Rixubis
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE RIXUBIS
Composizione:
Principio attivo: nonacog gamma*;
Ogni flaconcino contiene:
250 UI** di nonacog gamma, fattore della coagulazione del sangue umano ricombinante IX (rDNA), corrispondente a una concentrazione di 50 UI/ml dopo ricostituzione del medicinale con 5 ml di solvente;
500 UI** di nonacog gamma, fattore della coagulazione del sangue umano ricombinante IX (rDNA), corrispondente a una concentrazione di 100 UI/ml dopo ricostituzione del medicinale con 5 ml di solvente;
1000 UI** di nonacog gamma, fattore della coagulazione del sangue umano ricombinante IX (rDNA), corrispondente a una concentrazione di 200 UI/ml dopo ricostituzione del medicinale con 5 ml di solvente;
2000 UI** di nonacog gamma, fattore della coagulazione del sangue umano ricombinante IX (rDNA), corrispondente a una concentrazione di 400 UI/ml dopo ricostituzione del medicinale con 5 ml di solvente;
3000 UI** di nonacog gamma, fattore della coagulazione del sangue umano ricombinante IX (rDNA), corrispondente a una concentrazione di 600 UI/ml dopo ricostituzione del medicinale con 5 ml di solvente.
Eccipienti: L-istidina, cloruro di sodio, cloruro di calcio, mannitolo, saccarosio, polisorbato 80.
Ogni flaconcino di solvente contiene: acqua per preparazioni iniettabili – 5 ml.
_________________________________________________________________________
* Nonacog gamma (fattore della coagulazione del sangue ricombinante IX (rDNA)) è una glicoproteina purificata monomero costituita da 415 aminoacidi. Viene prodotto mediante tecnologia del DNA ricombinante in una linea cellulare derivata dall'ovaio di criceto cinese.
** L'attività (UI) è determinata mediante un saggio in un solo stadio per i fattori della coagulazione del sangue, in conformità con la Farmacopea Europea. L'attività specifica del medicinale RIXUBIS è approssimativamente compresa tra 200–390 UI/mg di proteina.
Forma farmaceutica. Polvere e solvente per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: polvere di colore bianco o quasi bianco; solvente – soluzione trasparente e incolore.
Gruppo farmacoterapeutico. Agenti antiedemorragici. Fattore della coagulazione del sangue IX.
Codice ATC B02BD04.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Il medicinale Rixubis contiene fattore della coagulazione del sangue IX ricombinante (nonacog gamma). Il fattore IX è una glicoproteina monocatenaria con un peso molecolare di circa 68.000 Dalton. È un fattore della coagulazione dipendente dalla vitamina K, prodotto nel fegato. Il fattore IX viene attivato dal fattore XIa nel sistema intrinseco della coagulazione e dal complesso fattore VII/fattore tissutale nel sistema estrinseco della coagulazione. Il fattore IX attivato, insieme al fattore VIII attivato, attiva il fattore X. Il fattore X attivato converte la protrombina in trombina. La trombina a sua volta converte il fibrinogeno in fibrina, portando così alla formazione del trombo.
Farmacodinamica
L'emofilia B è una patologia congenita legata al sesso del sistema della coagulazione, causata da livelli ridotti di fattore IX, che determina emorragie abbondanti nelle articolazioni, nei muscoli o negli organi interni, spontanee o in seguito a traumi accidentali o chirurgici. La terapia sostitutiva consente di aumentare i livelli plasmatici di fattore IX, correggendo temporaneamente la carenza e riducendo la tendenza a sviluppare emorragie.
Efficacia e sicurezza clinica
Prevenzione e controllo delle emorragie in pazienti di età pari o superiore a 12 anni, precedentemente trattati
L'efficacia del medicinale Rixubis è stata valutata nell'ambito della parte non controllata di uno studio combinato di fase 1/3 in aperto, in cui 73 pazienti di età compresa tra 12 e 59 anni, precedentemente trattati, hanno ricevuto Rixubis per la profilassi e/o per il trattamento degli episodi emorragici "su richiesta". Tutti i pazienti avevano una forma grave (livello di fattore IX < 1%) o moderatamente grave (livello di fattore IX < 2%) di emofilia B. Di questi, 59 pazienti hanno ricevuto Rixubis per la profilassi. I dati di 56 pazienti, trattati con Rixubis per almeno 3 mesi, sono stati inclusi nel set di dati destinato alla valutazione dell'efficacia della profilassi. Altri 14 pazienti hanno ricevuto Rixubis solo per il trattamento degli episodi emorragici. I pazienti del braccio di trattamento "su richiesta" dovevano avere almeno 12 episodi emorragici documentati, richiedenti trattamento, nei 12 mesi precedenti l'inclusione nello studio. La durata media del trattamento nei pazienti del braccio "su richiesta" è stata di 3,5 ± 1,00 mesi (mediana 3,4; intervallo da 1,2 a 5,1 mesi), con una frequenza media annuale di emorragie (annualised bleeding rate, ABR) pari a 33,9 ± 17,37, mediana 27,0 e intervallo da 12,9 a 73,1.
La mediana dell'ABR con Rixubis nella profilassi è stata di 2,0 per tutte le emorragie, di 0,0 per le emorragie spontanee e di 0,0 per le emorragie articolari. In 24 pazienti (42,9%) non si sono verificate emorragie.
Complessivamente, 249 episodi emorragici sono stati trattati con Rixubis, di cui 197 articolari e 52 extra-articolari (nei tessuti molli, nei muscoli, nelle cavità corporee, intracranici e altri). Dei 249 episodi emorragici, 163 erano di gravità moderata, 71 di lieve entità e 15 gravi. Il trattamento è stato effettuato tenendo conto delle caratteristiche individuali di ciascun paziente, considerando la gravità, la causa e la localizzazione dell'emorragia. La maggior parte (211 episodi; 84,7%) dei 249 episodi emorragici è stata risolta con 1-2 infusioni. L'efficacia emostatica nel controllo dell'emorragia è stata valutata come eccellente o elevata nel 96% di tutti gli episodi trattati.
Prevenzione e controllo delle emorragie in pazienti di età inferiore a 12 anni, precedentemente trattati
L'efficacia di Rixubis è stata valutata in uno studio combinato di fase 2/3, in cui 23 pazienti maschi di età compresa tra 1,8 e 11,8 anni (mediana di età: 7,10 anni), precedentemente trattati, di cui 11 con età inferiore a 6 anni, hanno ricevuto Rixubis per la profilassi e il controllo degli episodi emorragici. Tutti i pazienti avevano una forma grave (livello di fattore IX < 1%) o moderatamente grave (livello di fattore IX < 2%) di emofilia B. Tutti i 23 pazienti hanno ricevuto trattamento profilattico con Rixubis per almeno 3 mesi ed erano inclusi nell'analisi per la valutazione dell'efficacia della profilassi.
La mediana dell'ABR è stata di 2,0, di 0,0 per le emorragie spontanee e di 0,0 per le emorragie articolari.
In nove pazienti (39,1%) non si sono verificate emorragie.
Complessivamente, 26 episodi emorragici sono stati trattati con Rixubis, di cui 23 traumatici, 2 spontanei e 1 di origine sconosciuta. Di questi, 19 erano extra-articolari (nei tessuti molli, nei muscoli, nelle cavità corporee, intracranici e altri) e 7 articolari, uno dei quali in un'articolazione bersaglio. Dei 26 episodi emorragici, 15 erano lievi, 9 di gravità moderata e 2 gravi. Il trattamento è stato effettuato tenendo conto delle caratteristiche individuali di ciascun paziente, considerando la gravità, la causa e la localizzazione dell'emorragia. La maggior parte (23; 88,5%) è stata trattata con 1-2 infusioni. L'efficacia emostatica nel controllo dell'emorragia è stata valutata come eccellente o elevata nel 96,2% di tutti gli episodi trattati.
Trattamento perioperatorio
La sicurezza ed efficacia di Rixubis nel trattamento perioperatorio è stata valutata in uno studio prospettico, multicentrico, aperto e non controllato di fase 3, condotto su uomini con emofilia B grave o moderata, precedentemente trattati. L'analisi dell'efficacia per protocollo ha incluso 37 interventi chirurgici, comprendenti procedure maggiori e minori, interventi odontoiatrici o altre procedure chirurgiche invasive, effettuati su 27 pazienti di età compresa tra 17 e 57 anni. Ventuno interventi erano maggiori, compresi 13 interventi ortopedici e 3 interventi odontochirurgici. Diciassette interventi, comprese 10 estrazioni dentarie, sono stati considerati minori. I pazienti sottoposti a interventi maggiori dovevano aver effettuato uno studio di farmacocinetica (PK). Tutti i pazienti hanno ricevuto una dose basata sui dati più recenti di incremento della risposta dell'attività del fattore IX. La dose raccomandata iniziale di Rixubis doveva garantire un'attività del fattore IX mantenuta tra l'80-100% durante gli interventi chirurgici maggiori e tra il 30-60% durante gli interventi minori. Rixubis è stato somministrato mediante infusione endovenosa in bolo.
Durante l'intero periodo dello studio è stata mantenuta un'adeguata emostasi.
Farmacocinetica
Pazienti di età pari o superiore a 12 anni, precedentemente trattati
Uno studio di farmacocinetica randomizzato, in cieco e controllato, è stato condotto su uomini in assenza di emorragia (età ≥ 15 anni) nell'ambito di uno studio combinato centrale di fase 1/3. I pazienti hanno ricevuto uno dei due medicinali mediante singola infusione endovenosa. La dose media (± deviazione standard) e mediana di Rixubis nel set di dati analizzabile per protocollo (n = 25) è stata rispettivamente di 74,69 ± 2,37 e 74,25 UI/kg, con un intervallo compreso tra 71,27 e 79,38 UI/kg. I parametri farmacocinetici sono stati calcolati in base alle misurazioni dell'attività del fattore IX nei campioni ematici raccolti entro 72 ore da ciascuna infusione.
La valutazione della farmacocinetica di Rixubis è stata ripetuta in uno studio aperto e non controllato, condotto su uomini che avevano partecipato allo studio incrociato iniziale di PK e che avevano seguito una profilassi con Rixubis per 26 ± 1 settimane (media ± deviazione standard), con un periodo di trattamento totale con Rixubis di almeno 30 giorni. L'intervallo delle dosi di Rixubis utilizzate nel ripetuto studio di farmacocinetica è stato compreso tra 64,48 e 79,18 UI/kg (n = 23).
La Tabella 1 riporta i parametri farmacocinetici in tutti i pazienti i cui dati erano valutabili (analisi per protocollo).
Tabella 1
| Parametro |
Rixubis Studi incrociati iniziali (N = 25) |
Rixubis Valutazione ripetuta (N = 23) |
| AUC0-72 h (UI • h/dl)a Media ± deviazione standard (DS) Mediana (intervallo) |
1067,81 ± 238,42 1108,35 (696,07–1571,16) |
1156,15 ± 259,44 1170,26 (753,85–1626,81) |
| Aumento di recupero dell'attività a Cmax (UI/dl : UI/kg)b Media ± DS Mediana (intervallo) |
0,87 ± 0,22 0,88 (0,53–1,35) |
0,95 ± 0,25 0,93 (0,52–1,38) |
| Emivita (h) Media ± DS Mediana (intervallo) |
26,70 ± 9,55 24,58 (15,83–52,34) |
25,36 ± 6,86 24,59 (16,24–42,20) |
| Cmax (UI/dl) Media ± DS Mediana (intervallo) |
66,22 ± 15,80 68,10 (41,70–100,30) |
72,75 ± 19,73 72,40 (38,50–106,30) |
| Tempo medio di permanenza nel corpo (h) Media ± DS Mediana (intervallo) |
30,82 ± 7,26 28,93 (22,25–47,78) |
29,88 ± 4,16 29,04 (21,32–37,52) |
| Vssc (dl/kg) Media ± DS Mediana (intervallo) |
2,02 ± 0,77 1,72 (1,10–3,94) |
1,79 ± 0,45 1,74 (1,12–2,72) |
| Clearance (dl/kg•h) Media ± DS Mediana (intervallo) |
0,0644 ± 0,0133 0,0622 (0,0426–0,0912) |
0,0602 ± 0,0146 0,0576 (0,0413–0,0945) |
a Area sotto la curva concentrazione plasmatica-tempo da 0 a 72 ore dopo l'infusione.
b Calcolato come il rapporto (Cmax del fattore IX pre-trattamento) diviso per la dose in UI/kg, dove Cmax è il valore massimo di attività del fattore IX misurato dopo l'infusione.
c Volume di distribuzione allo stato stazionario.
L'aumento incrementale del recupero dell'attività del fattore IX a 30 minuti dall'infusione è stato determinato per tutti i pazienti arruolati nello studio combinato di Fase 1/3, al giorno 1 del trattamento, alle visite mediche alla settimana 5, 13 e 26 e al momento della conclusione dello studio o della cessazione della partecipazione allo studio, se non coincidente con la visita alla settimana 26. I dati ottenuti indicano che l'aumento incrementale del recupero è rimasto stabile per tutto il periodo (vedere Tabella 2).
Tabella 2
| Indicatore |
1° giorno di trattamento (N = 73) |
5a settimana (N = 71) |
13a settimana (N = 68) |
26a settimana (N = 55) |
Giorno di completamento/ interruzione dello studio b (N = 23) |
| Aumento di attività di recupero a 30 minuti dall'infusione (U/dl : U/kg) a Media ± DS Mediana (intervallo) |
0,79 ± 0,20 0,78 (0,26–1,35) |
0,83 ± 0,21 0,79 (0,46–1,48) |
0,85 ± 0,25 0,83 (0,14–1,47) |
0,89 ± 0,12 0,88 (0,52–1,29) |
0,87 ± 0,20 0,89 (0,52–1,32) |
a Calcolato come rapporto tra (livello di Fattore IX a 30 minuti prima dell'inizio del trattamento) diviso per la dose in UI/kg, dove C30 minuti è il livello di attività del Fattore IX misurato 30 minuti dopo l'infusione.
b Se non coincidente con il tempo della visita alla 26a settimana.
Popolazione pediatrica (maschi precedentemente trattati, di età inferiore ai 12 anni)
Nell'ambito dello studio combinato di Fase 2/3, è stata effettuata un'analisi farmacocinetica iniziale di Rixubis in 23 pazienti pediatrici di sesso maschile in assenza di emorragia. Per ridurre il disagio legato al frequente prelievo di campioni ematici in ciascun paziente, questi sono stati randomizzati in due gruppi con diversa sequenza di prelievo. La dose media (± deviazione standard) e la mediana della dose di Rixubis nell'insieme completo dei dati analizzati (n = 23) sono state rispettivamente di 75,50 ± 3,016 e 75,25 UI/kg, con un intervallo compreso tra 70,0 e 83,6 UI/kg. I parametri farmacocinetici sono stati calcolati sulla base delle misurazioni dell'attività del Fattore IX nei campioni ematici raccolti entro un periodo di 72 ore dall'infusione.
I parametri farmacocinetici per tutti i pazienti (insieme completo dei dati analizzati) sono riportati nella Tabella 3.
Tabella 3
| Parametro |
Fino a 6 anni (N = 11) |
Da 6 a 12 anni (N = 12) |
Tutti (N = 23) |
| AUCinf (MO • h/dl)а Media ± DS Mediana (intervallo) |
723,7 ± 119,00 717,2 (488–947) |
886,0 ± 133,66 863,7 (730–1138) |
808,4 ± 149,14 802,9 (488–1138) |
| Emivita (ore) Media ± DS Mediana (intervallo) |
27,67 ± 2,66 27,28 (24,0–32,2) |
23,15 ± 1,58 22,65 (21,8–27,4) |
25,31 ± 3,13 24,48 (21,8–32,2) |
| Tempo medio di permanenza nell'organismo (ore) Media ± DS Mediana (intervallo) |
30,62 ± 3,27 30,08 (26,2–36,2) |
25,31 ± 1,83 24,74 (23,7–30,3) |
27,85 ± 3,73 26,77 (23,7–36,2) |
| Vssb (dl/kg) Media ± DS Mediana (intervallo) |
3,22 ± 0,52 3,16 (2,65–4,42) |
2,21 ± 0,32 2,185 (1,70–2,70) |
2,7 ± 0,67 2,69 (1,70–4,42) |
| Clearance (dl/kg • h) Media ± DS Mediana (intervallo) |
0,1058 ± 0,01650 0,1050 (0,081–0,144) |
0,0874 ± 0,01213 0,0863 (0,069–0,108) |
0,0962 ± 0,01689 0,0935 (0,069–0,144) |
a Area sotto la curva concentrazione plasmatica-tempo da 0 ore a infinito.
b Volume di distribuzione allo stato stazionario.
L'incremento incrementale del recupero dell'attività del fattore IX a 30 minuti dopo l'infusione è stato determinato in tutti i pazienti arruolati nello studio combinato di Fase 2/3, alla valutazione dei parametri farmacocinetici iniziali (giorno 1 del trattamento), nei giorni delle visite mediche alle settimane 5, 13 e 26 e al termine dello studio o all'interruzione della partecipazione allo studio, se non coincidente con la visita medica alla settimana 26. I dati ottenuti indicano che l'incremento incrementale del recupero dell'attività rimane stabile nel tempo in tutte le fasce d'età dei pazienti pediatrici (vedere tabelle 4, 5, 6 riportate di seguito).
Tabella 4
Incremento incrementale del recupero dell'attività dopo somministrazione di Rixubis a 30 minuti dall'infusione nei pazienti pediatrici appartenenti a entrambe le fasce d'età
| Aumento del recupero dell'attività a 30 min dall'infusione |
FC (1° giorno di trattamento) Tutti (N = 22) |
5a settimana Tutti (N = 23) |
13a settimana Tutti (N = 21) |
26a settimana Tutti (N = 21) |
| (UI/dl: UI/kg)a Media ± DS Mediana (intervallo) |
0,67 ± 0,16 0,69 (0,31–1,00) |
0,68 ± 0,12 0,66 (0,48–0,92) |
0,71 ± 0,13 0,66 (0,51–1,00) |
0,72 ± 0,15 0,734 (0,51–1,01) |
a Calcolato come il valore (C30 min del fattore IX prima dell'inizio del trattamento), diviso per la dose in unità IU/kg, dove C30 min è il valore di misurazione dell'attività del fattore IX a 30 minuti dall'infusione.
Tabella 5
Incremento progressivo del ripristino dell'attività dopo somministrazione di Rixubis nei pazienti pediatrici di età inferiore ai 6 anni, misurato a 30 minuti dall'infusione.
| Augmento del recupero dell'attività a 30 minuti dopo l'infusione |
Fattore C (1° giorno di trattamento) Tutti (N = 10) |
5a settimana, Tutti (N = 11) |
13a settimana, Tutti (N = 10) |
26a settimana, Tutti (N = 10) |
| (U/ml: U/kg)a Media ± DS Mediana (intervallo) |
0,59 ± 0,13 0,59 (0,31–0,75) |
0,63 ± 0,10 0,6 (0,49–0,80) |
0,68 ± 0,12 0,66 (0,51–0,84) |
0,65 ± 0,13 0,61 (0,51–0,84) |
a Calcolato come il valore (C30 min del fattore IX prima dell'inizio del trattamento) diviso per la dose in unità UI/kg, dove C30 min è il valore di misurazione dell'attività del fattore IX a 30 minuti dopo l'infusione.
Tabella 6
Incremento progressivo del recupero dell'attività dopo l'uso del medicinale Rixubis a 30 minuti dall'infusione in pazienti pediatrici di età compresa tra 6 e 12 anni
| Aumento del recupero dell'attività a 30 minuti dall'infusione |
FC (1° giorno di trattamento) Tutti (N = 12) |
5a settimana, Tutti (N = 12) |
13a settimana, Tutti (N = 11) |
26a settimana, Tutti (N = 11) |
| (UI/dl: UI/kg)a Media ± DS Mediana (intervallo) |
0,73 ± 0,16 0,71 (0,51–1,00) |
0,73 ± 0,13 0,70 (0,48–0,92) |
0,73 ± 0,14 0,70 (0,54–1,00) |
0,8 ± 0,14 0,78 (0,56–1,01) |
a Calcolato come il valore (C30 min del fattore IX prima dell'inizio del trattamento) diviso per la dose in unità UI/kg, dove C30 min è il valore di misurazione dell'attività del fattore IX a 30 minuti dall'infusione.
Caratteristiche cliniche.
Indicazioni.
Per il trattamento e la prevenzione delle emorragie nei pazienti con emofilia B (deficit congenito del fattore IX).
Il medicinale Rixubis è indicato per i pazienti di tutte le fasce d'età.
Controindicazioni.
Ipersensibilità alla sostanza attiva o a uno qualsiasi degli eccipienti elencati nella sezione «Composizione».
Reazione allergica nota alle proteine del criceto.
Interazioni con altri medicinali ed altre forme di interazione.
Non sono state riportate interazioni tra medicinali a base di fattore di coagulazione del sangue umano IX (rDNA) e altri farmaci.
Caratteristiche particolari di impiego.
Tracciabilità
Per migliorare il tracciamento dei medicinali biologici, si raccomanda di indicare chiaramente il nome e il numero di lotto del prodotto somministrato, ad esempio nel proprio diario personale.
Ipersensibilità
Sono state riportate reazioni di ipersensibilità di natura allergica con l’uso di Rixubis. Il medicinale contiene una quantità residua di proteine di criceto. In caso di comparsa di sintomi di ipersensibilità, ai pazienti o alle persone che se ne prendono cura si deve raccomandare di interrompere immediatamente l’uso del medicinale e di consultare il medico. I pazienti devono essere informati sui sintomi iniziali di reazione di ipersensibilità, inclusi orticaria, orticaria generalizzata, senso di costrizione toracica, respiro sibilante, ipotensione e anafilassi.
Il rischio è più elevato nelle fasi iniziali del trattamento con concentrati di fattore IX nei pazienti mai trattati in precedenza, in particolare nei pazienti con mutazioni geniche ad alto rischio. Sono stati riportati in letteratura collegamenti tra la comparsa di inibitori del fattore IX e reazioni allergiche, specialmente nei pazienti con mutazioni geniche ad alto rischio. Per questo motivo, i pazienti con reazioni allergiche devono essere sottoposti a controllo per la presenza di tali inibitori.
In caso di insorgenza di shock, deve essere fornita la consueta terapia antishock.
Inibitori
Dopo il trattamento ripetuto con medicinali a base di fattore di coagulazione del sangue umano IX (rDNA), i pazienti devono essere sottoposti a controllo per la comparsa di anticorpi neutralizzanti (inibitori), la cui concentrazione deve essere determinata in unità Bethesda (BU) mediante un opportuno saggio biologico.
In letteratura sono stati riportati collegamenti tra la comparsa di inibitori del fattore IX e reazioni allergiche. Per questo motivo, i pazienti con reazioni allergiche devono essere sottoposti a controllo per la presenza di tali inibitori. I pazienti in cui si sviluppano inibitori del fattore IX presentano un rischio aumentato di anafilassi in caso di ulteriore trattamento con fattore IX.
A causa del rischio di reazioni allergiche con l’uso di concentrati di fattore IX, la prima somministrazione di fattore IX deve essere effettuata, a giudizio del medico curante, sotto supervisione medica, in modo da garantire l’immediata disponibilità di un’adeguata assistenza medica in caso di reazioni allergiche.
Sindrome nefrotica
Sono stati riportati casi di sindrome nefrotica in pazienti con emofilia B, precedentemente portatori di inibitori del fattore IX, dopo stimolazione dell’induzione della tolleranza immunitaria.
Tromboembolia
A causa del potenziale rischio di complicanze trombotiche, è necessario effettuare un monitoraggio clinico per rilevare precocemente i segni di trombosi e di coagulopatia consumptiva mediante opportuni esami biologici, quando questo medicinale viene somministrato a pazienti con malattia epatica, pazienti post-operatori, neonati o pazienti con rischio di sviluppare fenomeni trombotici o sindrome da coagulazione intravascolare disseminata (DIC). In ciascuno di questi casi, i benefici del trattamento con Rixubis devono essere attentamente valutati rispetto al rischio di tali complicanze.
Malattie cardiovascolari
Nei pazienti con malattie cardiovascolari preesistenti, la terapia sostitutiva con fattore IX può aumentare il rischio di patologia cardiovascolare.
Complicanze legate all’uso di catetere
Nel caso in cui sia necessario utilizzare un dispositivo venoso centrale (CVC), si devono considerare le possibili complicanze legate all’uso del CVC, inclusi infezioni locali, batteriemia e trombosi nel sito di cateterizzazione.
Considerazioni relative agli eccipienti
Dopo ricostituzione, questo medicinale contiene meno di 1 mmol (23 mg) di sodio per flaconcino, ovvero è praticamente privo di sodio. A seconda del peso corporeo del paziente e della dose prescritta, i pazienti possono ricevere più di un flaconcino di Rixubis, il che deve essere tenuto in considerazione in caso di dieta controllata per il contenuto di sodio.
Pazienti anziani
I pazienti di età pari o superiore a 65 anni non sono stati inclusi negli studi clinici con Rixubis. Non è noto se la loro risposta al trattamento differisca da quella dei pazienti più giovani. Come per tutti gli altri pazienti, la scelta della dose per il trattamento dei pazienti anziani deve essere effettuata su base individuale.
Bambini
Le avvertenze e le precauzioni sopra riportate si applicano sia al trattamento degli adulti che dei bambini.
Uso durante la gravidanza o l’allattamento.
Gravidanza
I dati sull’uso del fattore IX in donne in gravidanza sono assenti o limitati. Non sono stati condotti studi sugli animali sull’impatto del fattore IX sulla funzione riproduttiva.
Il fattore IX deve essere usato durante la gravidanza e l’allattamento solo se chiaramente indicato.
Allattamento
Non sono disponibili dati riguardo all’escrezione del fattore IX o dei suoi metaboliti nel latte umano.
Fertilità
Non sono disponibili informazioni sull’impatto del fattore IX sulla fertilità.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell’uso di macchinari.
Rixubis non influenza la capacità di guidare autoveicoli o di utilizzare macchinari.
Modalità e dosaggio di somministrazione
Il trattamento deve essere effettuato sotto la supervisione di un medico esperto nel trattamento dell'emofilia.
Monitoraggio durante il trattamento
Durante il trattamento si raccomanda un adeguato monitoraggio dei livelli del fattore IX come riferimento per la determinazione della dose e della frequenza delle infusioni ripetute. La risposta al fattore IX può variare da paziente a paziente, con differenti valori di emivita e grado di recupero. Pazienti con peso corporeo basso o elevato potrebbero necessitare di un'adeguata correzione della dose calcolata in base al peso corporeo. Nei casi di interventi chirurgici maggiori è estremamente importante garantire un rigoroso monitoraggio durante la terapia sostitutiva mediante analisi dei fattori della coagulazione (attività del fattore IX nel plasma sanguigno).
Per assicurare il raggiungimento dei livelli desiderati di attività del fattore IX nel plasma sanguigno, si raccomanda un attento monitoraggio mediante un metodo appropriato di determinazione dell'attività del fattore IX e, se necessario, una corrispondente correzione della dose e della frequenza delle infusioni ripetute. Quando si utilizza il metodo unistadio di analisi in vitro dei fattori della coagulazione basato sul tempo di tromboplastina parziale attivata (aPTT) per determinare l'attività del fattore IX nei campioni ematici dei pazienti, i risultati relativi all'attività del fattore IX possono dipendere notevolmente sia dal tipo di reagente aPTT sia dallo standard di riferimento utilizzato per l'analisi. Ciò risulta particolarmente importante in caso di cambio di laboratorio e/o dei reagenti utilizzati per l'analisi.
Dosaggio
La dose e la durata della terapia sostitutiva dipendono dal grado di carenza del fattore IX, dalla localizzazione e dalla gravità dell'emorragia, nonché dallo stato clinico, dall'età e dai parametri farmacocinetici del fattore IX nel paziente, come l'aumento del livello di attività del fattore IX e il tempo di emivita.
La quantità di fattore IX somministrata viene espressa in unità internazionali (UI), secondo lo standard attuale dell'OMS per i medicinali a base di fattore IX. L'attività del fattore IX nel plasma viene espressa in percentuale (rispetto al valore normale per il plasma umano) oppure in unità internazionali (secondo lo standard internazionale per il fattore IX nel plasma).
Un'unità internazionale (UI) di attività del fattore IX corrisponde all'attività del fattore IX presente in 1 ml di plasma umano normale.
Adulti
Trattamento "a richiesta"
Il calcolo della dose richiesta di fattore IX per pazienti di età pari o superiore a 12 anni si basa sull'osservazione empirica secondo cui l'applicazione di un'unità internazionale (UI) di fattore IX per kg di peso corporeo determina un aumento dell'attività del fattore IX nel plasma di 0,9 UI/dl (intervallo da 0,5 a 1,4 UI/dl) oppure dello 0,9 % dell'attività normale.
La dose richiesta viene determinata mediante la formula riportata di seguito.
| Quantità richiesta di unità |
= |
massa corporea (kg) |
× |
aumento desiderato del livello del fattore IX (% o UI/dl) |
× |
valore inverso del coefficiente di recupero del fattore IX registrato (dl/kg) |
Per un aumento progressivo del recupero del livello di attività di 0,9 UI/dl per 1 UI/kg, il dosaggio viene calcolato secondo la seguente formula:
| Quantità necessaria di unità |
= |
massa corporea (kg) |
× |
aumento desiderato del livello di fattore IX (% o UI/dl) |
× |
1,1 dl/kg |
La quantità della dose e la frequenza di somministrazione pianificata devono sempre essere orientate a garantire l'efficacia clinica in ciascun paziente specifico.
In caso di manifestazioni emorragiche riportate di seguito, i livelli di attività del fattore IX non devono essere inferiori al livello di attività plasmatica indicato (in % rispetto alla norma oppure UI/dl) per il periodo di tempo corrispondente. La tabella 7 può essere utilizzata per determinare la dose nei episodi emorragici e durante gli interventi chirurgici.
Tabella 7
| Gravità dell'emorragia/ tipo di intervento chirurgico |
Livello necessario di fattore IX, % o U/ml |
Frequenza di somministrazione (ore)/ durata del trattamento (giorni) |
| Emorragia Emoartrosi precoce, ematomi muscolari o emorragia orale |
20–40 |
Ripetere ogni 24 ore. Almeno 1 giorno, fino alla cessazione dell'emorragia, attestata dall'assenza di dolore, oppure fino alla cicatrizzazione della ferita. |
| Emoartrosi più marcata, ematomi muscolari o ematoma |
30–60 |
Ripetere l'infusione ogni 24 ore per 3–4 giorni o più a lungo, fino alla scomparsa del dolore e alla risoluzione dei gravi disturbi funzionali. |
| Emorragie pericolose per la vita |
60–100 |
Ripetere l'infusione ogni 8–24 ore fino alla scomparsa del pericolo. |
| Interventi chirurgici Interventi chirurgici minori, compresa l'estrazione dentaria |
30–60 |
Ogni 24 ore, almeno 1 giorno, fino alla cicatrizzazione della ferita. |
| Interventi chirurgici maggiori |
80–100 (prima e dopo l'intervento) |
Ripetere l'infusione ogni 8–24 ore fino a un adeguato rimarginamento della ferita; successivamente continuare il trattamento per almeno altri 7 giorni, mantenendo l'attività del fattore IX tra il 30 % e il 60 % (U/ml). |
Un attento monitoraggio della terapia sostitutiva è particolarmente importante in caso di intervento chirurgico maggiore o di emorragia potenzialmente letale.
Prevenzione
Per la profilassi a lungo termine delle emorragie nei pazienti di età pari o superiore a 12 anni con emofilia B grave, di solito si utilizzano dosi di 40-60 UI di fattore IX per kg di peso corporeo ogni 3-4 giorni. In alcuni casi, a seconda della farmacocinetica, dell'età, del fenotipo emorragico e del livello di attività fisica del paziente, potrebbe essere necessario ridurre gli intervalli tra le somministrazioni o aumentare la dose del medicinale.
Infusione continua
Non somministrare il medicinale Rixubis mediante infusione continua.
Modalità di somministrazione
Somministrazione endovenosa.
Nel caso in cui il medicinale venga somministrato dal paziente stesso o da una persona che si prende cura del paziente, è necessario che questa persona sia adeguatamente addestrata a eseguire tale procedura.
Il medicinale Rixubis deve essere somministrato alla velocità che risulti confortevole per il paziente, con un massimo di 10 ml/min.
Dopo la ricostituzione, la soluzione del medicinale è trasparente, incolore, priva di particelle estranee, con un pH compreso tra 6,8 e 7,2. L'osmolarità è superiore a 240 mosmoli/kg.
Per la somministrazione di questo medicinale devono essere utilizzate esclusivamente siringhe di plastica con connettore di tipo Luer.
Il medicinale Rixubis deve essere somministrato per via endovenosa dopo la ricostituzione della polvere con il solvente fornito.
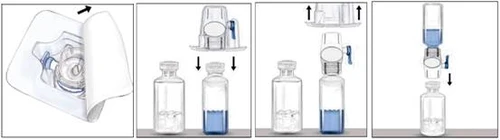
- Per la preparazione della soluzione del medicinale, utilizzare esclusivamente il solvente e il dispositivo di ricostituzione (BAXJECT II) forniti nella confezione.
- Per la somministrazione del medicinale deve essere utilizzata una siringa con connettore di tipo Luer.
- Non utilizzare il dispositivo BAXJECT II se risulta danneggiato, se il sistema sterile di protezione o la confezione sono compromessi o se sono presenti segni di deterioramento.
Ricostituzione
Applicare tecniche asettiche.
- Se il medicinale è conservato in frigorifero, estrarre i flaconcini con la polvere di Rixubis e con il solvente dal frigorifero e lasciarli riscaldare a temperatura ambiente (da 15 °C a 30 °C).
- Lavarsi accuratamente le mani con acqua calda e sapone.
- Rimuovere i tappi dai flaconcini contenenti la polvere e il solvente.
- Disinfettare i tappi con garze imbevute di alcol. Posizionare i flaconcini su una superficie piana e pulita.
- Aprire la confezione del dispositivo BAXJECT II rimuovendo il film di carta senza toccare la superficie interna del dispositivo (figura a). Non estrarre il dispositivo dalla confezione.
- Capovolgere la confezione e fissare il perno di plastica trasparente sul tappo del flaconcino contenente il solvente. Afferrare la confezione lungo i bordi e rimuovere la confezione dal dispositivo BAXJECT II (figura b). Non rimuovere il cappuccio di colore blu dal dispositivo BAXJECT II.
- Una volta che il dispositivo BAXJECT II è collegato al flaconcino contenente il solvente, capovolgere l'intero sistema in modo che il flaconcino con il solvente si trovi sopra il dispositivo. Fissare il perno di plastica bianco sul tappo del flaconcino contenente il medicinale Rixubis. Per effetto del vuoto, il solvente passerà nel flaconcino contenente Rixubis (figura c).
- Mescolare delicatamente fino a completo scioglimento della polvere. Il medicinale si dissolve rapidamente (entro 2 minuti). Assicurarsi che Rixubis sia completamente disciolto, altrimenti non tutto il medicinale ricostituito passerà attraverso il filtro del dispositivo. Prima dell'uso, ispezionare visivamente la soluzione ricostituita per verificare la presenza di particelle solide o variazioni di colore. La soluzione deve essere trasparente o leggermente opalescente. Non utilizzare soluzioni torbide o contenenti sedimenti.
| Figura a |
Figura b |
Figura c |
|
|
||
Non refrigerare il medicinale dopo la ricostituzione. Utilizzarlo immediatamente dopo la preparazione.
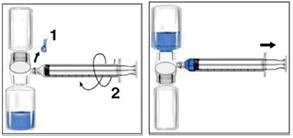
Somministrazione
Utilizzare tecniche asettiche.
- Rimuovere il tappo blu dal dispositivo BAXJECT II. Accertarsi di non aspirare aria nella siringa. Collegare la siringa al dispositivo BAXJECT II (figura d).
- Capovolgere il sistema (il flacone con la soluzione ricostituita deve trovarsi nella posizione superiore). Riempire lentamente la siringa con la soluzione ricostituita tirando indietro lo stantuffo (figura e).
- Scollegare la siringa.
- Applicare un ago a farfalla sulla siringa. Iniettare la soluzione in una vena. La soluzione deve essere somministrata lentamente, a una velocità tollerabile per il paziente, non più velocemente di 10 ml al minuto.
| Figura d |
Figura e |
|
|
|
Registri sempre il nome e il numero di lotto del medicinale Rixubis (ad esempio nel proprio diario personale) ogni volta che lo utilizza, al fine di conservare un registro dei medicinali impiegati.
Eventuali residui inutilizzati del medicinale o i suoi rifiuti devono essere smaltiti secondo le norme locali.
La stabilità chimica e fisica della soluzione medicinale diluita e pronta all'uso è di 3 ore a una temperatura non superiore a 30 °C. Dal punto di vista microbiologico, nonostante il metodo di ricostituzione escluda la contaminazione microbica, il medicinale deve essere utilizzato immediatamente. Se non viene utilizzato immediatamente, il periodo e le condizioni di conservazione sono sotto la responsabilità dell'utilizzatore. Non raffreddare.
Bambini
Pazienti di età compresa tra 12 e 17 anni
Il dosaggio è lo stesso per adulti e pazienti di età compresa tra 12 e 17 anni.
Pazienti di età inferiore a 12 anni
Trattamento "on demand"
Il calcolo della dose richiesta di fattore IX nei pazienti di età inferiore a 12 anni si basa sull'osservazione empirica che l'applicazione di un'unità internazionale (UI) di fattore IX per kg di peso corporeo determina un aumento dell'attività del fattore IX nel plasma pari a 0,7 UI/dl (intervallo da 0,31 a 1,0 UI/dl) oppure al 0,7 % dell'attività normale.
La dose richiesta è determinata mediante la seguente formula.
| Quantità necessaria di unità |
= |
massa corporea (kg) |
× |
aumento desiderato del livello di fattore IX (% o UI/dl) |
× |
valore inverso del tasso di recupero registrato (dl/kg) |
Per un incremento progressivo del livello di attività di 0,7 UI/ml per 1 UI/kg, il dosaggio viene calcolato secondo la seguente formula:
| Quantità necessaria di unità |
= |
massa corporea (kg) |
× |
aumento desiderato del livello di fattore IX (% o UI/dl) |
× |
1,4 dl/kg |
Per determinare la dose in caso di episodi emorragici e interventi chirurgici può essere utilizzata la stessa tabella prevista per gli adulti (vedere sopra la tabella 7).
Prevenzione
L’intervallo di dose raccomandato per il trattamento di pazienti pediatrici di età inferiore ai 12 anni è compreso tra 40 e 80 UI/kg ogni 3-4 giorni. In alcuni casi, a seconda della farmacocinetica, dell’età, del fenotipo emorragico e del livello di attività fisica del paziente, potrebbe rendersi necessario ridurre gli intervalli tra le somministrazioni o aumentare le dosi del medicinale.
Sovradosaggio
Gli effetti di dosi più elevate rispetto a quelle raccomandate di Rixubis non sono stati descritti.
Effetti indesiderati
Sono stati osservati rari casi di ipersensibilità o reazioni allergiche (che possono includere angioedema, sensazione di bruciore e prurito nel sito di infusione, brividi, vampate di calore, orticaria generalizzata, cefalea, eruzioni cutanee, ipotensione, letargia, nausea, stato di ansia, tachicardia, senso di oppressione al petto, acufene, vomito, respiro sibilante), che talvolta possono evolvere in una forma grave di anafilassi (incluso shock), verificatasi in stretta contiguità temporale con l'insorgenza di inibitori del fattore IX.
Dopo stimolazione per l'induzione della tolleranza immunologica in pazienti con emofilia B e con anamnesi di reazioni allergiche, in cui erano presenti inibitori del fattore IX, sono stati registrati casi di sindrome nefrotica.
Molto raramente sono stati osservati casi di formazione di anticorpi contro le proteine del criceto, associati all'insorgenza di reazioni di ipersensibilità.
Nei pazienti con emofilia B possono svilupparsi anticorpi neutralizzanti (inibitori) contro il fattore IX, con conseguente insufficiente efficacia del trattamento. In tali casi si raccomanda di rivolgersi a un centro specializzato per il trattamento dell'emofilia.
L'uso di medicinali contenenti fattore IX è associato a un potenziale rischio di episodi tromboembolici, con un rischio maggiore nell'impiego di prodotti con basso grado di purificazione. L'uso di preparazioni di fattore IX con basso grado di purific游戏副本
| Effetti indesiderati registrati negli studi clinici e nelle segnalazioni spontanee |
||
| Classe di sistemi e organi secondo la classificazione MedDRA |
Effetti indesiderati |
Frequenza, per paziente |
| Disturbi del sistema immunitario |
Ipersensibilità a) |
Sconosciuta |
| Disturbi del sistema nervoso |
Alterazione del gusto |
Comune |
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Dolore agli arti |
Comune |
a) PR (reazione avversa) è spiegata più dettagliatamente nel paragrafo seguente.
Descrizione delle singole reazioni avverse
Ipersensibilità
Sono state osservate reazioni di tipo allergico, manifestatesi come dispnea, prurito, orticaria generalizzata ed eruzioni cutanee.
Bambini
Ci si aspetta che la frequenza, il tipo e la gravità delle reazioni avverse nei bambini siano simili a quelli negli adulti. Tuttavia, non sono disponibili dati riguardo ai pazienti non trattati in precedenza, poiché negli studi clinici sono stati inclusi solo pazienti precedentemente trattati. Per questo motivo, non sono stati condotti studi sull'immunogenicità in relazione alla formazione di inibitori in questo gruppo a rischio.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l'autorizzazione del medicinale è di grande importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, così come i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il Sistema Informativo Automatizzato di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità.
3 anni.
Condizioni di conservazione.
Conservare a una temperatura non superiore a 30 °C. Non congelare.
Conservare in un luogo inaccessibile ai bambini.
Incompatibilità.
A causa della mancanza di studi sulla compatibilità, questo medicinale non deve essere mescolato con altri medicinali.
Quando si utilizza questo medicinale, si devono impiegare esclusivamente siringhe di plastica con connettore di tipo Luer. A causa dell'assorbimento del fattore IX della coagulazione del sangue umano sulle superfici interne di alcuni dispositivi per infusione, il dosaggio del medicinale potrebbe risultare impreciso.
Confezione.
1 flaconcino di polvere (250 UI, 500 UI, 1000 UI, 1500 UI, 2000 UI o 3000 UI) in combinazione con 1 flaconcino di solvente (5 ml di acqua per preparazioni iniettabili) e 1 dispositivo per la ricostituzione BAXJECT II per confezione.
Categoria di rilascio.
Sotto prescrizione medica.
Produttore.
Baxalta Belgium Manufacturing SA / Baxalta Belgium Manufacturing SA.
Sede del produttore e indirizzo del luogo dell'attività.
Boulevard Rene Branquart 80, Lessines, 7860, Belgio / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.