un trattamento appropriato.
Formazione di anticorpi
L’uso troppo frequente del medicinale aumenta il rischio di formazione di anticorpi, con conseguente possibile mancata risposta al trattamento (vedi sezione «Modalità e dosi di somministrazione»).
Il rischio di formazione di anticorpi può essere minimizzato somministrando la dose efficace più bassa possibile e con gli intervalli massimi clinicamente indicati tra le iniezioni.
Indicazioni
Spasmo blefarico
È opportuno evitare iniezioni vicino al muscolo elevatore della palpebra superiore per ridurre il rischio di sviluppare ptosi. La diffusione della neurotossina botulinica di tipo A nel muscolo obliquo inferiore può causare diplopia. Evitando iniezioni mediali nella palpebra inferiore, si può ridurre la frequenza di questo effetto indesiderato.
A causa degli effetti anticolinergici della neurotossina botulinica di tipo A, Xeomin deve essere usato con cautela nei pazienti a rischio di glaucoma ad angolo chiuso.
Per prevenire l’ectropion, si devono evitare iniezioni nella palpebra inferiore e trattare tempestivamente qualsiasi difetto epiteliale. A tale scopo possono essere necessarie lacrime artificiali, unguenti, lenti a contatto terapeuthe morbide o la chiusura dell’occhio con una benda o altri metodi.
La riduzione del numero di battiti delle palpebre dopo l’iniezione di Xeomin nel muscolo orbicolare dell’occhio può portare all’esposizione della cornea, difetti epiteliali persistenti e formazione di ulcere corneali, in particolare nei pazienti con alterazioni dei nervi cranici (nervo facciale). Nei pazienti sottoposti in precedenza a interventi agli occhi, è necessario effettuare un accurato esame della sensibilità corneale.
Ecchimosi possono facilmente svilupparsi nei tessuti molli della palpebra. Premendo delicatamente sul sito di iniezione immediatamente dopo l’iniezione, si riduce questo rischio.
Torticollis spastico
Xeomin deve essere iniettato con cautela in aree vicine a strutture sensibili, come l’arteria carotide, l’apice del polmone o l’esofago.
Ai pazienti con acinesia o con stile di vita sedentario si deve raccomandare di riprendere gradualmente l’attività fisica dopo l’iniezione di Xeomin.
I pazienti devono essere informati che le iniezioni di Xeomin per il trattamento del torticollis spastico possono causare disfagia da lieve a grave, con rischio di aspirazione e dispnea. In questi casi può essere necessario un intervento medico (ad esempio, alimentazione tramite sonda gastrica) (vedi anche sezione «Effetti indesiderati»). Limitare la dose somministrata al muscolo sterno-cleido-mastoideo a 100 unità riduce il rischio di disfagia. I pazienti con ridotta massa muscolare del collo o quelli che richiedono iniezioni bilaterali nel muscolo sterno-cleido-mastoideo hanno un rischio maggiore. La disfagia è attribuita alla diffusione dell’effetto farmacologico di Xeomin, dovuta alla dispersione della neurotossina nei muscoli dell’esofago.
Spasticità dell’arto superiore
Xeomin deve essere iniettato con cautela in aree vicine a strutture sensibili, come l’arteria carotide, l’apice del polmone o l’esofago.
Ai pazienti con acinesia o con stile di vita sedentario si deve raccomandare di riprendere gradualmente l’attività fisica dopo l’iniezione di Xeomin.
Xeomin per il trattamento della spasticità focale è stato studiato in combinazione con le normali terapie riabilitative e non deve essere considerato un sostituto di tali trattamenti. Xeomin probabilmente non sarà efficace nel migliorare l’ampiezza di movimento articolare in presenza di contrattura fissa.
Sono stati riportati episodi nuovi o ricorrenti, generalmente in pazienti predisposti a tali eventi. La relazione esatta tra questi episodi e l’iniezione di tossina botulinica non è stata stabilita.
Uso durante la gravidanza o l’allattamento.
Gravidanza
Non esistono dati adeguati sull’uso della neurotossina botulinica di tipo A in donne in gravidanza. Studi sugli animali hanno dimostrato effetti tossici sulla funzione riproduttiva (vedi anche sezione «Dati di sicurezza preclinici»). Il rischio potenziale nell’uomo è sconosciuto. Pertanto, Xeomin non deve essere usato durante la gravidanza, salvo in caso di stretta necessità e solo se il beneficio potenziale giustifica il rischio.
Allattamento
Non è noto se la neurotossina botulinica di tipo A sia escreta nel latte materno. Pertanto, Xeomin non deve essere usato durante l’allattamento.
Funzione riproduttiva
Non esistono dati clinici sull’impatto della neurotossina botulinica di tipo A sulla funzione riproduttiva. Nei conigli non è stato osservato alcun effetto dannoso sulla fertilità di maschi o femmine (vedi anche sezione «Dati di sicurezza preclinici»).
Capacità di guidare veicoli o di usare macchinari.
Xeomin ha un effetto lieve o moderato sulla capacità di guidare veicoli o di usare macchinari. I pazienti devono essere informati che, in caso di comparsa di astenia, debolezza muscolare, vertigini, disturbi visivi o ptosi palpebrale, devono evitare di guidare veicoli o svolgere altre attività potenzialmente pericolose.
Modalità e dosi di somministrazione.
Le unità di misura dell'attività del prodotto Xeomin non sono intercambiabili con quelle di altri prodotti a base di tossina botulinica a causa delle differenze nei metodi di determinazione dell'attività.
Per informazioni dettagliate sugli studi clinici di confronto tra Xeomin e i complessi tradizionali di tossina botulinica di tipo A (900 kD), si prega di consultare la sezione «Farmacodinamica».
Il prodotto Xeomin deve essere somministrato esclusivamente da medici specializzati e con esperienza nel trattamento con tossina botulinica di tipo A.
Il dosaggio ottimale, la frequenza e il numero di punti di iniezione intramuscolare devono essere stabiliti dal medico per ogni singolo paziente. La dose deve essere titolata con attenzione.
Non si devono superare le dosi raccomandate per singola somministrazione di Xeomin.
La soluzione ricostituita di Xeomin viene somministrata per via intramuscolare.
Dopo la ricostituzione, Xeomin deve essere utilizzato immediatamente, in un’unica procedura e per un solo paziente.
Rughe verticali tra le sopracciglia, evidenti durante la massima contrazione (rughe glabellari)
Dosaggio
Dopo la ricostituzione di Xeomin (50 unità/1,25 ml), il volume raccomandato per l'iniezione è di 0,1 ml (4 unità) in ciascuno dei 5 punti di iniezione: due iniezioni in ciascun muscolo corrugatore delle sopracciglia (corrugator) e una iniezione nel muscolo procerus, corrispondente alla dose standard di 20 unità. Il medico può aumentare la dose fino a un massimo di 30 unità, se necessario per soddisfare le esigenze individuali del paziente, con intervalli tra le sedute di trattamento di almeno 3 mesi.
La riduzione dell'intensità delle rughe verticali tra le sopracciglia (rughe glabellari) si verifica generalmente entro 2-3 giorni, con effetto massimo raggiunto entro il giorno 30. L'effetto dura fino a 4 mesi dopo l'iniezione.
Modalità di somministrazione
La soluzione ricostituita di Xeomin viene iniettata utilizzando un ago sterile sottile (ad esempio un ago calibro 30-33G / diametro 0,20-0,30 mm / lunghezza 13 mm).
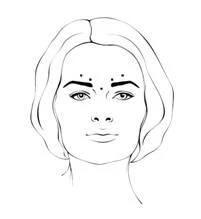
Prima e durante l'iniezione, il pollice o l'indice devono premere saldamente al di sotto del margine orbitale per evitare la diffusione della soluzione in questa zona. L'ago deve essere diretto verso l'alto e medialmente. Per ridurre il rischio di blefaroptosi, si devono evitare iniezioni vicino al muscolo elevatore della palpebra superiore e alla porzione craniale del muscolo orbicolare dell'occhio. Le iniezioni nel muscolo corrugatore delle sopracciglia devono essere effettuate nella parte centrale del muscolo e almeno 1 cm al di sopra degli archi sopracciliari.
Rughe periorbitali laterali, evidenti durante il massimo sorriso ("zampe di gallina")
Dosaggio
Dopo la ricostituzione di Xeomin (50 unità/1,25 ml), il volume raccomandato per l'iniezione è di 0,1 ml (4 unità) in ciascuno dei 3 punti di iniezione su entrambi i lati. Un'iniezione di 0,1 ml viene effettuata approssimativamente a 1 cm lateralmente rispetto al margine osseo orbitale. Le altre due iniezioni (0,1 ml ciascuna) devono essere effettuate rispettivamente a circa 1 cm sopra e sotto il punto della prima iniezione.
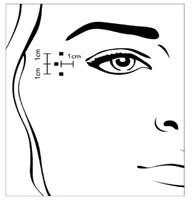
La dose raccomandata standard totale per una singola procedura di trattamento è di 12 unità per lato (dose totale somministrata: 24 unità).
La riduzione delle rughe periorbitali laterali, evidenti durante il massimo sorriso ("zampe di gallina"), si verifica generalmente entro i primi 6 giorni, con effetto massimo raggiunto entro il giorno 30. L'effetto dura fino a 4 mesi dopo l'iniezione.
Attualmente non sono disponibili dati sull'efficacia e sulla sicurezza di più di due iniezioni con intervallo di 4 mesi per il trattamento delle rughe periorbitali laterali evidenti durante il massimo sorriso.
Modalità di somministrazione
La soluzione ricostituita di Xeomin viene iniettata utilizzando un ago sterile sottile (ad esempio un ago calibro 30-33G / diametro 0,20-0,30 mm / lunghezza 13 mm). L'iniezione deve essere effettuata per via intramuscolare nel muscolo orbicolare dell'occhio, appena sotto il derma, per evitare la diffusione di Xeomin. Si devono evitare iniezioni troppo vicine al muscolo massetere per prevenire il ptosi (abbassamento) del labbro.
Per il trattamento delle rughe glabellari e delle "zampe di gallina", gli intervalli tra le sedute di trattamento devono essere di almeno 3 mesi. Se il trattamento risulta inefficace o l'effetto delle iniezioni ripetute diminuisce, si devono considerare metodi terapeutici alternativi.
Rughe orizzontali della fronte, evidenti durante la massima contrazione del muscolo frontale
Dosaggio
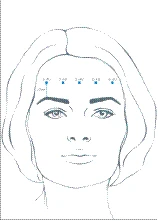
In base alle esigenze individuali dei pazienti, il range di dosaggio raccomandato varia da 10 a 20 unità, con un intervallo di almeno 3 mesi tra le sedute di trattamento. Dopo la ricostituzione, la dose totale di Xeomin da 10 a 20 unità viene iniettata nel muscolo frontale in 5 punti di iniezione allineati orizzontalmente, posizionati almeno a 2 cm sopra il margine osseo orbitale. A ciascun punto di iniezione vengono somministrate rispettivamente 2, 3 o 4 unità di prodotto.
La riduzione dell'intensità delle rughe della fronte durante la massima contrazione del muscolo frontale si osserva generalmente entro 7 giorni, con effetto massimo raggiunto entro il giorno 30. L'effetto dura fino a 4 mesi dopo l'iniezione.
Modalità di somministrazione
La soluzione ricostituita di Xeomin viene iniettata utilizzando un ago sterile sottile (ad esempio un ago calibro 30-33G / diametro 0,20-0,30 mm / lunghezza 13 mm). Per prevenire il ptosi delle sopracciglia, si devono evitare iniezioni di Xeomin vicino al margine orbitale, poiché ciò potrebbe causare paralisi delle fibre muscolari inferiori.
Gli intervalli tra le sedute di trattamento devono essere di almeno 3 mesi. Se il trattamento risulta inefficace o l'effetto delle iniezioni ripetute diminuisce, si devono considerare metodi terapeutici alternativi.
Blefarospasmo
Dosaggio
La dose iniziale raccomandata è di 1,25-2,5 unità per punto di iniezione. La dose iniziale non deve superare le 25 unità per occhio. La dose totale non deve superare le 100 unità per occhio per seduta di trattamento. Il trattamento ripetuto può generalmente essere effettuato non più frequentemente di ogni 12 settimane. Gli intervalli tra le procedure terapeutiche devono essere determinati in base alla reale necessità clinica del singolo paziente.
L'effetto del prodotto inizia in media entro 4 giorni dall'iniezione. L'effetto terapeutico di Xeomin dura generalmente da 3 a 4 mesi, sebbene possa durare significativamente più a lungo o più brevemente. Se necessario, il trattamento può essere ripetuto.
Se l'effetto della dose iniziale è stato insufficiente, la dose può essere raddoppiata nelle procedure successive. Tuttavia, non è stato dimostrato alcun beneficio aggiuntivo dall'iniezione di più di 5,0 unità in un singolo punto.
Modalità di somministrazione
Dopo la ricostituzione, Xeomin viene somministrato con un ago sterile appropriato (ad esempio un ago calibro 27-30G / diametro 0,30-0,40 mm / lunghezza 12,5 mm). Il controllo elettromiografico durante la procedura non è necessario. Il volume raccomandato per singola iniezione è di circa 0,05-0,1 ml.
Xeomin viene iniettato nelle parti mediale e laterale del muscolo orbicolare della palpebra superiore e nella parte laterale del muscolo orbicolare della palpebra inferiore. Se la vista è compromessa a causa degli spasmi, possono essere effettuate iniezioni aggiuntive nell'area della fronte, nelle regioni laterali del muscolo orbicolare dell'occhio e nella parte superiore del viso.
Torticollo spastico
Dosaggio
Nel trattamento del torticollo spastico, il dosaggio di Xeomin deve essere personalizzato per ogni paziente in base alla posizione della testa e del collo, alla localizzazione del dolore, all'ipertrofia muscolare, al peso corporeo e alla risposta individuale alle iniezioni.
La dose raccomandata per singolo punto di iniezione è fino a 50 unità, mentre la dose massima per la prima procedura è di 200 unità. Il medico può prescrivere dosi fino a 300 unità nelle sedute successive, in base alla risposta individuale del paziente.
Generalmente, l'effetto del prodotto inizia entro 7 giorni dall'iniezione. L'effetto di ogni seduta di trattamento con Xeomin dura circa 3-4 mesi, sebbene possa durare significativamente più a lungo o più brevemente. L'intervallo raccomandato tra le procedure è di almeno 10 settimane. Gli intervalli tra le sedute terapeutiche devono essere determinati in base alla reale necessità clinica del singolo paziente.
Modalità di somministrazione
Per le iniezioni nei muscoli superficiali si utilizzano aghi sterili appropriati, ad esempio aghi calibro 25-30G / diametro 0,30-0,50 mm / lunghezza 37 mm, mentre per i muscoli profondi si utilizzano aghi di dimensioni maggiori, ad esempio calibro 22G / diametro 0,70 mm / lunghezza 75 mm. Il volume raccomandato per singola iniezione in un punto è di circa 0,1-0,5 ml.
Il trattamento del torticollo spastico prevede l'iniezione di Xeomin nel muscolo sternocleidomastoideo, nel muscolo elevatore della scapola, nei muscoli scaleni, nel muscolo romboide e/o nei muscoli trapezi. Questo elenco non è esaustivo, poiché qualsiasi muscolo coinvolto nel controllo della posizione della testa può essere coinvolto nel processo patologico e richiedere trattamento. In caso di difficoltà nell'identificazione dei singoli muscoli coinvolti, può essere necessaria l'elettromiografia o l'ecografia. La massa muscolare e il grado di ipertrofia o atrofia sono fattori da considerare nella scelta della dose appropriata.
L'esecuzione di iniezioni in più punti consente a Xeomin di distribuirsi uniformemente nelle aree di innervazione muscolare soggette a distonia (in particolare durante l'iniezione in muscoli di grandi dimensioni). Il numero ottimale di punti di iniezione dipende dall'entità del muscolo da denervare chimicamente.
Non si devono effettuare iniezioni bilaterali nel muscolo sternocleidomastoideo, poiché esiste un rischio aumentato di effetti indesiderati (in particolare disfagia) se le iniezioni bilaterali o le dosi somministrate in questo muscolo superano complessivamente 100 unità.
Spasticità dell'arto superiore
Dosaggio
Il dosaggio preciso e il numero di punti di iniezione devono essere stabiliti individualmente per ogni paziente in base alle dimensioni, al numero e alla localizzazione dei muscoli coinvolti, alla gravità della spasticità e alla presenza di debolezza muscolare localizzata.
Dosi terapeutiche raccomandate per tipo di muscolo:
| Struttura clinica |
Unità (intervallo) |
Numero di punti di iniezione in un singolo muscolo |
|
| Polso flesso |
|||
| Flessore ulnare del carpo (Flexor carpi ulnaris) |
20−100 |
1−2 |
|
| Mano a pugno chiuso |
|||
| Flessore superficiale delle dita (Flexor digitorum superficialis) |
25−100 |
2 |
|
| Flessore profondo delle dita (Flexor digitorum profundus) |
25−100 |
2 |
|
| Gomito flesso |
|||
| Brachioradiale |
25−100 |
1−3 |
|
| Bicipite |
50−200 |
1−4 |
|
| Brachiale |
25−100 |
1−2 |
|
| Avambraccio pronato |
|||
| Pronatore quadrato (Pronator quadratus) |
10−50 |
1 |
|
| Pronatore rotondo (Pronator teres) |
25−75 |
1−2 |
|
| Pollice premuto sul palmo |
|||
| Flessore lungo del pollice (Flexor pollicis longus) |
10−50 |
1 |
|
| Adduttore del pollice (Adductor pollicis) |
5−30 |
1 |
|
| Flessore breve del pollice (Flexor pollicis brevis/ Opponens pollicis) |
5−30 |
1 |
|
| Rotazione interna della spalla/spalla abdotta/spalla addotta |
|||
| Muscolo deltoide, parte clavicolare (Deltoideus, pars clavicularis) |
20−150 |
1−3 |
|
| Muscolo grande dorsale (Latissimus dorsi) |
25−150 |
1−4 |
|
| Muscolo grande pettorale (Pectoralis major) |
20−200 |
1−6 |
|
| Muscolo sottoscapolare (Subscapularis) |
15−100 |
1−4 |
|
| Muscolo terzo rotondo (Teres major) |
20−100 |
1−2 |
|
La dose totale massima per il trattamento della spasticità degli arti superiori non deve superare 500 unità per singola sessione di trattamento, mentre la dose per i muscoli della spalla non deve superare le 250 unità.
L'effetto del medicinale inizia, secondo le segnalazioni dei pazienti, già 4 giorni dopo l'iniezione. Il massimo effetto, considerato un miglioramento del tono muscolare, si manifesta entro 4 settimane. In generale, l'effetto terapeutico dura 12 settimane, anche se può protrarsi per un periodo significativamente più lungo o più breve. Di norma, il trattamento ripetuto può essere effettuato non più spesso di ogni 12 settimane. Gli intervalli tra le procedure terapeutiche devono essere determinati in base alle effettive esigenze cliniche del singolo paziente.
Modalità di somministrazione
La soluzione ricostituita del medicinale Xeomin viene somministrata per iniezione utilizzando un ago sterile appropriato (ad esempio un ago calibro 26G/diametro 0,45 mm/lunghezza 37 mm per i muscoli superficiali e un ago più lungo, ad esempio calibro 22G/diametro 0,7 mm/lunghezza 75 mm, per i muscoli profondi).
Qualora sorgano difficoltà nell'identificare i singoli muscoli coinvolti, le iniezioni devono essere effettuate sotto controllo elettromiografico o ecografico. L'esecuzione di iniezioni in più punti consente al medicinale Xeomin di distribuirsi uniformemente nell'area di innervazione del muscolo, il che è particolarmente importante quando si inietta nei muscoli più grandi.
Gruppi di pazienti particolari
I dati clinici ottenuti negli studi di fase III sull'uso del medicinale Xeomin nei pazienti di età pari o superiore a 65 anni sono limitati. Fino al completamento di ulteriori studi in questa fascia di età, Xeomin non è raccomandato per l'uso nei pazienti di età superiore a 65 anni.
Tutte le indicazioni
Per il trattamento delle rughe glabellari, delle "zampe di gallina" e delle rughe orizzontali della fronte
In assenza di risposta al trattamento entro un mese dalla prima iniezione, si devono adottare le seguenti misure:
- analizzare le possibili cause della mancata risposta al trattamento, ad esempio: dose troppo bassa, tecnica di iniezione inadeguata, possibile formazione di anticorpi neutralizzanti nei confronti della neurotossina;
- aggiustare la dose in base all'analisi del precedente tentativo di trattamento fallito;
- rivalutare l'idoneità del trattamento con neurotossina botulinica di tipo A;
- se durante il primo ciclo di trattamento non sono stati osservati effetti indesiderati, è possibile somministrare un ulteriore ciclo di trattamento, purché sia rispettato un intervallo minimo di 3 mesi tra il ciclo iniziale e quello ripetuto.
Per il trattamento del blefarospasmo, della torticollis spasmodica e della spasticità dell'arto superiore
In assenza di risposta al trattamento entro un mese dalla prima iniezione, si devono adottare le seguenti misure:
- riesaminare la conferma clinica dell'effetto della neurotossina sul muscolo in cui è stato iniettato il medicinale, ad esempio effettuando un'elettromiografia presso un centro specializzato;
- analizzare le possibili cause della mancata risposta al trattamento, ad esempio: scarsa delimitazione dei muscoli da iniettare, dose troppo bassa, tecnica di iniezione inadeguata, contrattura fissa, antagonista troppo debole, possibile sviluppo di anticorpi;
- riesaminare l'idoneità del trattamento con neurotossina botulinica di tipo A;
- se durante il primo ciclo di trattamento non sono stati osservati effetti indesiderati, è possibile somministrare un ulteriore ciclo di trattamento alle seguenti condizioni: 1) aggiustamento della dose in base all'analisi del precedente tentativo di trattamento fallito; 2) localizzazione dei muscoli coinvolti mediante elettromiografia; 3) rispetto dell'intervallo minimo raccomandato tra il ciclo iniziale e quello ripetuto.
Ricostituzione e smaltimento del medicinale non utilizzato
Prima dell'uso, Xeomin deve essere ricostituito con soluzione di cloruro di sodio 9 mg/ml (0,9%) per iniezioni. La ricostituzione e la diluizione devono essere effettuate secondo le raccomandazioni della buona pratica clinica, in particolare per quanto riguarda l'asepsi.
Le operazioni di ricostituzione del contenuto della fiala e di preparazione della siringa devono essere eseguite su asciugamani di carta con sottofondo in plastica per evitare schizzi. Nella siringa si aspira la quantità appropriata di solvente (vedere sotto). Dopo aver inserito verticalmente l'ago attraverso il tappo di gomma, il solvente viene introdotto delicatamente nella fiala, evitando la formazione di schiuma. Per la ricostituzione della soluzione si raccomanda l'uso di un ago corto e conico di calibro 20-27G. La fiala deve essere scartata se la mancanza di vuoto impedisce l'introduzione del solvente. Si rimuove la siringa dalla fiala e si mescola Xeomin con il solvente, ruotando e capovolgendo delicatamente la fiala oppure semplicemente capovolgendola (non agitare energeticamente). Se necessario, l'ago utilizzato per la ricostituzione può rimanere nella fiala, ma la quantità necessaria di soluzione deve essere prelevata con una nuova siringa sterile per iniezioni.
Il medicinale Xeomin ricostituito è una soluzione limpida, incolore, priva di particelle solide.
Il medicinale Xeomin non deve essere utilizzato se, dopo la ricostituzione, la soluzione risulta torbida o contiene particelle visibili o fiocchi.
È necessario determinare con attenzione il volume corretto di solvente per la quantità prescelta di unità di dose del medicinale, al fine di evitare un eventuale sovradosaggio accidentale. Se durante una singola procedura iniettiva vengono utilizzate fiale di Xeomin di diversa capacità, è necessario determinare con attenzione la corretta quantità di solvente durante la ricostituzione di una determinata quantità di unità per 0,1 ml. La quantità di solvente varia a seconda della dose prescelta del medicinale, da 50 a 100 unità di Xeomin. Ogni siringa deve essere opportunamente etichettata.
Per il trattamento delle rughe glabellari, delle "zampe di gallina" e delle rughe orizzontali della fronte
Soluzioni possibili del medicinale Xeomin con potenza di 50 e 100 unità
| Dose somministrata (unità per 0,1 ml) |
Quantità di solvente (cloruro di sodio 9 mg/ml (0,9 %) per iniezione) |
|
| Flaconcino da 50 unità |
Flaconcino da 100 unità |
|
| 5 unità |
1 ml |
2 ml |
| 4 unità |
1,25 ml |
2,5 ml |
Per il trattamento del blefarospasmo, della torcicollo spasmodico e della spasticità dell'arto superiore
Possibili concentrazioni del farmaco Xeomin con potenza di 50 e 100 unità
| Dose ottenuta (unità per 0,1 ml) |
Quantità di solvente (cloruro di sodio 9 mg/ml (0,9%) per iniezione) |
|
| Flaconcino contenente 50 unità |
Flaconcino contenente 100 unità |
|
| 20 unità |
0,25 ml |
0,5 ml |
| 10 unità |
0,5 ml |
1 ml |
| 8 unità |
0,625 ml |
1,25 ml |
| 5 unità |
1 ml |
2 ml |
| 4 unità |
1,25 ml |
2,5 ml |
| 2,5 unità |
2 ml |
4 ml |
| 2 unità |
2,5 ml |
5 ml |
| 1,25 unità |
4 ml |
Non applicabile |
Dal punto di vista microbiologico, il medicinale deve essere utilizzato immediatamente. Se non viene utilizzato immediatamente, l'utente è responsabile della durata e delle condizioni di conservazione del contenitore aperto. Generalmente, la conservazione non deve superare le 24 ore a una temperatura compresa tra 2 °C e 8 °C, a meno che il ricostituente non sia stato aggiunto in condizioni asettiche controllate e validate.
La soluzione iniettabile conservata per più di 24 ore e qualsiasi soluzione iniettabile non utilizzata devono essere smaltite.
Procedura per lo smaltimento sicuro di fiale, siringhe e materiali utilizzati
Le fiale non utilizzate, i residui di soluzione ricostituita nella fiala e/o le siringhe devono essere sterilizzati mediante autoclave. In alternativa, i residui del medicinale preparato Xeomin possono essere inattivati aggiungendo una delle seguenti soluzioni: etanolo al 70 %, isopropanolo al 50 %, laurilsolfato sodico (SDS) allo 0,1 % (detergente anionico), soluzione diluita di idrossido di sodio (0,1 N NaOH) o soluzione diluita di ipoclorito di sodio (almeno 0,1 % di NaOCl).
Dopo l'inattivazione, le fiale, le siringhe e i materiali utilizzati non devono essere svuotati; devono essere collocati in appositi contenitori e smaltiti secondo le norme locali vigenti.
Raccomandazioni in caso di incidente durante la manipolazione della tossina botulinica di tipo A
- Il medicinale versato deve essere immediatamente rimosso: utilizzando materiale assorbente imbevuto di una delle soluzioni sopra elencate, nel caso di polvere, oppure materiale assorbente asciutto, nel caso di soluzione ricostituita;
- le superfici contaminate devono essere pulite con materiale assorbente imbevuto di una delle soluzioni sopra descritte e successivamente asciugate;
- se la fiala si rompe, procedere come descritto sopra, raccogliere con attenzione i frammenti di vetro e rimuovere il medicinale versato o disperso, evitando lesioni cutanee;
- in caso di contatto del medicinale con la pelle, lavare abbondantemente la zona interessata con acqua;
- in caso di contatto con gli occhi, sciacquare accuratamente con abbondante acqua o soluzione per il lavaggio degli occhi;
- in caso di contatto con ferite, tagli o aree cutanee danneggiate, lavare abbondantemente con acqua e adottare le misure mediche appropriate in base alla dose somministrata.
È necessario seguire scrupolosamente le istruzioni per la manipolazione del medicinale e per la sua corretta procedura di smaltimento.
Pediatria.
La sicurezza ed efficacia del medicinale Xeomin nei bambini e negli adolescenti di età inferiore ai 17 anni non sono state ancora stabilite. Pertanto, l'uso di Xeomin in questa categoria di pazienti non è raccomandato fino a quando non saranno disponibili nuovi dati.
Sovradosaggio.
Sintomi da sovradosaggio
Dosi elevate di tossina botulinica di tipo A possono causare un marcato paralisi muscolare con sintomi in sedi distanti dal sito di iniezione. Tali sintomi comprendono, in particolare, debolezza generale, ptosi, diplopia, difficoltà respiratorie, difficoltà del linguaggio, nonché paralisi dei muscoli respiratori e difficoltà di deglutizione, che possono portare allo sviluppo di polmonite da aspirazione.
Misure da adottare in caso di sovradosaggio
In caso di sovradosaggio, il paziente deve essere tenuto sotto stretta osservazione medica per monitorare l'eventuale insorgenza di sintomi di eccessiva debolezza muscolare o paralisi muscolare. Il paziente potrebbe necessitare di un trattamento sintomatico. In caso di paralisi dei muscoli respiratori, è necessario un supporto respiratorio.
Effetti indesiderati.
Di solito gli effetti indesiderati si verificano durante la prima settimana dopo il trattamento e sono di natura transitoria. Gli effetti indesiderati possono essere correlati al principio attivo, alla procedura di iniezione o ad entrambi i fattori.
Reazioni avverse indipendenti dall'indicazione
Reazioni correlate all'iniezione del medicinale
Dolore localizzato, infiammazione, parestesia, ipoestesia, dolore, sensibilità, gonfiore, edema, eritema, prurito, infezione localizzata, ematoma, emorragia e/o ecchimosi possono essere correlate all'esecuzione dell'iniezione.
Il dolore correlato all'ago e/o la sensazione di ansia possono provocare reazioni vascolari parasimpatiche, ad esempio ipotensione sintomatica transitoria, nausea, ronzio alle orecchie e perdita di coscienza.
Effetti indesiderati della classe delle sostanze della tossina botulinica di tipo A
La debolezza muscolare localizzata rappresenta uno degli effetti farmacologici attesi della tossina botulinica.
Diffusione della tossina
Durante il trattamento con tossine botuliniche per altre indicazioni, sono stati riportati molto raramente effetti indesiderati correlati alla diffusione della tossina in aree distanti dal sito di iniezione, che hanno causato sintomi corrispondenti agli effetti della tossina botulinica di tipo A (aumentata debolezza muscolare, disfagia e polmonite da aspirazione con esito fatale in alcuni casi) (vedere il paragrafo «Proprietà farmacodinamiche»).
Gli effetti indesiderati sopra elencati non possono essere completamente esclusi con l'uso del medicinale Xeomin.
Reazioni di ipersensibilità
Raramente sono stati riportati casi di gravi e/o immediate reazioni di ipersensibilità, inclusa anafilassi, malattia da siero, orticaria, edema dei tessuti molli e dispnea. Alcune di queste reazioni sono state osservate dopo l'uso del complesso tradizionale della tossina botulinica di tipo A, da solo o in combinazione con altre sostanze note per la loro capacità di indurre reazioni simili.
Reazioni avverse osservate nella pratica clinica
Di seguito sono riportate informazioni sulla frequenza degli effetti indesiderati osservati nella pratica clinica per singole indicazioni. La frequenza è definita come segue: molto frequente (≥ 1/10); frequente (da ≥ 1/100 a < 1/10); non frequente (da ≥ 1/1.000 a < 1/100); raro (da ≥ 1/10.000 a < 1/1.000); molto raro (< 1/10.000); non nota (non può essere stimata sulla base dei dati disponibili).
Solchi verticali tra le sopracciglia (rughe glabellari)
Infezioni e infestazioni
Non frequente: bronchite, nasofaringite, malattia simil-influenzale.
Disturbi psichiatrici
Non frequente: depressione, insonnia.
Disturbi del sistema nervoso
Frequente: cefalea.
Non frequente: paralisi facciale (ptosi delle sopracciglia).
Disturbi dell'occhio
Non frequente: edema palpebrale, ptosi palpebrale, visione offuscata.
Disturbi della cute e del tessuto sottocutaneo
Non frequente: prurito, noduli cutanei, ptosi delle sopracciglia.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo
Frequente: «sopracciglia di Mefistofele» (sollevamento laterale delle sopracciglia).
Non frequente: scosse muscolari, spasmi muscolari, asimmetria facciale (asimmetria delle sopracciglia), sensazione di pesantezza.
Disturbi generali e condizioni in sede di iniezione
Non frequente: ematoma in sede di iniezione, dolore in sede di iniezione, dolore locale, debolezza, affaticamento, malessere (sensazione di pesantezza di palpebre/sopracciglia).
Disturbi vascolari
Non frequente: ematoma.
Solchi periorbitali laterali visibili durante il massimo sorriso («zampe di gallina»)
Disturbi dell'occhio
Frequente: edema palpebrale, secchezza oculare.
Disturbi generali e condizioni in sede di iniezio
Frequente: ematoma in sede di iniezione.
Rughe nella parte superiore del viso
Disturbi del sistema nervoso
Molto frequente: cefalea.
Frequente: ipoestesia.
Disturbi generali e condizioni in sede di iniezione
Frequente: ematoma in sede di iniezione, dolore in sede di iniezione, eritema in sede di iniezione, malessere (sensazione di pesantezza nella regione frontale).
Disturbi dell'occhio
Frequente: ptosi palpebrale, secchezza oculare.
Disturbi della cute e del tessuto sottocutaneo
Frequente: ptosi delle sopracciglia.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo
Frequente: asimmetria facciale, «sopracciglia di Mefistofele» (sollevamento laterale delle sopracciglia).
Disturbi del sistema gastrointestinale
Frequente: nausea.
Blefarospasmo
Disturbi del sistema nervoso
Frequente: cefalea, paralisi facciale.
Disturbi dell'occhio
Molto frequente: ptosi palpebrale, secchezza oculare.
Frequente: vista offuscata, disturbi visivi, diplopia, aumento della lacrimazione.
Disturbi del sistema gastrointestinale
Frequente: secchezza orale, disfagia.
Disturbi della cute e del tessuto sottocutaneo
Frequente: eruzione cutanea.
Disturbi sistemici e condizioni in sede di iniezione
Frequente: dolore in sede di iniezione, affaticamento.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo
Frequente: debolezza muscolare.
Torticollis spastico
Disturbi del sistema nervoso
Frequente: cefalea, stato presincopale, vertigini.
Non frequente: disturbi del linguaggio.
Disturbi respiratori, toracici e mediastinici
Non frequente: disfonia, dispnea.
Disturbi del sistema gastrointestinale
Molto frequente: disfagia.
Frequente: secchezza orale, nausea.
Disturbi della cute e del tessuto sottocutaneo
Frequente: iperidrosi.
Non frequente: eruzione cutanea.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo
Frequente: dolore al collo, debolezza muscolare, dolore muscolare, spasmi muscolari, rigidità muscoloscheletrica.
Disturbi sistemici e condizioni in sede di iniezione
Frequente: dolore in sede di iniezione, astenia.
Infezioni e infestazioni
Frequente: infezioni delle vie respiratorie superiori.
Il trattamento del torticollis spastico può provocare lo sviluppo di disfagia di grado variabile di gravità con rischio di aspirazione, che talvolta può richiedere assistenza medica. La disfagia può persistere per 2-3 settimane dopo l'iniezione, tuttavia è stato riportato un caso di disfagia durata cinque mesi.
Spasticità dell'arto superiore
Disturbi del sistema nervoso
Non frequente: cefalea, ipoestesia.
Disturbi del sistema gastrointestinale
Frequente: secchezza orale.
Non frequente: disfagia, nausea.
Disturbi del sistema muscoloscheletrico e del tessuto connettivo
Non frequente: debolezza muscolare, dolore agli arti, dolore muscolare.
Disturbi sistemici e condizioni in sede di iniezione
Non frequente: astenia.
Frequente: dolore in sede di iniezione, sensazione di calore.
Non noto: dolore in sede di iniezione.
Esperienza post-commercializzazione
Sono stati riportati sviluppo di sintomi simil-influenzali e reazioni di ipersensibilità, come gonfiore, edema (anche in sedi distanti dal sito di iniezione), eritema, prurito, eruzione cutanea (locale e generalizzata) e dispnea. Inoltre, con frequenza «non nota», sono stati riportati effetti indesiderati a carico del sistema muscoloscheletrico indipendentemente dall'indicazione: atrofia muscolare.
Segnalazione delle reazioni avverse sospette
È molto importante segnalare le reazioni avverse sospette dopo l'autorizzazione del medicinale. Tale segnalazione consente un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari sono invitati a segnalare qualsiasi reazione avversa sospetta attraverso il sistema nazionale di segnalazione.
Periodo di validità
3 anni.
La soluzione ricostituita può essere conservata a una temperatura compresa tra 2 °C e 8 °C per 24 ore.
Condizioni di conservazione.
Conservare a una temperatura non superiore a 25 °C. Conservare in un luogo non accessibile ai bambini.
Incompatibilità.
Questo medicinale non deve essere miscelato con altri medicinali, salvo quelli menzionati nel paragrafo «Modalità e via di somministrazione».
Confezionamento.
Polvere per soluzione iniettabile, 50 unità LD50 o 100 unità LD50 per flaconcino. 1 flaconcino per confezione di cartone.
Categoria farmaceutica.
Sotto prescrizione medica.
Produttore.
Merz Pharma GmbH & Co. KGaA.
Sede del produttore e indirizzo del luogo di esercizio dell'attività.
Ludwigstrasse 22, 64354 Reinheim, Germania.