Vegovi FlexTach
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE Vegovi FlexTach (Vegovi FlexTouch)
Composizione:
Principio attivo: semaglutide;
0,25 mg
ogni penna preriempita contiene 1 mg di semaglutide* in 1,5 ml di soluzione, 1 ml di soluzione contiene 0,68 mg di semaglutide*, 1 penna preriempita contiene 4 dosi da 0,25 mg;
0,5 mg
ogni penna preriempita contiene 2 mg di semaglutide* in 1,5 ml di soluzione, 1 ml di soluzione contiene 1,34 mg di semaglutide*, 1 penna preriempita contiene 4 dosi da 0,5 mg;
1 mg
ogni penna preriempita contiene 4 mg di semaglutide* in 3 ml di soluzione, 1 ml di soluzione contiene 1,34 mg di semaglutide*, 1 penna preriempita contiene 4 dosi da 1 mg;
\u>1,7 mg
ogni penna preriempita contiene 6,8 mg di semaglutide* in 3 ml di soluzione, 1 ml di soluzione contiene 2,27 mg di semaglutide*, 1 penna preriempita contiene 4 dosi da 1,7 mg;
\u>2,4 mg
ogni penna preriempita contiene 9,6 mg di semaglutide* in 3 ml di soluzione, 1 ml di soluzione contiene 3,2 mg di semaglutide*, 1 penna preriempita contiene 4 dosi da 2,4 mg;
Eccipienti: fosfato disodico diidrato; propilenglicole; fenolo; acido cloridrico (per la correzione del pH); idrossido di sodio (per la correzione del pH); acqua per preparazioni iniettabili.
* Analogo del peptide-1 simile al glucagone umano (GLP-1), prodotto mediante tecnologia del DNA ricombinante in Saccharomyces cerevisiae.
Forma farmaceutica. Soluzione iniettabile.
Principali proprietà fisico-chimiche: soluzione isotonica trasparente e incolore; pH = 7,4.
Gruppo farmacoterapeutico. Agenti ipoglicemizzanti, esclusa l'insulina. Analoghi del peptide-1 simile al glucagone (GLP-1). Codice ATC A10BJ06.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Il semaglutide è un analogo del GLP-1 con una omologia del 94 % rispetto al GLP-1 umano. Il semaglutide agisce come agonista recettoriale del GLP-1, legandosi e attivando in modo selettivo i recettori del GLP-1, che sono il bersaglio del GLP-1 nativo.
Il GLP-1 è un regolatore fisiologico dell'appetito e dell'assunzione calorica, e i recettori del GLP-1 sono presenti in diverse aree del cervello coinvolte nella regolazione dell'appetito.
Studi sugli animali mostrano che il semaglutide agisce nel cervello attraverso il recettore del GLP-1. Il semaglutide ha un effetto diretto su aree del cervello coinvolte nella regolazione omeostatica dell'assunzione di cibo nell'ipotalamo e nel tronco encefalico. Il semaglutide può influenzare il sistema edonico della ricompensa attraverso un effetto diretto e indiretto su aree del cervello, inclusi il setto, il talamo e l'amigdala.
Gli studi clinici mostrano che il semaglutide riduce l'assunzione energetica, aumenta la sensazione di sazietà, di pienezza e di controllo sull'assunzione di cibo, riduce la sensazione di fame, nonché la frequenza e l'intensità della voglia di mangiare. Inoltre, il semaglutide riduce la voglia di cibi ricchi di grassi.
Il semaglutide regola i contributi omeostatici ed edonici con funzione esecutiva per modulare l'assunzione calorica, l'appetito, la ricompensa e la scelta degli alimenti.
Inoltre, studi clinici hanno dimostrato che il semaglutide riduce la glicemia agendo in modo dipendente dal glucosio, stimolando la secrezione di insulina e inibendo la secrezione di glucagone in condizioni di glicemia elevata. Il meccanismo di riduzione della glicemia è accompagnato anche da un lieve ritardo dello svuotamento gastrico nella fase postprandiale precoce. In caso di ipoglicemia, il semaglutide riduce la secrezione di insulina e non interferisce con la secrezione di glucagone.
L'espressione dei recettori del GLP-1 avviene anche nel cuore, nel sistema vascolare, nel sistema immunitario e nei reni.
Durante gli studi clinici, il semaglutide ha avuto un effetto positivo sui livelli lipidici nel plasma, riducendo la pressione arteriosa sistolica e l'infiammazione. Inoltre, studi sugli animali hanno mostrato che il semaglutide inibisce lo sviluppo dell'aterosclerosi e ha un effetto antinfiammatorio sul sistema cardiovascolare.
Effetti farmacodinamici
Appetito, assunzione energetica e scelta degli alimenti
Il semaglutide riduce l'appetito, potenziando la sensazione di sazietà e di pienezza, riducendo contemporaneamente la sensazione di fame e la tendenza a mangiare. Nello studio di fase 1, l'assunzione energetica durante un pasto ad libitum era del 35 % inferiore con semaglutide rispetto al placebo dopo 20 settimane di trattamento. Questo effetto è stato favorito da un miglioramento del controllo sull'assunzione di cibo, da una riduzione della voglia di mangiare e da una minore attrazione per cibi ricchi di grassi. La voglia di mangiare è stata ulteriormente valutata nello studio STEP 5 mediante il questionario CoEQ (Control of Eating Questionnaire). Alla settimana 104, la differenza calcolata nel trattamento ha favorito il semaglutide sia per il controllo della voglia di mangiare che per la voglia di cibi salati, mentre non si è osservato un effetto chiaro sulla voglia di cibi dolci.
Lipidi a digiuno e nel periodo postprandiale
Rispetto al placebo, il semaglutide alla dose di 1 mg riduceva le concentrazioni di trigliceridi a digiuno e di lipoproteine a densità molto bassa (VLDL) a digiuno rispettivamente del 12 % e del 21 %. I livelli postprandiali di trigliceridi e VLDL in risposta all'assunzione di cibi ricchi di grassi diminuivano di oltre il 40 %.
Efficacia e sicurezza clinica
L'efficacia e la sicurezza del semaglutide nel controllo del peso corporeo, come complemento a una dieta ipocalorica e a un'aumentata attività fisica, sono state valutate in quattro studi randomizzati in doppio cieco, controllati con placebo, di fase III, della durata di 68 settimane (STEP 1-4). In totale, 4684 pazienti (2652 randomizzati al trattamento con semaglutide) sono stati inclusi in questi studi. Inoltre, l'efficacia e la sicurezza del semaglutide su un periodo di due anni sono state valutate in uno studio randomizzato in doppio cieco, controllato con placebo, di fase IIIb (STEP 5), che ha coinvolto 304 pazienti (152 pazienti trattati con semaglutide).
Il trattamento con semaglutide ha dimostrato una riduzione del peso corporeo superiore, clinicamente significativa e sostenuta, rispetto al placebo in pazienti con obesità (IMC ≥ 30 kg/m²) o con sovrappeso (IMC ≥ 27 kg/m² fino a < 30 kg/m²) e almeno una comorbidità correlata al peso. Inoltre, durante gli studi una percentuale maggiore di pazienti ha raggiunto una perdita di peso pari o superiore al 5 %, 10 %, 15 % e 20 % con semaglutide rispetto al placebo. La riduzione del peso corporeo si è verificata indipendentemente dalla presenza di sintomi gastrointestinali come nausea, vomito o diarrea.
Il trattamento con semaglutide ha inoltre mostrato miglioramenti statisticamente significativi nella circonferenza della vita, nella pressione arteriosa sistolica e nel livello di attività fisica rispetto al placebo.
L'efficacia è stata dimostrata indipendentemente dall'età, sesso, razza, origine etnica, livello iniziale di peso corporeo, IMC, presenza di diabete mellito di tipo 2 o grado di compromissione renale. Le variazioni nell'efficacia sono state osservate in tutte le sottopopolazioni di pazienti. Una perdita di peso relativamente maggiore è stata osservata nelle donne e nei pazienti senza diabete mellito di tipo 2, nonché nei pazienti con un peso corporeo iniziale più basso rispetto a quelli con un peso più elevato.
Studio STEP 1. Controllo del peso corporeo
In uno studio clinico randomizzato in doppio cieco della durata di 68 settimane, 1961 pazienti con obesità (IMC ≥ 30 kg/m²) o con sovrappeso (IMC ≥ 27 kg/m² fino a < 30 kg/m²) e con almeno una comorbidità correlata al peso sono stati randomizzati a ricevere semaglutide o placebo. Tutti i pazienti hanno seguito una dieta ipocalorica e un'aumentata attività fisica per tutta la durata dello studio.
La riduzione del peso corporeo è avvenuta precocemente e si è mantenuta per tutta la durata dello studio. Alla fine del trattamento (settimana 68), la riduzione del peso corporeo è risultata migliore e clinicamente significativa rispetto al placebo (vedi tabella 1 e figura 1). Inoltre, una percentuale maggiore di pazienti ha raggiunto una perdita di peso pari o superiore al 5 %, 10 %, 15 % e 20 % con semaglutide rispetto al placebo (vedi tabella 1). Tra i pazienti con prediabete all'inizio dello studio, una percentuale maggiore di pazienti ha mostrato uno stato glicemico normale alla fine del trattamento con semaglutide rispetto al placebo (84,1 % contro 47,8 %).
Tabella 1. Studio STEP 1: risultati alla settimana 68
| Indice |
Vegovi FlexTach |
Placebo |
| Numero di pazienti (N) |
1306 |
655 |
| Peso corporeo (kg) |
||
| Livello iniziale (kg) |
105,4 |
105,2 |
| Variazione (%) rispetto al livello iniziale1,2 |
-14,9 |
-2,4 |
| Differenza (%) rispetto al placebo1 [95 % IC] |
-12,4 [-13,4; -11,5]* |
- |
| Variazione (kg) rispetto al livello iniziale |
-15,3 |
-2,6 |
| Differenza (kg) rispetto al placebo1 [95 % IC] |
-12,7 [-13,7; -11,7] |
- |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 5 %3 |
83,5* |
31,1 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 10 %3 |
66,1* |
12,0 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 15 %3 |
47,9* |
4,8 |
| Circonferenza vita (cm) |
||
| Livello iniziale |
114,6 |
114,8 |
| Variazione rispetto al livello iniziale1 |
-13,5 |
-4,1 |
| Differenza rispetto al placebo1 [95 % IC] |
-9,4 [-10,3; -8,5]* |
- |
| Pressione arteriosa sistolica (mmHg) |
||
| Livello iniziale |
126 |
127 |
| Variazione rispetto al livello iniziale1 |
-6,2 |
-1,1 |
| Differenza rispetto al placebo1 [95 % IC] |
-5,1 [-6,3; -3,9]* |
- |
* p < 0,0001 (bilateral unadjusted) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 17.1% and 22.4% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional obesity treatment, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -16.9% and -2.4% for semaglutide 2.4 mg and placebo, respectively.
3 Estimated using a binary regression model based on the same imputation procedure as in the primary analysis.
HTMLIMG0END
Observed values for patients completing each scheduled visit and estimates with multiple imputation (MI) of data obtained from patients who dropped out.
Fig. 1. STEP 1: Mean change in body weight (%) from baseline to week 68
After the 68-week study, a 52-week treatment-free period was conducted, involving 327 patients who completed the main study period on maintenance dose of semaglutide or placebo. During the treatment-free period from week 68 to week 120, mean body weight increased in both treatment groups. However, in patients who had received semaglutide during the main study period, body weight remained 5.6% lower than baseline compared to 0.1% in the placebo group.
Study STEP 2. Weight control in patients with type 2 diabetes
In a double-blind, 68-week clinical trial, 1210 patients with overweight or obesity (BMI ≥ 27 kg/m²) and type 2 diabetes were randomized to receive once-weekly semaglutide 2.4 mg, semaglutide 1 mg, or placebo. Patients enrolled in the study had inadequately controlled type 2 diabetes (HbA1c 7–10%) and were either on diet and exercise alone or on 1–3 oral antidiabetic agents. All patients followed a reduced-calorie diet and increased physical activity throughout the study.
Treatment with semaglutide for 68 weeks resulted in significant and clinically meaningful reductions in body weight and HbA1c compared to placebo (see Table 2 and Figure 2).
Table 2. Study STEP 2: Results at week 68
| Indicatore |
Vegovi FlexTach |
Placebo |
| Numero di pazienti (N) |
404 |
403 |
| Peso corporeo (kg) |
||
| Livello iniziale (kg) |
99,9 |
100,5 |
| Variazione (%) rispetto al livello iniziale1,2 |
-9,6 |
-3,4 |
| Differenza (%) rispetto al placebo1 [95 % IC] |
-6,2 [-7,3; -5,2]* |
- |
| Variazione (kg) rispetto al livello iniziale |
-9,7 |
-3,5 |
| Differenza (kg) rispetto al placebo1 [95 % IC] |
-6,1 [-7,2; -5,0] |
- |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 5 %3 |
67,4* |
30,2 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 10 %3 |
44,5* |
10,2 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 15 %3 |
25,0* |
4,3 |
| Circonferenza vita (cm) |
||
| Livello iniziale |
114,5 |
115,5 |
| Variazione rispetto al livello iniziale1 |
-9,4 |
-4,5 |
| Differenza rispetto al placebo1 [95 % IC] |
-4,9 [-6,0; -3,8]* |
- |
| Pressione arteriosa sistolica (mmHg) |
||
| Livello iniziale |
130 |
130 |
| Variazione rispetto al livello iniziale1 |
-3,9 |
-0,5 |
| Differenza rispetto al placebo1 [95 % IC] |
-3,4 [-5,6; -1,3]** |
- |
| HbA1c (mmol/mol (%)) |
||
| Livello iniziale |
65,3 (8,1) |
65,3 (8,1) |
| Variazione rispetto al livello iniziale1 |
-17,5 (-1,6) |
-4,1 (-0,4) |
| Differenza rispetto al placebo1 [95 % IC] |
-13,5 [-15,5; -11,4] (-1,2 [-1,4; -1,1])* |
- - |
* p < 0,0001 (bilateral non corretto) a favore; ** p < 0,05 (bilateral non corretto) a favore.
1 Valutato utilizzando un modello ANCOVA con aggiustamento multiplo basato su tutti i dati, indipendentemente dall’interruzione del trattamento randomizzato o dall’inizio di altri farmaci contro l’obesità o chirurgia bariatrica.
2 Durante lo studio, il trattamento è stato definitivamente interrotto nell’11,6% e nel 13,9% dei pazienti randomizzati rispettivamente a semaglutide 2,4 mg e placebo. Assumendo che tutti i pazienti randomizzati abbiano continuato il trattamento e non abbiano ricevuto ulteriore terapia per l’obesità, le variazioni stimate dalla randomizzazione alla settimana 68 per il peso corporeo, basate su un modello misto per misurazioni ripetute, includendo tutte le osservazioni fino al primo interruzione, sono state rispettivamente -10,6% e -3,1% per semaglutide 2,4 mg e placebo.
3 Valutato con un modello di regressione binaria basato sulla stessa procedura di aggiustamento utilizzata nell’analisi primaria.
**
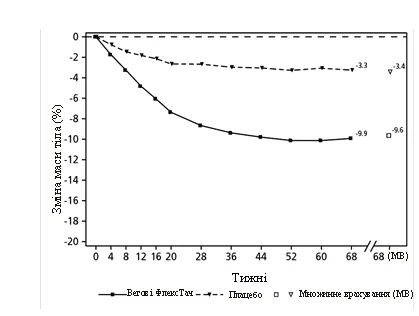
**
Valori osservati nei pazienti che completano ogni visita pianificata e stime con aggiustamento multiplo (AM) dei dati provenienti dai pazienti che hanno abbandonato lo studio.
Fig. 2. STEP 2: Variazione media del peso corporeo (%) dal basale alla settimana 68
Studio STEP 3. Controllo del peso con terapia comportamentale intensiva
In uno studio clinico in doppio cieco della durata di 68 settimane, 611 pazienti con obesità (BMI ≥ 30 kg/m²) o con sovrappeso (BMI ≥ 27 kg/m² e < 30 kg/m²) e con almeno una comorbidità correlata al peso corporeo sono stati randomizzati a ricevere semaglutide o placebo. Tutti i pazienti hanno ricevuto una terapia comportamentale intensiva (TCI), costituita da una dieta fortemente ipocalorica, aumento dell’attività fisica e consulenza comportamentale per tutta la durata dello studio.
Il trattamento con semaglutide e TCI per 68 settimane ha determinato una riduzione del peso corporeo significativa e clinicamente rilevante rispetto al placebo (vedere Tabella 3).
Tabella 3. Studio STEP 3: risultati alla settimana 68
| Indice |
Vegovi FlexTach |
Placebo |
| Numero di pazienti (N) |
407 |
204 |
| Peso corporeo (kg) |
||
| Livello iniziale (kg) |
106,9 |
103,7 |
| Variazione (%) rispetto al livello iniziale1,2 |
-16,0 |
-5,7 |
| Differenza (%) rispetto al placebo1 [95 % IC] |
-10,3 [-12,0; -8,6]* |
- |
| Variazione (kg) rispetto al livello iniziale |
-16,8 |
-6,2 |
| Differenza (kg) rispetto al placebo1 [95 % IC] |
-10,6 [-12,5; -8,8] |
- |
| Pazienti (%) che hanno raggiunto una perdita di peso corporeo ≥ 5 %3 |
84,8* |
47,8 |
| Pazienti (%) che hanno raggiunto una perdita di peso corporeo ≥ 10 %3 |
73,0* |
27,1 |
| Pazienti (%) che hanno raggiunto una perdita di peso corporeo ≥ 15 %3 |
53,5* |
13,2 |
| Circonferenza vita (cm) |
||
| Livello iniziale |
113,6 |
111,8 |
| Variazione rispetto al livello iniziale1 |
-14,6 |
-6,3 |
| Differenza rispetto al placebo1 [95 % IC] |
-8,3 [-10,1; -6,6]* |
- |
| Pressione arteriosa sistolica (mmHg) |
||
| Livello iniziale |
124 |
124 |
| Variazione rispetto al livello iniziale1 |
-5,6 |
-1,6 |
| Differenza rispetto al placebo1 [95 % IC] |
-3,9 [-6,4; -1,5]* |
- |
*p < 0,0001 (bilateral, non-adjusted) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 16.7% and 18.6% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from randomization to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -17.6% and -5.0% for semaglutide 2.4 mg and placebo, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
Study STEP 4. Maintenance of Body Weight Control
In a double-blind, 68-week clinical trial, 902 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and at least one weight-related comorbidity were enrolled. All patients followed a reduced-calorie diet and increased physical activity throughout the study. From week 0 to week 20 (run-in period), all patients received semaglutide. At week 20 (baseline), patients who had reached the maintenance dose of 2.4 mg were randomized to continue treatment or switch to placebo. At week 0 (start of run-in period), patients had a mean body weight of 107.2 kg and a mean BMI of 38.4 kg/m².
Patients who reached the maintenance dose of 2.4 mg at week 20 (baseline) and continued semaglutide treatment for 48 weeks (weeks 20–68) continued to lose body weight and achieved greater and clinically meaningful weight reduction compared to those who switched to placebo (see Table 4 and Figure 3). Body weight steadily increased from week 20 to week 68 in patients who switched to placebo at week 20 (baseline). However, the observed mean body weight at week 68 was still lower than at the start of the run-in period (week 0) (see Figure 3). Patients who received semaglutide from week 0 (run-in) to week 68 (end of treatment) achieved a mean change in body weight of -17.4%, with 87.8% achieving weight loss ≥ 5%, 78.0% achieving ≥ 10%, 62.2% achieving ≥ 15%, and 38.6% achieving ≥ 20%.
Table 4. Study STEP 4: Outcomes from Week 20 to Week 68
| Indicatore |
Vegovi FlexTach |
Placebo |
| Numero di pazienti (N) |
535 |
268 |
| Peso corporeo (kg) |
||
| Livello basale1 (kg) |
96,5 |
95,4 |
| Variazione (%) rispetto al livello basale1,2,3 |
-7,9 |
6,9 |
| Differenza (%) rispetto al placebo2 [95 % IC] |
-14,8 [-16,0; -13,5]* |
- |
| Variazione (kg) rispetto al livello basale |
-7,1 |
6,1 |
| Differenza (kg) rispetto al placebo2 [95 % IC] |
-13,2 [-14,3; -12,0] |
- |
| Circonferenza vita (cm) |
||
| Livello basale |
105,5 |
104,7 |
| Variazione rispetto al livello basale1 |
-6,4 |
3,3 |
| Differenza rispetto al placebo2 [95 % IC] |
-9,7 [-10,9; -8,5]* |
- |
| Pressione arteriosa sistolica (mmHg) |
||
| Livello basale |
121 |
121 |
| Variazione rispetto al livello basale1,2 |
0,5 |
4,4 |
| Differenza rispetto al placebo2 [95 % IC] |
-3,9 [-5,8; -2,0]* |
- |
* p < 0,0001 (bilateral non-adjusted) for superiority.
1 Baseline = week 20.
2 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
3 During the study, treatment was permanently discontinued in 5.8% and 11.6% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from randomization to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -8.1% and -6.5% for semaglutide 2.4 mg and placebo, respectively.
**
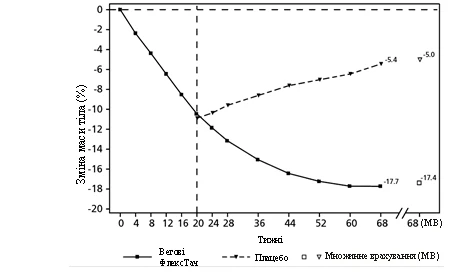
**
Observed values for patients completing each scheduled visit and multiple imputation (MI) estimates for data obtained from patients who discontinued.
Fig. 3. STEP 4: Mean change in body weight (%) from week 0 to week 68
Study STEP 5. 2-year data
In a double-blind clinical trial lasting 104 weeks, 304 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and with at least one weight-related comorbidity were randomized to receive semaglutide or placebo. All patients followed a reduced-calorie diet and increased physical activity throughout the study. At baseline, patients had a mean BMI of 38.5 kg/m² and a mean body weight of 106.0 kg.
Treatment with semaglutide over 104 weeks resulted in significant and clinically meaningful weight reduction compared to placebo. Mean body weight decreased from baseline to week 68 with semaglutide, after which a plateau was reached. With placebo, mean body weight decreased to a lesser extent and plateaued after approximately 20 weeks of treatment (see Table 5 and Figure 4). Patients receiving semaglutide achieved a mean change in body weight of -15.2%, with 74.7% of these patients losing ≥ 5% body weight, 59.2% losing ≥ 10%, and 49.7% losing ≥ 15%. 80% and 37% of patients with prediabetes at baseline achieved normal glycaemic status at the end of treatment with semaglutide and placebo, respectively.
Table 5. Study STEP 5: Results at week 104
| Indicatore |
Vegovi FlexTach |
Placebo |
| Numero di pazienti (N) |
152 |
152 |
| Peso corporeo (kg) |
||
| Livello iniziale (kg) |
105,6 |
106,5 |
| Variazione (%) rispetto al livello iniziale1,2 |
-15,2 |
-2,6 |
| Differenza (%) rispetto al placebo1 [95 % CI] |
-12,6 [-15,3; -9,8]* |
- |
| Variazione (kg) rispetto al livello iniziale |
-16,1 |
-3,2 |
| Differenza (kg) rispetto al placebo1 [95 % CI] |
-12,9 [‑16,1; ‑9,8] |
- |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 5 %3 |
74,7* |
37,3 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 10 %3 |
59,2* |
16,8 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 15 %3 |
49,7* |
9,2 |
| Circonferenza della vita (cm) |
||
| Livello iniziale |
115,8 |
115,7 |
| Variazione rispetto al livello iniziale1 |
-14,4 |
5,2 |
| Differenza rispetto al placebo1 [95 % CI] |
-9,2 [-12,2; -6,2]* |
- |
| Pressione arteriosa sistolica (mmHg) |
||
| Livello iniziale |
126 |
125 |
| Variazione rispetto al livello iniziale1 |
-5,7 |
-1,6 |
| Differenza rispetto al placebo1 [95 % CI] |
-4,2 [-7,3; -1,0]* |
- |
* p < 0,0001 (bilateral non-adjusted) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 13.2% and 27.0% of patients randomized to receive semaglutide and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -16.7% and -0.6% for semaglutide and placebo, respectively.
3 Estimated using a binary regression model based on the same imputation procedure as in the primary analysis.
HTMLIMG3END
Observed values for patients completing each scheduled visit and estimates with multiple imputation (MI) of data obtained from patients who dropped out.
Fig. 4. STEP 5: Mean change in body weight (%) from week 0 to week 104
Study STEP 8. Comparison of semaglutide with liraglutide
In a randomized, open-label, pairwise placebo-controlled trial lasting 68 weeks, 338 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and with at least one weight-related comorbidity were randomized to receive once-weekly semaglutide, once-daily liraglutide 3 mg, or placebo. Administration of once-weekly semaglutide and liraglutide 3 mg was open-label, but each active treatment group was double-blind versus placebo administered at the same dosing frequency. At baseline, patients had a mean BMI of 37.5 kg/m² and a mean body weight of 104.5 kg.
Treatment with once-weekly semaglutide over 68 weeks resulted in significant and clinically meaningful weight reduction compared to liraglutide. Mean body weight decreased from baseline to week 68 with semaglutide treatment. With liraglutide treatment, mean body weight decreased to a lesser extent (see Table 6). 37.4% of patients receiving semaglutide lost ≥ 20% of body weight, compared to 7.0% of patients receiving liraglutide. Table 6 shows the results for confirmatory endpoints of ≥ 10%, ≥ 15%, and ≥ 20% body weight loss.
Table 6. Study STEP 8: Results at week 68 comparing semaglutide and liraglutide treatment
| Indicatore |
Vegovi FlexTach |
Liraglutide 3 mg |
| Numero di pazienti (N) |
126 |
127 |
| Peso corporeo (kg) |
||
| Livello iniziale (kg) |
102,5 |
103,7 |
| Variazione (%) rispetto al livello iniziale1,2 |
-15,8 |
-6,4 |
| Differenza (%) rispetto al liraglutide1 [IC 95 %] |
‑9,4 [‑12,0;‑6,8]* |
- |
| Variazione (kg) rispetto al livello iniziale |
‑15,3 |
‑6,8 |
| Differenza (kg) rispetto al liraglutide1 [IC 95 %] |
‑8,5 [‑11,2;‑5,7] |
- |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 10 %3 |
69,4* |
27,2 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 15 %3 |
54,0* |
13,4 |
| Pazienti (%) che hanno raggiunto una perdita di peso ≥ 20 %3 |
37,4* |
7,0 |
* p < 0,0001 (bilateral unadjusted) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 13.5% and 27.6% of patients randomized to semaglutide and liraglutide, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -16.7% and -6.7% for semaglutide and liraglutide, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
Effects on body composition
In a sub-study of STEP 1 (N = 140), body composition was measured using dual-energy X-ray absorptiometry (DEXA). DEXA assessments showed that treatment with semaglutide was associated with a greater reduction in fat mass than in lean body mass, resulting in improved body composition compared to placebo at 68 weeks. Furthermore, reduction in total fat mass was accompanied by a reduction in visceral fat. These results indicate that the majority of overall body weight loss was related to a reduction in adipose tissue, including visceral fat.
Improvement in physical functioning
Semaglutide demonstrated a modest improvement in measures of physical functioning. Physical functioning was assessed using both the generic health-related quality of life questionnaire, Short Form-36v2 Health Survey, Acute Version (SF-36), and the Impact of Weight on Quality of Life Clinical Trials Version (IWQOL-Lite-CT).
Cardiovascular assessment
In the SUSTAIN 6 trial, 3297 patients with inadequately controlled type 2 diabetes and high cardiovascular risk were randomized to receive subcutaneous semaglutide at a dose of 0.5 mg or 1 mg once weekly or placebo, in addition to standard of care. The treatment duration was 104 weeks. The mean age of patients was 65 years, and the mean BMI was 33 kg/m².
The primary endpoint was time from randomization to first occurrence of a major adverse cardiovascular event (MACE): cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke. The total MACE count was 254, including 108 (6.6%) with semaglutide and 146 (8.9%) with placebo.
Cardiovascular safety of treatment with semaglutide 0.5 or 1 mg was confirmed, as the hazard ratio (HR) for semaglutide compared to placebo was 0.74 [0.58, 0.95] [95% CI], driven by a reduction in the incidence of non-fatal myocardial infarction and non-fatal stroke, with no difference in the rate of cardiovascular death (see Figure 5).
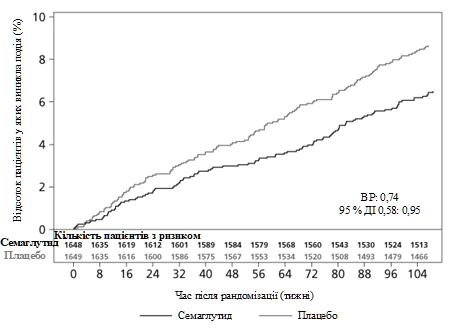
Figure 5. Kaplan-Meier plot of time to first occurrence of the composite endpoint: cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke (SUSTAIN 6 clinical trial)
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Vegovi FlexTach in one or more subsets of the paediatric population for body weight control (see section "Posology and method of administration" for information on paediatric use).
Study STEPTEENS: Body weight control in patients aged 12–18 years
In a 68-week double-blind trial, 201 pubertal adolescents aged 12 to 18 years with obesity or overweight and at least one weight-related comorbidity were randomized 2:1 to receive semaglutide or placebo. All patients followed a reduced-calorie diet and increased physical activity throughout the study.
At the end of treatment (week 68), improvement in BMI with semaglutide was greater and clinically meaningful compared to placebo (see Table 6 and Figure 6). Furthermore, a higher proportion of patients achieved ≥ 5%, ≥ 10%, and ≥ 15% body weight loss with semaglutide compared to placebo (see Table 6).
Table 6. STEPTEENS: Results at week 68
| Indicatore |
Vegovi FlexTach |
Placebo |
| Numero di pazienti (N) |
134 |
67 |
| IMC |
||
| Livello iniziale (IMC) |
37,7 |
35,7 |
| Variazione (%) rispetto al livello iniziale1,2 |
-16,1 |
0,6 |
| Differenza (%) rispetto al placebo1 [95 % IC] |
-16,7 [‑20,3; ‑13,2] |
- |
| Livello iniziale (IMC ISV (indice di deviazione standard)) |
3,4 |
3,1 |
| Variazione rispetto al livello iniziale IMC ISV1 |
-1,1 |
-0,1 |
| Differenza rispetto al placebo1 [95 % IC] |
-1,0 [‑1,3; ‑0,8] |
- |
| Massa corporea |
||
| Livello iniziale (kg) |
109,9 |
102,6 |
| Variazione (%) rispetto al livello iniziale1 |
-14,7 |
2,8 |
| Differenza (%) rispetto al placebo1 [95 % IC] |
-17,4 [-21,1; -13,8] |
- |
| Variazione (kg) rispetto al livello iniziale1 |
-15,3 |
2,4 |
| Differenza (kg) rispetto al placebo1 [95 % IC] |
-17,7 [-21,8; -13,7] |
- |
| Pazienti (%) che hanno raggiunto una perdita di massa corporea ≥ 5 %3 |
72,5* |
17,7 |
| Numero di pazienti (N) |
134 |
67 |
| Pazienti (%) che hanno raggiunto una perdita di massa corporea ≥ 10 %3 |
61,8 |
8,1 |
| Pazienti (%) che hanno raggiunto una perdita di massa corporea ≥ 15 %3 |
53,4 |
4,8 |
| Circonferenza della vita (cm) |
||
| Livello iniziale |
111,9 |
107,3 |
| Variazione rispetto al livello iniziale1 |
-12,7 |
-0,6 |
| Differenza rispetto al placebo1 [95 % IC] |
-12,1 [-15,6; -8,7]* |
- |
| Pressione arteriosa sistolica (mmHg) |
||
| Livello iniziale |
120 |
120 |
| Variazione rispetto al livello iniziale1 |
-2,7 |
-0,8 |
| Differenza rispetto al placebo1 [95 % IC] |
-1,9 [-5,0; -1,1]* |
- |
*p < 0,0001 (bilateral unadjusted) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 10.4% and 10.4% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measures, including all observations up to first discontinuation, were -17.9% and -0.6% for semaglutide 2.4 mg and placebo, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
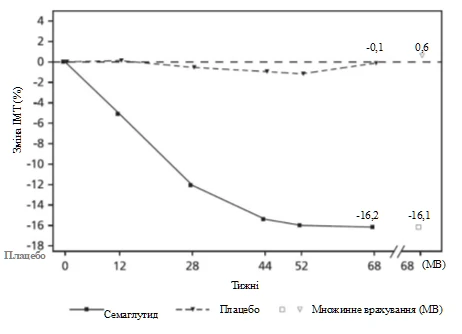
Observed values for patients completing each scheduled visit and multiple imputation (MI) of data from patients who discontinued.
Fig. 6. STEP TEENS: Mean change in BMI (%) from baseline to week 68
Pharmacokinetics.
Compared to native GLP-1, semaglutide has a prolonged elimination half-life of approximately 1 week, allowing for once-weekly subcutaneous administration. The primary mechanism behind this prolonged action is albumin binding, which reduces renal clearance and protects against metabolic degradation. Additionally, semaglutide is stabilized against degradation by the enzyme DPP-4.
Absorption
The mean steady-state concentration of semaglutide after subcutaneous administration of its maintenance dose was approximately 75 nmol/L in patients with overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) or obesity (BMI ≥ 30 kg/m²), based on data from phase 3 trials, where 90% of patients had concentrations ranging between 51 nmol/L and 110 nmol/L. Following doses ranging from 0.25 mg to 2.4 mg once weekly, steady-state exposure to semaglutide increased in a dose-dependent manner. Steady-state exposure over time was stable, as determined up to week 68. Similar exposure was achieved following subcutaneous administration of semaglutide in the anterior abdominal wall, thigh, or upper arm. The absolute bioavailability of semaglutide after subcutaneous administration was 89%.
Distribution
The mean volume of distribution of semaglutide after subcutaneous administration in patients with overweight or obesity was approximately 12.4 L. Semaglutide is highly bound to plasma albumin (>99%).
Metabolism/Biotransformation
Prior to excretion, semaglutide is actively metabolized via proteolytic cleavage of the peptide backbone and subsequent beta-oxidation of the fatty acid side chain. Neutral endopeptidase (NEP) has been identified as one of the active metabolic enzymes.
Elimination
Material related to semaglutide is primarily excreted in urine and feces. Approximately 3% of the administered dose was excreted in urine as unchanged semaglutide. The clearance of semaglutide in patients with overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) or obesity (BMI ≥ 30 kg/m²) was approximately 0.05 L/h. With an elimination half-life of approximately 1 week, semaglutide will remain in circulation for about 7 weeks after the last 2.4 mg dose.
Special patient groups
Elderly patients
Patient age does not affect the pharmacokinetics of semaglutide, according to data from phase 3 trials involving patients aged 18–86 years.
Gender, race, and ethnicity
Patient gender, race (Caucasian, African, or Asian), and ethnicity (Hispanic or Latino, non-Hispanic or non-Latino) do not affect the pharmacokinetics of semaglutide, according to data from phase 3a trials.
Body weight
Patient body weight affects semaglutide exposure. Higher body weight leads to lower drug exposure; a 20% difference in body weight between patients results in nearly an 18% difference in exposure. The 2.4 mg once-weekly dose of semaglutide provides adequate systemic exposure across a body weight range of 54.4–245.6 kg, as evaluated for exposure-response relationships in clinical trials.
Renal impairment
Renal impairment did not have a clinically significant effect on the pharmacokinetics of semaglutide. This was demonstrated after a single 0.5 mg dose of semaglutide administered to patients with varying degrees of renal impairment (mild, moderate, severe, and dialysis patients) compared to patients with normal renal function. This was also confirmed by data from phase 3a clinical trials in patients with overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) or obesity (BMI ≥ 30 kg/m²) and mild to moderate renal impairment.
Hepatic impairment
Hepatic impairment had no effect on semaglutide exposure. The pharmacokinetics of semaglutide were evaluated in patients with varying degrees of hepatic impairment (mild, moderate, severe) compared to patients with normal liver function in a study using a single 0.5 mg dose of semaglutide.
Pre-diabetes and diabetes
Pre-diabetes and diabetes had no clinically relevant effect on semaglutide exposure based on phase 3 trial data.
Immunogenicity
The development of anti-semaglutide antibodies during treatment with semaglutide was infrequent (see section "Adverse reactions"), and the immune response apparently did not affect semaglutide pharmacokinetics.
Children
The pharmacokinetic properties of semaglutide were evaluated in a clinical trial involving adolescent patients (aged 12 to <18 years) with obesity or overweight and at least one comorbidity related to body weight (124 patients, body weight 61.6–211.9 kg). Semaglutide exposure in children was similar to that in adults with obesity or overweight.
The safety and efficacy of semaglutide in children under 12 years of age have not been studied.
Preclinical safety data
Preclinical data based on studies of pharmacological safety, repeat-dose toxicity, and genotoxicity revealed no risk for humans.
Non-lethal tumors originating from C-cells of the thyroid gland observed in rodents are effects typical of the class of GLP-1 receptor agonists. In a 2-year carcinogenicity study in rats and mice, semaglutide caused the development of thyroid C-cell tumors at clinically relevant exposure levels. No other treatment-related tumors were observed. Rodent C-cell tumors are due to a non-genotoxic, GLP-1 receptor-mediated mechanism to which rodents are partially sensitive. The relevance of this mechanism to humans is considered low but cannot be entirely excluded.
In fertility studies in rats, semaglutide did not affect mating performance or fertility in males. In female rats, prolonged estrous cycle duration and a slight reduction in corpora lutea (ovulations) were observed at doses associated with body weight loss.
In embryo-fetal development studies in rats, semaglutide caused embryotoxic effects at exposures below clinically relevant levels. Semaglutide caused marked body weight loss in females and reduced embryo survival and growth. Fetuses showed significant skeletal and visceral malformations, including changes in long bones, ribs, spine, tail bones, blood vessels, and brain ventricles. Mechanistic evaluation indicated that the embryotoxic effect involves GLP-1 receptor-mediated disruption of nutrient supply to the embryo via the yolk sac in rats. Due to anatomical and functional differences in yolk sac structure between species and the lack of GLP-1 receptor expression in the yolk sac of non-human primates, this mechanism is considered unlikely in humans. However, the possibility of a direct effect of semaglutide on the fetus cannot be ruled out.
In developmental toxicity studies in rabbits and cynomolgus monkeys, at clinically relevant exposure levels, increased pregnancy loss and a slight increase in fetal abnormalities were observed. These findings coincided with marked body weight loss in females, reaching up to 16%. It is unknown whether these effects are related to reduced food intake in females due to the direct action of GLP-1.
Postnatal growth and development were assessed in cynomolgus monkeys. Offspring were slightly smaller at birth, but body weight normalized during the lactation period.
In young rats, semaglutide caused delayed sexual maturation in both males and females. This delay did not affect fertility or reproductive capacity in either sex, nor the ability of females to maintain pregnancy.
Caratteristiche cliniche.
Indicazioni.
Adulti
Il medicinale Vegovi FlexTach è indicato per il controllo del peso corporeo come complemento a una dieta ipocalorica e a un aumento dell'attività fisica in pazienti adulti con un indice di massa corporea (IMC) iniziale
- superiore a 30 kg/m² (obesità) oppure
- compreso tra 27 e 30 kg/m² (sovrappeso) in presenza di almeno una patologia concomitante correlata al peso corporeo, ad esempio disglicemia (prediabete o diabete di tipo 2), ipertensione, dislipidemia, apnea ostruttiva del sonno o malattia cardiovascolare.
Bambini ≥ 12 anni
Il medicinale Vegovi FlexTach è indicato per il controllo del peso corporeo come complemento a una dieta ipocalorica e a un aumento dell'attività fisica in bambini di età pari o superiore a 12 anni con
- obesità* e
- peso corporeo superiore a 60 kg.
L'uso di Vegovi FlexTach deve essere interrotto e rivalutato se nei bambini l'IMC non si riduce di almeno il 5% dopo 12 settimane di trattamento con 2,4 mg o con la dose massima tollerata.
* Obesità (IMC ≥ 95° percentile), come definito nei grafici di crescita dell'IMC per sesso ed età (CDC.gov) (vedere tabella 7).
Tabella 7. Valori soglia dell'IMC per l'obesità (≥ 95° percentile) per sesso ed età nei bambini di età pari o superiore a 12 anni (criteri CDC)
| Età (anni) |
IMC (kg/m2) al 95° percentile |
|
| Sesso maschile |
Sesso femminile |
|
| 12 |
24,2 |
25,2 |
| 12,5 |
24,7 |
25,7 |
| 13 |
25,1 |
26,3 |
| 13,5 |
25,6 |
26,8 |
| 14 |
26,0 |
27,2 |
| 14,5 |
26,4 |
27,7 |
| 15 |
26,8 |
28,1 |
| 15,5 |
27,2 |
28,5 |
| 16 |
27,5 |
28,9 |
| 16,5 |
27,9 |
29,3 |
| 17 |
28,2 |
29,6 |
| 17,5 |
28,6 |
30,0 |
Controindicazioni.
Ipersensibilità al principio attivo o agli eccipienti del medicinale (vedere la sezione «Composizione»).
Interazioni con altri medicinali e altre forme di interazione.
Il semaglutide rallenta lo svuotamento gastrico e potenzialmente può influenzare la velocità di assorbimento dei medicinali orali assunti contemporaneamente. Non è stato osservato alcun effetto clinicamente significativo sullo svuotamento gastrico dopo l’uso di semaglutide alla dose di 2,4 mg, probabilmente a causa di un effetto di tolleranza. Il semaglutide deve essere usato con cautela nei pazienti che assumono medicinali orali che richiedono un rapido assorbimento nel tratto gastrointestinale.
Paracetamolo
Sulla base dei risultati della valutazione della farmacocinetica del paracetamolo durante un test standardizzato con cibo, il semaglutide rallenta la velocità di svuotamento gastrico. Dopo somministrazione concomitante di paracetamolo e semaglutide alla dose di 1 mg, i valori di AUC0-60min e Cmax del paracetamolo sono diminuiti rispettivamente del 27% e del 23%. L’esposizione totale al paracetamolo (AUC0-5h) non è cambiata. Non è stato osservato alcun effetto clinicamente significativo del semaglutide sul paracetamolo. Non è necessaria alcuna correzione della dose quando il paracetamolo viene assunto contemporaneamente al semaglutide.
Contraccettivi orali
Non si prevede che il semaglutide riduca l’efficacia dei contraccettivi orali. In particolare, quando somministrato contemporaneamente a un contraccettivo orale combinato (etinilestradiolo 0,03 mg / levonorgestrel 0,15 mg), il semaglutide non ha avuto effetti clinicamente significativi sull’esposizione totale a etinilestradiolo e levonorgestrel. L’esposizione a etinilestradiolo non è stata alterata; l’esposizione a levonorgestrel è aumentata del 20% a stato stazionario. La Cmax di nessuna delle due sostanze è stata modificata.
Atorvastatina
Il semaglutide non ha modificato l’esposizione totale all’atorvastatina dopo somministrazione di una dose singola di atorvastatina (40 mg). La Cmax dell’atorvastatina è diminuita del 38%. Tale riduzione è stata considerata clinicamente non significativa.
Digossina
Il semaglutide non ha modificato l’esposizione totale o la Cmax della digossina dopo somministrazione di una dose singola di digossina (0,5 mg).
Metformina
Il semaglutide non ha modificato l’esposizione totale o la Cmax della metformina dopo somministrazione di metformina 500 mg due volte al giorno per 3,5 giorni.
Warfarina
Il semaglutide non ha modificato l’esposizione totale o la Cmax della warfarina R- e S- dopo somministrazione di una dose singola di warfarina (25 mg), e gli effetti farmacodinamici della warfarina, misurati tramite il rapporto normalizzato internazionale (INR), non sono risultati clinicamente significativi. Tuttavia, all’inizio del trattamento con semaglutide, nei pazienti che assumono warfarina o altri derivati delle cumarine, si raccomanda un monitoraggio frequente dell’INR.
Bambini
Gli studi sull’interazione sono stati condotti solo su pazienti adulti.
Caratteristiche particolari di impiego.
Tracciabilità
Ai fini del miglioramento della tracciabilità dei medicinali biologici, il nome e il numero di lotto del medicinale somministrato devono essere chiaramente registrati sull’imballaggio.
Disidratazione
L’uso di agonisti del recettore GLP-1 può essere associato a reazioni avverse gastrointestinali che possono causare disidratazione, che in rari casi può portare a un peggioramento della funzionalità renale. I pazienti devono essere informati del potenziale rischio di disidratazione in relazione alle reazioni avverse gastrointestinali e devono essere adottate misure per prevenire la disidratazione.
Pancreatite acuta
Sono stati osservati casi di pancreatite acuta durante l’uso di agonisti del recettore GLP-1 (vedere il paragrafo «Effetti indesiderati»). I pazienti devono essere informati sui sintomi tipici della pancreatite acuta. In caso di sospetto di pancreatite, il trattamento con semaglutide deve essere interrotto; se la pancreatite viene confermata, il trattamento con semaglutide non deve essere ripreso.
La semaglutide deve essere usata con cautela nei pazienti con anamnesi di pancreatite.
In assenza di altri segni e sintomi di pancreatite acuta, un solo aumento dei livelli degli enzimi pancreatici non è un indicatore prognostico di pancreatite acuta.
Diabete mellito di tipo 2
La semaglutide non deve essere utilizzata come sostituto dell’insulina nei pazienti con diabete mellito di tipo 2.
La semaglutide non deve essere usata in associazione con altri agonisti del recettore GLP-1. Tale combinazione non è stata valutata e si ritiene probabile un aumento del rischio di reazioni avverse legate a sovradosaggio.
Ipo glicemia nei pazienti con diabete mellito di tipo 2
È noto che l’insulina e le sulfoniluree possono causare ipoglicemia. Nei pazienti trattati con semaglutide in combinazione con sulfoniluree o insulina, il rischio di ipoglicemia può aumentare. Tale rischio può essere ridotto riducendo la dose di sulfonilurea o insulina all’inizio del trattamento con l’agonista del recettore GLP-1. L’aggiunta del medicinale Vegovi FlexTach al trattamento di pazienti in terapia con insulina non è stata valutata.
Retinopatia diabetica nei pazienti con diabete mellito di tipo 2
Nei pazienti con retinopatia diabetica trattati con semaglutide è stato osservato un aumento del rischio di complicanze della retinopatia diabetica (vedere il paragrafo «Effetti indesiderati»). Un rapido miglioramento del controllo glicemico è stato associato a un peggioramento transitorio della retinopatia diabetica, tuttavia non si può escludere la possibilità di altri meccanismi d’azione. I pazienti con retinopatia diabetica devono essere attentamente monitorati e trattati in conformità con le raccomandazioni cliniche. Non esiste esperienza nell’uso del medicinale Vegovi FlexTach in pazienti con diabete mellito di tipo 2 con retinopatia diabetica non controllata o potenzialmente instabile. L’uso del medicinale Vegovi FlexTach in tali pazienti non è raccomandato.
Popolazioni non studiate
Non sono stati studiati la sicurezza e l’efficacia del medicinale Vegovi FlexTach nei seguenti pazienti:
- che assumono altri medicinali per il controllo del peso corporeo,
– con diabete mellito di tipo 1,
- con grave compromissione della funzionalità renale (vedere il paragrafo «Posologia e modo di somministrazione»),
- con grave insufficienza epatica (vedere il paragrafo «Posologia e modo di somministrazione»),
- con insufficienza cardiaca congestizia di classe IV secondo la classificazione della New York Heart Association (NYHA).
L’uso in questi pazienti non è raccomandato.
L’esperienza nell’uso del medicinale Vegovi FlexTach è limitata nei seguenti pazienti:
- di età pari o superiore a 75 anni (vedere il paragrafo «Posologia e modo di somministrazione»),
- con compromissione epatica lieve o moderata (vedere il paragrafo «Posologia e modo di somministrazione»),
– con malattie infiammatorie intestinali,
- con gastroparesi diabetica.
In tali pazienti, il medicinale Vegovi FlexTach deve essere usato con cautela.
Contenuto di sodio
Questo medicinale contiene meno di 1 mmol di sodio (23 mg)/dose, pertanto può essere considerato praticamente privo di sodio.
Uso durante la gravidanza o l’allattamento.
Donne in età fertile
Alle donne in età fertile si raccomanda di usare un metodo contraccettivo durante il trattamento con semaglutide (vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione»).
Gravidanza
Studi sugli animali hanno dimostrato tossicità riproduttiva (vedere il paragrafo «Dati preclinici di sicurezza»). I dati sull’uso della semaglutide in donne in gravidanza sono limitati. Pertanto, non si deve usare semaglutide durante la gravidanza. Se una paziente pianifica una gravidanza o è in stato di gravidanza, il trattamento con semaglutide deve essere interrotto. A causa del lungo emivita della semaglutide, il suo uso deve essere interrotto almeno 2 mesi prima della pianificazione di una gravidanza (vedere il paragrafo «Farmacocinetica»).
Allattamento
Negli studi sugli animali, la semaglutide è stata escreta nel latte durante l’allattamento nei ratti. Non si può escludere un rischio per il neonato allattato al seno. Non si deve usare semaglutide durante l’allattamento.
Fertilità
L’effetto della semaglutide sulla fertilità nell’uomo non è noto. L’uso di semaglutide nei maschi di ratto non ha influenzato la fertilità. Nelle femmine di ratto è stato osservato un allungamento del ciclo estrale e una riduzione del numero di ovulazioni alle dosi associate alla perdita di peso corporeo.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell’uso di macchinari.
La semaglutide non ha alcun effetto o ha un effetto trascurabile sulla capacità di guidare autoveicoli o di usare macchinari. Tuttavia, può verificarsi vertigine, soprattutto durante il periodo di aumento della dose. In caso di vertigini, si deve prestare attenzione durante la guida di autoveicoli o l’uso di macchinari.
Pazienti con diabete mellito di tipo 2
Nei pazienti trattati con semaglutide in combinazione con sulfoniluree o insulina, si deve raccomandare di adottare misure di sicurezza per prevenire l’ipoglicemia durante la guida di autoveicoli o l’uso di macchinari (vedere il paragrafo «Caratteristiche particolari di impiego»).
Modalità e dosi di somministrazione
Dosaggio
Adulti
La dose di mantenimento di semaglutide di 2,4 mg una volta alla settimana si ottiene a partire da una dose iniziale di 0,25 mg. Per ridurre la probabilità di reazioni avverse a carico del tratto gastrointestinale, la dose deve essere aumentata gradualmente durante un periodo di 16 settimane fino alla dose di mantenimento di 2,4 mg una volta alla settimana (vedere tabella 8). In caso di reazioni gastrointestinale significative, si dovrà considerare un ritardo nell’aumento della dose o una riduzione alla dose precedente finché le condizioni non migliorino. Dosi superiori a 2,4 mg alla settimana non sono raccomandate.
Tabella 8. Schema di aumento della dose
| Aumento della dose |
Dose settimanale |
| Settimana 1–4 |
0,25 mg |
| Settimana 5–8 |
0,5 mg |
| Settimana 9–12 |
1 mg |
| Settimana 13–16 |
1,7 mg |
| Dose di mantenimento |
2,4 mg |
Bambini
Nei bambini di età pari o superiore a 12 anni, si deve applicare lo stesso schema di aumento della dose previsto per gli adulti (vedere tabella 8). La dose deve essere aumentata fino a 2,4 mg (dose di mantenimento) o fino alla massima dose tollerata. Non si raccomandano dosi settimanali superiori a 2,4 mg.
Pazienti con diabete mellito di tipo 2
All’inizio del trattamento con semaglutide nei pazienti con diabete mellito di tipo 2, si deve considerare la possibilità di ridurre la dose di insulina o di stimolatori della secrezione insulinica (come le sulfoniluree) somministrati contemporaneamente, al fine di ridurre il rischio di ipoglicemia; vedere il paragrafo «Particolari avvertenze e precauzioni per l’uso».
Dose dimenticata
Se una dose viene dimenticata, deve essere somministrata il più presto possibile entro 5 giorni dalla dimenticanza. Se sono trascorsi più di 5 giorni, la dose non somministrata deve essere saltata e la successiva dose deve essere somministrata nel giorno previsto dal programma. In questo modo, in ogni caso, i pazienti possono riprendere il loro regolare schema settimanale di somministrazione. Se sono state dimenticate più dosi, si deve considerare la possibilità di ridurre la dose iniziale per riavviare il trattamento.
Popolazioni particolari
Pazienti anziani (oltre i 65 anni)
Non è necessaria alcuna modifica della dose in base all’età del paziente. L’esperienza nel trattamento di pazienti di età ≥ 75 anni è limitata e non si può escludere una maggiore sensibilità in alcuni anziani.
Compromissione renale
Nei pazienti con compromissione renale lieve o moderata non è necessaria alcuna modifica della dose. L’esperienza nell’uso di semaglutide nei pazienti con compromissione renale grave è limitata. L’uso di semaglutide non è raccomandato nei pazienti con compromissione renale grave (GFR < 30 ml/min/1,73 m²), compresi i pazienti con malattia renale terminale (vedere i paragrafi «Particolari avvertenze e precauzioni per l’uso», «Effetti indesiderati» e «Farmacocinetica»).
Compromissione epatica
Nei pazienti con compromissione epatica lieve o moderata non è necessaria alcuna modifica della dose. L’esperienza nell’uso di semaglutide nei pazienti con compromissione epatica grave è limitata. L’uso di semaglutide non è raccomandato nei pazienti con compromissione epatica grave; nei pazienti con compromissione epatica lieve o moderata deve essere usato con cautela (vedere i paragrafi «Particolari avvertenze e precauzioni per l’uso» e «Farmacocinetica»).
Modalità di somministrazione
Somministrazione sottocutanea
Vegovi FlexTach deve essere somministrato una volta alla settimana in qualsiasi momento della giornata, indipendentemente dai pasti.
Il medicinale deve essere iniettato per via sottocutanea nell’area dell’addome anteriore, nella coscia o nella spalla. Il sito di iniezione può essere variato. Il medicinale non deve essere somministrato per via endovenosa o intramuscolare.
Se necessario, il giorno settimanale di somministrazione può essere modificato, purché l’intervallo tra due dosi sia di almeno 3 giorni (> 72 ore). Dopo aver scelto un nuovo giorno di somministrazione, si deve continuare a seguire lo schema settimanale.
Quando si somministra il medicinale con una penna preriempita Vegovi FlexTach, la penna deve essere premuta saldamente contro la pelle fino a quando la striscia gialla smette di muoversi. L’iniezione dura circa 5–10 secondi.
Prima dell’uso del medicinale, si deve consigliare ai pazienti di leggere attentamente le istruzioni per l’uso della penna pre-riempita Vegovi FlexTach contenute nel foglio illustrativo.
Per ulteriori informazioni prima dell’uso, vedere il paragrafo «Istruzioni per l’uso della penna pre-riempita».
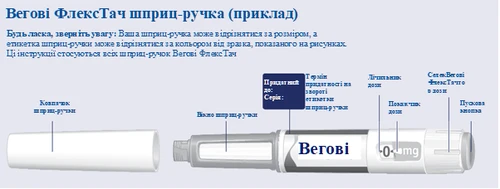
| Istruzioni per l'uso della penna preriempita Vegovi FlexTach |
|||
| Prima di iniziare a usare la penna preriempita Vegovi FlexTach una volta alla settimana, leggere attentamente queste istruzioni ogni volta e parlare con il proprio medico, infermiere o farmacista su come somministrare correttamente Vegovi FlexTach. Vegovi FlexTach è una penna preriempita con dosatore che contiene quattro dosi prescritte, corrispondenti a quattro somministrazioni settimanali. Si prega di utilizzare la tabella situata all'interno del coperchio della confezione di cartone per tenere traccia del numero di dosi già utilizzate e di quelle ancora disponibili nella penna. Vegovi FlexTach è disponibile in cinque diverse penne preriempite, ognuna contenente una delle seguenti dosi prescritte di semaglutide:
Controllare sempre l'etichetta della propria penna per assicurarsi che contenga la dose prescritta di Vegovi FlexTach La penna è progettata per essere utilizzata con aghi monouso 30G, 31G e 32G, lunghi fino a 8 mm. La confezione contiene:
|
|||
|
|
|||
|
|||
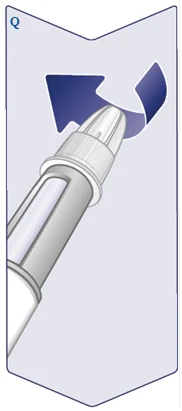
| Verificare il nome e la dose della propria penna per assicurarsi che contenga la dose prescritta del medicinale Vegovi FlexTach. Rimuovere il tappo della penna. (Vedere figura A). |
|
||
| Assicurarsi che la soluzione nella penna sia limpida e incolore. Guardare attraverso la finestra della penna. Se la soluzione è torbida o colorata, non utilizzare la penna. (Vedere figura B). |
|
||
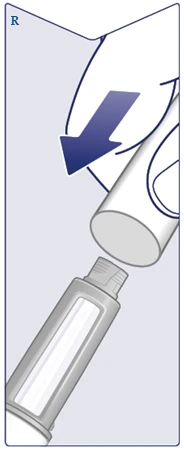
| Utilizzare sempre un ago nuovo per ogni iniezione. Prendere un ago quando si è pronti a effettuare l'iniezione. Verificare che la membrana di carta e il tappo esterno dell'ago non siano danneggiati, poiché ciò potrebbe compromettere la sterilità. In caso di danni, utilizzare un ago nuovo. Rimuovere la membrana di carta. (Vedere figura C). |
|
||
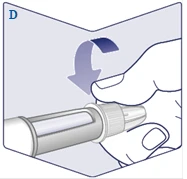
| Avvitare l'ago sulla penna. Ruotarlo fino a quando non sarà ben fissato. (Vedere figura D). |
|
||
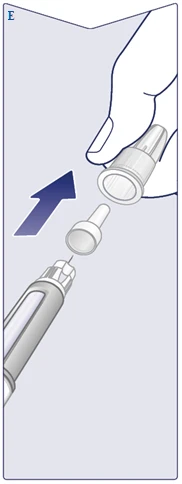
| L'ago è protetto da due tappi. È necessario rimuovere entrambi. Se si dimentica di rimuovere entrambi i tappi, non sarà possibile somministrare Vegovi FlexTach. Rimuovere il tappo esterno dell'ago e conservarlo. Sarà necessario dopo l'iniezione per rimuovere in sicurezza l'ago dalla penna. Rimuovere il tappo interno dell'ago e gettarlo via. Potrebbe apparire una goccia di soluzione sulla punta dell'ago. Tuttavia, è necessario verificare il flusso solo al primo utilizzo di una nuova penna. Vedere il punto «Verifica del funzionamento di ogni nuova penna». Non utilizzare mai un ago piegato o danneggiato. Per ulteriori informazioni sulla gestione degli aghi, vedere il punto «Circa i vostri aghi» in fondo a queste istruzioni. (Vedere figura E). |
|
||
| Verifica del funzionamento di ogni nuova penna |
|||
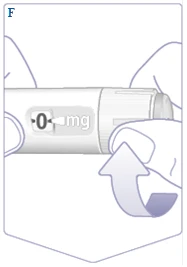
| Se si è già utilizzata la penna Vegovi FlexTach, passare al punto 2. «Impostazione della dose». Soltanto prima del primo utilizzo di ogni nuova penna Vegovi FlexTach, verificare il flusso. Ruotare il selettore della dose finché il contatore della dose non mostra il simbolo di verifica del flusso (). (Vedere figura F). |
|
||
| Assicurarsi che il simbolo di verifica del flusso sia allineato con l'indicatore della dose. (Vedere figura G). |
|
||
| Verifica del flusso |
|||
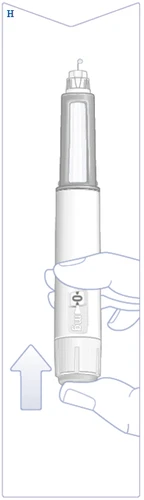
| Tenere la penna con l'ago rivolto verso l'alto. Premere e tenere premuto il pulsante di erogazione finché il contatore della dose non torna al simbolo . Il simbolo deve essere allineato con l'indicatore della dose. Sulla punta dell'ago deve apparire una goccia di soluzione. Questa goccia indica che la penna è pronta per l'uso. Se la goccia non appare, ripetere la verifica del flusso. Questo può essere fatto solo due volte. Se la goccia non appare ancora, sostituire l'ago e ripetere la verifica del flusso. Non utilizzare la penna se non appare ancora una goccia di soluzione. (Vedere figura H). |
|
||
|
|||
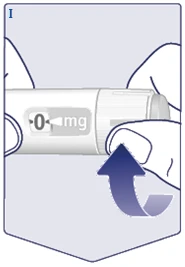
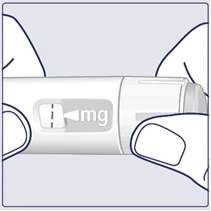
| Ruotare il selettore della dose finché il contatore della dose non si ferma e mostra la dose prescritta. (Vedere figura I). |
|
||
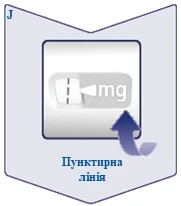
| La linea tratteggiata () sul contatore della dose aiuta a identificare la dose. Il selettore della dose emette un clic diverso quando viene ruotato avanti, indietro o dopo la dose. Si sentirà un clic ogni volta che si ruota il selettio della dose. Non impostare la dose contando i clic uditi. (Vedere figura J). |
|
||
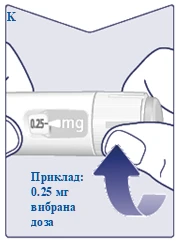
| Quando la dose prescritta è allineata con l'indicatore della dose, la dose è stata selezionata. In questa figura, la dose è mostrata come esempio. Se il contatore della dose si ferma prima di raggiungere la dose prescritta, consultare il punto «È sufficiente Vegovi FlexTach?» in fondo a queste istruzioni. (Vedere figura K). |
|
||
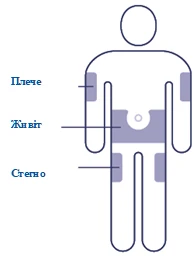
| Scegliere il sito di iniezione Scegliere un punto sulla spalla, coscia o addome (mantenendo una distanza di 5 cm dall'ombelico). È possibile effettuare l'iniezione nella stessa area del corpo ogni settimana, ma assicurarsi che non sia esattamente nello stesso punto della volta precedente. |
|
||
|
|||
| Inserire l'ago sotto la pelle. Assicurarsi di vedere il contatore della dose. Non coprirlo con le dita, altrimenti l'iniezione potrebbe interrompersi. (Vedere figura L). |
|
||
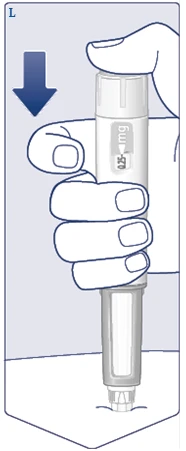
| Premere e tenere premuto il pulsante di erogazione finché il contatore della dose non mostra . (Vedere figura M). Mantenere l'ago sotto la pelle anche dopo che il contatore della dose è tornato a 0 e contare lentamente fino a 6. deve essere allineato con l'indicatore della dose. Successivamente, si potrebbe sentire o percepire un clic quando il contatore della dose torna a . (Vedere figura N). |
|
||
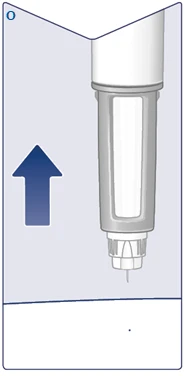
| Rimuovere l'ago dalla pelle. Se si estrae l'ago troppo presto, si potrebbe vedere fuoriuscire della soluzione dalla punta dell'ago e la dose completa non verrà somministrata. Se appare una goccia di sangue nel sito di iniezione, premere leggermente per fermare il sanguinamento. Potrebbe apparire una goccia di soluzione sulla punta dell'ago dopo l'iniezione. Questo è normale e non influenza il volume della dose somministrata. (Vedere figura O). |
|
||
|
|||
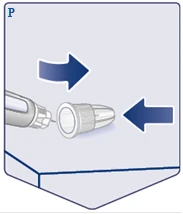
| Posizionare il tappo esterno dell'ago su una superficie piana e infilare il tappo esterno sull'ago, senza toccare né l'ago né il tappo esterno. Non appena l'ago è coperto, premere delicatamente fino in fondo sul tappo esterno dell'ago. (Vedere figura P). |
|
||
| Svitare l'ago e smaltirlo con attenzione seguendo le indicazioni del medico, dell'infermiere, del farmacista o delle autorità locali. Non tentare mai di rimettere il tappo interno sull'ago. Si potrebbe ferire con l'ago. Rimuovere sempre l'ago dalla penna immediatamente dopo ogni iniezione per evitare l'occlusione dell'ago, contaminazione, infezione e dosaggio impreciso. Non conservare mai la penna con l'ago applicato. (Vedere figura Q). |
|
||
| Rimettere il tappo sulla penna dopo ogni utilizzo per proteggere la soluzione dall'esposizione alla luce. (Vedere figura R). |
|
||
| Quando la penna è vuota, smaltirla senza l'ago seguendo le indicazioni del medico, dell'infermiere, del farmacista o delle autorità locali. Il tappo della penna e la confezione vuota possono essere gettati con i rifiuti domestici. |
|||
| Circa i vostri aghi |
|||
| Come riconoscere un ago ostruito o danneggiato
Cosa fare con un ago ostruito
|
|||
| Cura della propria penna |
|||
| Trattare la penna con cura. Un uso improprio o un maneggiamento scorretto potrebbe causare la somministrazione di una dose errata. In tal caso, non si otterrebbe l'effetto desiderato dal medicinale.
|
|||
| È sufficiente Vegovi FlexTach? |
|||
| Se il contatore della dose si ferma prima della dose prescritta, non c'è abbastanza soluzione per somministrare la dose completa. Smaltire questa penna e utilizzare una nuova penna Vegovi FlexTach. |
|
||
| Informazioni importanti |
|||
|
|||








Bambini
Nei bambini a partire dai 12 anni di età non è necessaria alcuna correzione della dose. La sicurezza e l'efficacia dell'uso di semaglutide nei bambini al di sotto dei 12 anni di età non sono state stabilite.
Sovradosaggio
Il sovradosaggio di semaglutide può essere associato a disturbi gastrointestinali, che possono portare a disidratazione. In caso di sovradosaggio, si deve osservare il paziente per rilevare eventuali segni clinici e iniziare un appropriato trattamento di supporto.
Effetti indesiderati.
Riepilogo del profilo di sicurezza
In quattro studi clinici di fase 3a, 2650 pazienti hanno ricevuto il medicinale Vegovi FlexTach. La durata degli studi è stata di 68 settimane. Gli effetti indesiderati più frequentemente riportati erano di origine gastrointestinale, inclusi nausea, diarrea, costipazione e vomito.
Effetti indesiderati
Di seguito sono riportati gli effetti indesiderati osservati negli studi clinici di fase 3. La frequenza di insorgenza degli effetti indesiderati è basata sui dati degli studi clinici di fase 3.
Gli effetti indesiderati sono classificati per sistemi e organi e per frequenza di insorgenza. La valutazione della frequenza è stata effettuata secondo la seguente scala: molto comune (≥ 1/10), comune (da ≥ 1/100 a < 1/10), non comune (da ≥ 1/1000 a < 1/100), raro (da ≥ 1/10000 a < 1/1000), molto raro (< 1/10000).
Disturbi del sistema immunitario: raro – reazione anafilattica.
Disturbi del metabolismo e della nutrizione: comune – ipoglicemia in pazienti con diabete mellito di tipo 2a.
Disturbi del sistema nervoso: molto comune – cefalea b; comune – capogiri b.
Disturbi della vista: comune – retinopatia diabetica in pazienti con diabete mellito di tipo 2a.
Disturbi cardiaci: non comune – ipotensione, ipotensione ortostatica, aumento della frequenza cardiaca a,c.
Disturbi gastrointestinali: molto comune – vomito a,b, diarrea a,b, costipazione a,b, nausea a,b, dolore addominale b,c; comune – gastrite b,c, malattia da reflusso gastroesofageo b, dispepsia b, eruttazione b, meteorismo b, distensione addominale b; non comune – pancreatite acuta a, ritardo dello svuotamento gastrico.
Disturbi epatici e delle vie biliari: comune – calcolosi biliare a.
Disturbi della cute e del tessuto sottocutaneo: comune – perdita di capelli a; raro – angioedema.
Disturbi generali e reazioni nel sito di somministrazione: molto comune – affaticamento b,c; comune – reazioni nel sito di somministrazione c.
Esami di laboratorio: non comune – aumento del livello di amilasi, aumento del livello di lipasi c.
a Descrizione di singoli effetti indesiderati riportata di seguito.
b In prevalenza osservato durante il periodo di aumento del dosaggio.
c Termini preferenziali raggruppati.
Descrizione di singoli effetti indesiderati
Effetti indesiderati gastrointestinali
Negli studi della durata di 68 settimane, la nausea si è verificata nel 43,9% dei pazienti trattati con semaglutide (nel 16,1% di quelli trattati con placebo), la diarrea nel 29,7% (nel 15,9% con placebo) e il vomito nel 24,5% (nel 6,3% con placebo). La maggior parte dei casi era di gravità da lieve a moderata e di breve durata. La costipazione si è verificata nel 24,2% dei pazienti trattati con semaglutide (nell’11,1% nel gruppo placebo) ed era di gravità lieve o moderata ma più prolungata. Nei pazienti trattati con semaglutide, la durata media della nausea era di 8 giorni, del vomito di 2 giorni, della diarrea di 3 giorni e della costipazione di 47 giorni.
I pazienti con compromissione renale di grado moderato (FGR ≥ 30 ml/min/1,73 m²) possono manifestare effetti gastrointestinali più intensi durante il trattamento con semaglutide.
Gli effetti indesiderati gastrointestinali hanno portato all’interruzione definitiva del trattamento nel 4,3% dei pazienti.
Pancreatite acuta
La frequenza di episodi di pancreatite acuta confermata da esperti negli studi clinici di fase 3 è stata dello 0,2% con semaglutide e < 0,1% con placebo.
Calcolosi biliare acuta / calcolosi biliare
La calcolosi biliare è stata registrata nel 1,6% dei pazienti e ha portato a colecistite nell’0,6% dei pazienti trattati con semaglutide. Calcolosi biliare e colecistite sono stati osservati rispettivamente nell’1,1% e 0,3% dei pazienti trattati con placebo.
Perdita di capelli
È stata riportata perdita di capelli nel 2,5% dei pazienti trattati con semaglutide e nell’1,0% di quelli trattati con placebo. La reazione era prevalentemente di gravità lieve e la maggior parte dei pazienti si è ripresa durante il proseguimento del trattamento. La perdita di capelli si è verificata più frequentemente nei pazienti con una maggiore perdita di peso corporeo (≥ 20%).
Aumento della frequenza cardiaca
Negli studi clinici di fase 3, nei pazienti trattati con semaglutide si è osservato un aumento medio della frequenza cardiaca di 3 battiti al minuto (bpm) rispetto al valore medio iniziale di 72 bpm. La percentuale di pazienti con un aumento della frequenza cardiaca di almeno 10 bpm rispetto al valore iniziale in qualsiasi momento durante il periodo di trattamento è stata del 67,0% nel gruppo semaglutide contro il 50,1% nel gruppo placebo.
Immunogenicità
Data la potenziale immunogenicità dei medicinali contenenti proteine o peptidi, nei pazienti trattati con semaglutide possono formarsi anticorpi. La percentuale di pazienti con risultato positivo al test per la presenza di anticorpi anti-semaglutide in qualsiasi momento dopo l’inizio del trattamento è stata bassa (2,9%) e nessun paziente ha sviluppato anticorpi neutralizzanti contro la semaglutide o anticorpi con effetto neutralizzante sul GLP-1 alla fine degli studi clinici. Durante il trattamento, alte concentrazioni di semaglutide potrebbero aver ridotto la sensibilità dei test, pertanto non si può escludere il rischio di risultati falsamente negativi. Tuttavia, nei pazienti con risultato positivo al test per gli anticorpi, la presenza di anticorpi è stata temporanea e senza effetto visibile sull’efficacia e sulla sicurezza.
Ipoglicemia nei pazienti con diabete mellito di tipo 2
Nello studio STEP 2, l’ipoglicemia clinicamente significativa si è verificata nel 6,2% (0,1 episodi per paziente-anno) dei pazienti trattati con semaglutide, rispetto al 2,5% (0,03 episodi per paziente-anno) dei pazienti trattati con placebo. L’ipoglicemia con semaglutide si è verificata sia con che senza l’uso concomitante di sulfoniluree. Un episodio (0,2% dei pazienti, 0,002 episodi per paziente-anno) è stato registrato come grave in un paziente che non assumeva sulfoniluree in concomitanza. Il rischio di ipoglicemia aumentava con l’uso concomitante di semaglutide e sulfoniluree.
Complicanze della retinopatia diabetica
In uno studio clinico della durata di 2 anni su semaglutide 0,5 mg e 1 mg rispetto a placebo, sono stati arruolati 3297 pazienti con diabete mellito di tipo 2, alto rischio cardiovascolare, lunga durata del diabete e controllo inadeguato della glicemia. In questo studio, le complicanze della retinopatia diabetica si sono sviluppate in un numero maggiore di pazienti trattati con semaglutide (3,0%) rispetto a quelli trattati con placebo (1,8%). Questo è stato osservato in pazienti con retinopatia diabetica confermata che assumevano insulina. La differenza tra i regimi di trattamento è emersa precocemente e si è mantenuta per tutta la durata dello studio. Nello studio STEP 2, le malattie della retina sono state riportate nel 6,9% dei pazienti trattati con Vegovi FlexTach, nel 6,2% dei pazienti trattati con semaglutide 1 mg e nel 4,2% dei pazienti trattati con placebo. La maggior parte dei casi è stata registrata come retinopatia diabetica (4,0%, 2,7% e 2,7% rispettivamente) e retinopatia non proliferativa (0,7%, 0%, 0% rispettivamente).
Bambini
In uno studio clinico condotto su bambini di età compresa tra 12 e 18 anni con obesità o sovrappeso e almeno una comorbidità associata al peso corporeo, 133 pazienti hanno ricevuto Vegovi FlexTach. La durata dello studio è stata di 68 settimane.
In generale, la frequenza, il tipo e la gravità degli effetti indesiderati nei bambini sono stati simili a quelli negli adulti. La calcolosi biliare è stata registrata nel 3,8% dei pazienti trattati con Vegovi FlexTach e nello 0% dei pazienti trattati con placebo.
Dopo 68 settimane di trattamento, non è stato osservato alcun effetto sulla crescita o sullo sviluppo puberale.
Segnalazione degli effetti indesiderati
La segnalazione degli effetti indesiderati dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette un monitoraggio continuo del rapporto rischio/beneficio del medicinale. I professionisti sanitari e farmaceutici, così come i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di effetti indesiderati e di mancata efficacia del medicinale attraverso il Sistema Informativo Automatizzato di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità. 3 anni.
Dopo il primo utilizzo: 6 settimane. Conservare a temperatura inferiore a 30 °C o in frigorifero (a temperatura di 2 °C – 8 °C).
Condizioni di conservazione.
Conservare in frigorifero (2 °C – 8 °C). Tenere lontano dagli elementi congelanti. Non congelare.
Conservare la penna preriempita con il tappo applicato per proteggerla dalla luce.
Conservare in un luogo inaccessibile ai bambini.
Incompatibilità.
Questo medicinale non deve essere miscelato con altri medicinali poiché non sono stati effettuati studi di compatibilità.
Confezione.
0,25, 0,5 mg
Cartuccia in vetro (vetro tipo I) da 1,5 ml, chiusa da un lato con un pistone in gomma (clorobutile) e dall’altro con un tappo in alluminio con guarnizione laminata in gomma (bromobutile/poliiisoprene). La cartuccia è inserita in una penna preriempita monouso realizzata in polipropilene, poliossimetilene, policarbonato e acrilonitrile butadiene stirene.
1 penna preriempita e 4 aghi monouso NovoFine® Plus in una confezione di cartone.
1 mg, 1,7 mg
Cartuccia in vetro (vetro tipo I) da 3 ml, chiusa da un lato con un pistone in gomma (clorobutile) e dall’altro con un tappo in alluminio con guarnizione laminata in gomma (bromobutile/poliiisoprene). La cartuccia è inserita in una penna preriempita monouso realizzata in polipropilene, poliossimetilene, policarbonato e acrilonitrile butadiene stirene.
1 penna preriempita e 4 aghi monouso NovoFine® Plus in una confezione di cartone.
2,4 mg
Cartuccia in vetro (vetro tipo I) da 3 ml, chiusa da un lato con un pistone in gomma (clorobutile) e dall’altro con un tappo in alluminio con guarnizione laminata in gomma (bromobutile/poliiisoprene). La cartuccia è inserita in una penna preriempita monouso realizzata in polipropilene, poliossimetilene, policarbonato e acrilonitrile butadiene stirene.
1 o 3 penne preriempite e 4 o 12 aghi monouso NovoFine® Plus in una confezione di cartone.
Categoria di rilascio. Sotto prescrizione medica.
Richiedente/Produttore.
A/T Novo Nordisk.
Indirizzo del richiedente/produttore e sede operativa.
Novo Allé
2880, Bagsværd
Danimarca.