Nuwiq®
Ucraina
Indice
- ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE NUWIQ®
- Composizione:
- Proprietà farmacologiche.
- Caratteristiche cliniche.
- Caratteristiche particolari di utilizzo.
- Modalità di somministrazione e dosi
- Effetti indesiderati
- La frequenza si basa su studi con tutti i farmaci del fattore VIII che includevano pazienti con emofilia A grave.
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE NUWIQ®
Composizione:
sostanza attiva: simoctocog alfa (fattore della coagulazione del sangue VIII ricombinante);
1 flaconcino di polvere per soluzione iniettabile contiene simoctocog alfa (fattore della coagulazione del sangue VIII ricombinante) 1500 UI, oppure 2500 UI, oppure 3000 UI, oppure 4000 UI;
sostanze eccipienti: cloruro di sodio; saccarosio; cloridrato di L-arginina; cloruro di calcio diidrato; polossamero 188; citrato di sodio diidrato.
Sostanza per la ricostituzione: acqua per preparazioni iniettabili.
Forma farmaceutica. Polvere e solvente per soluzione iniettabile.
Principali caratteristiche fisico-chimiche:
Polvere: grumo bianco. Può essere presente una piccola quantità di polvere bianca.
Solvente: liquido limpido, incolore, privo di particelle.
Gruppo farmacoterapeutico. Agenti antiedemorragici. Fattore della coagulazione del sangue VIII.
Codice ATC B02BD02.
Proprietà farmacologiche.
Farmacodinamica.
Il fattore della coagulazione del sangue VIII si lega al fattore di von Willebrand nella circolazione del paziente. Il fattore VIII attivato agisce come cofattore per il fattore IX attivato, riducendo il tempo di trasformazione del fattore X in fattore X attivato. Il fattore X attivato converte la protrombina in trombina. La trombina successivamente converte il fibrinogeno in fibrina e si forma il coagulo.
L'emofilia A è una malattia ereditaria legata al sesso, causata da una carenza congenita del fattore di coagulazione VIII:C, che porta a emorragie nelle articolazioni, nei muscoli e negli organi interni, che si verificano spontaneamente o in seguito a traumi accidentali o chirurgici. Nella terapia sostitutiva, i livelli plasmatici di fattore VIII aumentano, correggendo temporaneamente la carenza del fattore di coagulazione del sangue VIII e la tendenza alle emorragie.
Popolazione adulta e pazienti di età compresa tra 12 e 65 anni.
Prevenzione: In uno studio clinico condotto su 32 pazienti adulti con emofilia A grave, la dose media profilattica del medicinale Nuwiq® è stata di 468,7 UI/kg/mese. Trattamento dell'emorragia: La dose media per il trattamento delle emorragie nei pazienti in terapia profilattica è stata di 33,0 UI/kg. In un altro studio clinico, 22 pazienti adulti hanno ricevuto trattamento su richiesta. In totale, in 986 episodi emorragici è stata somministrata una dose media di 30,9 UI/kg. In generale, le emorragie lievi hanno richiesto dosi medie inferiori, mentre quelle gravi hanno richiesto dosi medie triplicate.
Profilassi individualizzata: La profilassi individualizzata basata sul farmacocinetico (PK) è stata valutata in 66 pazienti adulti precedentemente trattati (PTPs) con emofilia A grave. Dopo una fase standard di 1-3 mesi di profilassi (dose somministrata ogni due giorni o tre volte alla settimana), 44 pazienti (67%) sono passati a un regime di dosaggio basato sulla valutazione del loro profilo PK, e 40 pazienti hanno completato un periodo di 6 mesi di profilassi secondo le dosi e le modalità di somministrazione prescritte. Di questi, 34 (85%) ricevevano il trattamento due volte alla settimana o meno. 33 pazienti (82,5%) non hanno avuto alcuna emorragia e 36 (90,0%) non hanno avuto emorragie spontanee. La frequenza media annuale delle emorragie (calcolata su base annua), espressa come media + DS, è stata di 1,2 + 3,9, mentre la dose media + DS è stata di 52,2 + 12,2 UI/kg per iniezione e di 99,7 + 25,6 UI/kg settimanali.
Va sottolineato che la frequenza annuale delle emorragie (ABR) non è comparabile tra diversi concentrati di fattore e tra diversi studi clinici.
Popolazione pediatrica
I dati sono stati ottenuti da 29 bambini precedentemente trattati di età compresa tra 2 e 5 anni, 31 bambini di età compresa tra 6 e 12 anni e un adolescente di 14 anni. La dose media profilattica per iniezione è stata di 37,8 UI/kg. Venti pazienti hanno ricevuto dosi medie superiori a 45 UI/kg. La dose media mensile profilattica del medicinale Nuwiq® è stata di 521,9 UI/kg. Per il trattamento delle emorragie, ai bambini è stata somministrata una dose media maggiore di Nuwiq® (43,9 UI/kg) rispetto agli adulti (33,0 UI/kg), così come una dose media più elevata per il trattamento sia delle emorragie minori che di quelle maggiori (78,2 UI/kg contro 41,7 UI/kg). In generale, ai bambini più piccoli sono state necessarie dosi medie maggiori (6-12 anni: 43,9 UI/kg; 2-5 anni: 52,6 UI/kg). Questi dati sono stati confermati da un monitoraggio a lungo termine di 49 di questi bambini, trattati per un periodo medio aggiuntivo di circa 30 mesi (intervallo da 9,5 a 52 mesi); durante questo periodo, il 45% dei bambini non ha avuto emorragie spontanee.
I dati relativi a 108 pazienti con emofilia A grave (˂ 1% FVIII:C) precedentemente non trattati sono stati ottenuti in uno studio clinico prospettico aperto. Nella maggior parte dei pazienti, il trattamento profilattico è stato avviato dopo la prima emorragia che ha richiesto un trattamento.
Farmacocinetica.
Popolazione adulta
Tabella 1.
Parametri farmacocinetici del medicinale Nuwiq® (dose di 50 UI/kg) in adulti di età compresa tra 18 e 65 anni con emofilia A grave, precedentemente trattati (n=20).
| Parametri farmacocinetici |
Analisi cromogenica |
|
| Media ± DS |
Mediana (limiti) |
|
| AUC (h*MO/ml) |
22,6 ± 8,0 |
22,3 (8,4 – 38,1) |
| T 1/2 (h) |
14,7 ± 10,4 |
12,5 (5,4 – 55,6) |
| IVR (%/MO/kg) |
2,5 ± 0,4 |
2,5 (1,7 – 3,2) |
| CL (ml/h/kg) |
3,0 ± 1,2 |
2,7 (1,5 – 6,4) |
AUC - area sotto la curva (FVIII:C), T½ - emivita
IVR - recupero dei parametri in vivo, CL - clearance, SD - deviazione standard
Tabella 2.
Parametri farmacocinetici del medicinale Nuwiq® (dose 50 UI/kg) in bambini di età compresa tra 6 e 12 anni con emofilia A grave, già trattati in precedenza (n=12).
| Parametri farmacocinetici |
Analisi cromogenica |
|
| Media ± DS |
Mediana (limiti) |
|
| AUC (h*MO/ml) |
13,2 ± 3,4 |
12,8 (7,8 – 19,1) |
| T 1/2 (h) |
10,0 ± 1,9 |
9,9 (7,6 – 14,1) |
| IVR (%/MO/kg) |
1,9 ± 0,4 |
1,9 (1,2 – 2,6) |
| CL (ml/h/kg) |
4,3 ± 1,2 |
4,2 (2,8 – 6,9) |
AUC - area sotto la curva (FVIII:C), T½ - emivita
IVR - recupero in vivo, CL - clearance, SD - deviazione standard
Tabella 3.
Parametri farmacocinetici del medicinale Nuwiq® (dose di 50 UI/kg) in bambini di età compresa tra 2 e 5 anni con emofilia A grave, già trattati in precedenza (n = 13).
| Parametri farmacocinetici |
Analisi cromogenica |
|
| Media ± DS |
Mediana (limiti) |
|
| AUC (h*MO/ml) |
11,7 ± 5,3 |
10,5 (4,9 – 23,8) |
| T 1/2 (h) |
9,5 ± 3,3 |
8,2 (4,3 – 17,3) |
| IVR (%/MO/kg) |
1,9 ± 0,3 |
1,8 (1,5 – 2,4) |
| CL (ml/h/kg) |
5,4 ± 2,4 |
5,1 (2,3 – 10,9) |
AUC - area sotto la curva (FVIII:C), T½ - emivita
IVR - recupero dei parametri in vivo, CL - clearance, SD - deviazione standard
Popolazione pediatrica
Sulla base dei dati pubblicati, i parametri di recupero e l'emivita risultano inferiori nei bambini più giovani rispetto agli adulti, mentre il tasso di eliminazione è più elevato; ciò può essere in parte spiegato dal maggiore volume plasmatico per chilogrammo di peso corporeo nei pazienti più giovani.
Sottogruppi per peso corporeo
Tabella 4.
Parametri farmacocinetici del medicinale Nuwiq® (dose di 50 UI/kg) in adulti di età compresa tra 18 e 65 anni con emofilia A grave, precedentemente trattati (n = 20), in base al peso corporeo del paziente
| Parametri farmacocinetici |
Tutti i pazienti (n =20) |
Peso corporeo normale (n =14) |
Condizione pre-obesità (n =4) |
Obesità (n =2) |
| Analisi cromogenica, media ± DS |
||||
| AUC (h*UI/ml) |
22,6 ± 8,0 |
20,4 ± 6,9 |
24,9 ± 8,9 |
33,5 ± 6,5 |
| T 1/2 (h) |
14,7 ± 10,4 |
14,7 ± 12,1 |
13,4 ± 5,9 |
17,2 ± 4,8 |
| IVR (%/UI/kg) |
2,5 ± 0,4 |
2,4 ± 0,4 |
2,7 ± 0,4 |
2,8 ± 0,3 |
| CL (ml/h/kg) |
3,0 ± 1,2 |
3,2 ± 1,3 |
2,6 ± 1,0 |
1,8 ± 0,4 |
| Analisi cromogenica, mediana (limiti) |
||||
| AUC (h*UI/ml) |
22,3 (8,4 – 38,1) |
21,2 (8,4– 32,6) |
23,3 (17,4 – 35,5) |
33,5 (28,9 – 38,1) |
| T 1/2 (h) |
12,5 (5,4 – 55,6) |
12,3 (5,4– 55,6) |
11,2 (9,3 – 22,0) |
17,2 (13,8 – 20,6) |
| IVR (%/UI/kg) |
2,5 (1,7 – 3,2) |
2,4 (1,7 – 3,1) |
2,8 (2,3 – 3,2) |
2,8 (2,6 – 3,0) |
| CL (ml/h/kg) |
2,7 (1,5 – 6,4) |
2,8 (1,7 – 6,4) |
2,5 (1,6 – 3,7) |
1,8 (1,5 – 2,0) |
Peso corporeo normale BMI - 18,5 – 25 kg/m2, Peso corporeo elevato: BMI 25 – 30 kg/m2, Obesità: BMI > 30 kg/m2, SD - deviazione standard
Caratteristiche cliniche.
Indicazioni.
Trattamento e profilassi delle emorragie in pazienti con emofilia A (deficit congenito del fattore VIII).
Il medicinale Nuwiq® può essere utilizzato in pazienti di tutte le fasce d'età.
Controindicazioni.
Ipersensibilità alla sostanza attiva o a qualsiasi componente del medicinale.
Interazioni con altri medicinali ed altre forme di interazione.
Non sono stati effettuati studi sulle interazioni del medicinale Nuwiq® con altri medicinali.
Caratteristiche particolari di utilizzo.
Tracciabilità
Per migliorare la tracciabilità dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto (serie) del prodotto somministrato.
L'attività specifica del medicinale Nuwiq® è di circa 9500 UI/mg di proteina.
Simoctocog alfa (fattore della coagulazione del sangue VIII (rDNA)) è una proteina pura costituita da 1440 amminoacidi. La sequenza amminoacidica è simile alla forma 90+80 kDa del fattore VIII del plasma umano (cioè con il dominio B rimosso). Nuwiq® è prodotto mediante tecnologia del DNA ricombinante a partire da cellule renali di embrione umano (HEK) 293F geneticamente modificate. Durante il processo produttivo e nel prodotto finito non vengono aggiunti materiali di origine animale o umana.
Ipersensibilità
Come con qualsiasi altro prodotto proteico somministrato per via endovenosa, esiste il rischio di sviluppare reazioni allergiche e ipersensibilità. Nuwiq® contiene tracce di proteine cellulari umane diverse dal fattore VIII. In caso di comparsa di sintomi di ipersensibilità, l'assunzione del medicinale deve essere immediatamente interrotta e si deve ricorrere all'assistenza medica. I pazienti devono essere informati sui segni precoci di reazioni di ipersensibilità, come orticaria, orticaria generalizzata, sensazione di oppressione toracica, difficoltà respiratorie, sibili, ipotensione arteriosa e anafilassi.
In caso di insorgenza di shock, deve essere avviato il trattamento medico standard per lo stato di shock.
Inibitori
La formazione di anticorpi neutralizzanti (inibitori) contro il fattore VIII è una complicanza nota nel trattamento dei pazienti con emofilia A. Tali inibitori sono generalmente immunoglobuline di tipo IgG la cui azione è diretta contro l'attività procoagulante del fattore VIII e la cui concentrazione viene quantificata in unità Bethesda (UB) per 1 ml di plasma, mediante test modificato. Il rischio di formazione di inibitori è correlato alla gravità della malattia e dipende dall'esposizione al fattore VIII, risultando massimo nei primi 50 giorni di esposizione, ma persiste per tutta la vita, anche se raro.
Sono stati osservati casi di ri-comparsa di inibitori (titolo basso) in pazienti precedentemente trattati con un prodotto di fattore VIII ricombinante per oltre 100 giorni e con anamnesi positiva per la formazione di inibitori, in seguito al passaggio da un prodotto di fattore VIII ricombinante a un altro. Per questo motivo, si raccomanda un rigoroso monitoraggio di tutti i pazienti per la comparsa di inibitori dopo ogni modifica del medicinale.
L'importanza clinica della formazione di inibitori dipende dal titolo dell'inibitore: gli inibitori a basso titolo, temporaneamente presenti o con titolo costantemente basso, rappresentano un rischio minore di risposta clinica inadeguata rispetto agli inibitori ad alto titolo.
È necessario un rigoroso monitoraggio di tutti i pazienti in trattamento con fattori ricombinanti della coagulazione del sangue VIII per la comparsa di inibitori, mediante adeguata osservazione clinica e analisi di laboratorio. Se i livelli attesi di attività del fattore VIII nel plasma non vengono raggiunti o se il sanguinamento non si arresta nonostante l'uso della dose appropriata del prodotto, si devono effettuare analisi per determinare la presenza di inibitori del fattore VIII. Nei pazienti con titoli elevati di inibitori, la terapia con fattore VIII può risultare inefficace; in tal caso, si devono considerare altre opzioni terapeutiche, come l'induzione della tolleranza immunologica (ITI). Il trattamento di tali pazienti deve essere effettuato da medici esperti nella gestione dell'emofilia e della comparsa di inibitori del fattore VIII.
Complicanze cardiovascolari
Nei pazienti con fattori di rischio cardiovascolari preesistenti, la terapia sostitutiva con FVIII può aumentare il rischio di eventi cardiovascolari.
Complicanze associate all'uso di catetere
Nel caso in cui sia necessario utilizzare un dispositivo per l'accesso venoso centrale (CVAD), si devono considerare i rischi associati al suo utilizzo, inclusi infezioni locali, batteriemia e trombosi nel sito del catetere.
Si raccomanda fortemente di registrare, ad ogni somministrazione di Nuwiq®, il nome e il numero di lotto (serie) del medicinale, al fine di stabilire un eventuale legame tra lo stato del paziente e il lotto del prodotto somministrato.
Tutte le avvertenze si applicano sia agli adulti che ai bambini.
Informazioni sugli eccipienti (contenuto di sodio)
1 ml di soluzione diluita contiene 7,35 mg (18,4 mg di sodio per flaconcino), ovvero, sostanzialmente, «privo di sodio».
Tuttavia, a seconda del peso corporeo e della dose, il paziente può ricevere più di un flaconcino. Tale informazione deve essere tenuta in considerazione nei pazienti sottoposti a dieta con contenuto controllato di sodio.
Uso durante la gravidanza o l'allattamento.
Non sono stati effettuati studi sugli effetti di Nuwiq® sulla funzione riproduttiva negli animali. Poiché l'emofilia A è rara nelle donne, non esistono dati sull'uso di Nuwiq® durante la gravidanza e l'allattamento. Pertanto, Nuwiq® può essere somministrato durante la gravidanza e l'allattamento solo se strettamente necessario. Non sono disponibili dati sugli effetti sulla fertilità.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell'uso di macchinari.
Non sono stati osservati effetti sulla capacità di guidare autoveicoli o di lavorare con macchinari.
Modalità di somministrazione e dosi
Il trattamento deve essere effettuato sotto la supervisione di un medico esperto nella gestione dell'emofilia.
Monitoraggio del trattamento
Durante il corso del trattamento, si raccomanda di determinare in modo appropriato i livelli del fattore VIII al fine di controllare la dose necessaria da somministrare e la frequenza delle infusioni ripetute. Singoli pazienti possono presentare una risposta diversa all'applicazione del fattore VIII, mostrando differenti emivite e tempi di recupero (ripristino). La dose basata sul peso corporeo potrebbe richiedere un aggiustamento (correzione) in caso di peso corporeo insufficiente o eccessivo. In caso di intervento chirurgico esteso, è obbligatorio un monitoraggio accurato della terapia sostitutiva mediante analisi della coagulazione del sangue (attività del fattore VIII nel plasma).
Quando si utilizza un'analisi in una sola fase dell'attività coagulante del sangue basata sul tempo di tromboplastina parziale attivato (aPTT) in vitro per determinare l'attività del fattore VIII nei campioni di sangue dei pazienti, sia il tipo di reagente aPTT sia il campione standard utilizzato nell'analisi possono influenzare significativamente i risultati dell'attività del fattore VIII nel plasma. Inoltre, possono verificarsi scostamenti significativi tra i risultati ottenuti con l'analisi in una sola fase basata sull'aPTT e quelli ottenuti con l'analisi cromogenica secondo la Farmacopea Europea. Ciò risulta particolarmente importante, specialmente quando si cambia laboratorio e/o i reagenti utilizzati nell'analisi.
Modalità di somministrazione
La dose e la durata della terapia sostitutiva dipendono dalla gravità della carenza di fattore VIII, dalla sede e dall'entità dell'emorragia, nonché dallo stato clinico del paziente.
La quantità di unità di fattore VIII somministrata viene espressa in unità internazionali (UI), riferite allo standard attivo dell'OMS per i farmaci contenenti fattore VIII. L'attività del fattore VIII nel plasma viene determinata in percentuale (rispetto al plasma normale umano) oppure prevalentemente in unità internazionali (secondo lo standard internazionale per il fattore VIII nel plasma).
1 unità internazionale (UI) di attività del fattore VIII corrisponde alla quantità di fattore VIII presente in 1 ml di plasma normale umano.
Trattamento su richiesta
I calcoli della dose necessaria di fattore VIII si basano su dati empirici che indicano come 1 unità internazionale (UI) di fattore VIII per 1 kg di peso corporeo aumenti l'attività del fattore VIII nel plasma sanguigno di circa il 2% rispetto all'attività normale, ovvero di circa 2 UI/dl. La dose necessaria può essere calcolata mediante la seguente formula:
I.
Unità necessarie = peso corporeo (kg) × incremento desiderato del fattore VIII (%) (UI/dl) × 0,5 (UI/kg per UI/dl)
II.
| Crescita prevista del fattore VIII (% del normale) = |
2 × UI somministrate |
| peso corporeo (kg) |
II.
La dose da somministrare e la frequenza di somministrazione devono sempre essere adattate all'efficacia clinica nel singolo caso.
Nei casi emorragici indicati di seguito, l'attività del fattore VIII non deve scendere al di sotto del livello plasmatico indicato (in % del valore normale o U/ml) durante il periodo corrispondente. La Tabella 5 può essere utilizzata per determinare il dosaggio durante episodi emorragici e interventi chirurgici.
Tabella 5
| Grado di emorragia/ tipo di procedura chirurgica |
Livello richiesto di fattore VIII (%) (UI/dl) |
Frequenza di somministrazione (ore)/ durata della terapia (giorni) |
| Emorragia Emartrosi precoce, emorragia muscolare o orale |
20–40 |
Ripetere ogni 12 – 24 ore per almeno 1 giorno finché l’emorragia accompagnata da dolore non si arresta o fino alla completa guarigione |
| Emartrosi più estesa, emorragia muscolare o ematoma |
30–60 |
Ripetere ogni 12 – 24 ore per 3 – 4 giorni o più, finché il dolore e le alterazioni acute non scompaiono |
| Emorragia potenzialmente letale |
60–100 |
Ripetere le iniezioni ogni 8 – 24 ore finché il pericolo non scompare |
| Chirurgia Intervento minore, compresa l’estrazione dentale Intervento maggiore |
30–60 |
Ogni 24 ore per almeno 1 giorno fino alla completa guarigione |
| 80–100 (prima e dopo l’intervento chirurgico) |
Ripetere le iniezioni ogni 8 – 24 ore fino alla significativa cicatrizzazione della ferita; successivamente continuare la terapia per almeno 7 giorni per mantenere l’attività del fattore VIII tra il 30% e il 60% (UI/dl) |
Prevenzione
Per la profilassi a lungo termine dell'emorragia in pazienti con emofilia A grave, la dose abituale è generalmente compresa tra 20 e 40 UI di fattore VIII per kg di peso corporeo ogni 2-3 giorni. In alcuni casi, specialmente nei pazienti più giovani, può essere necessario aumentare la dose o la frequenza di somministrazione del medicinale.
Durante il trattamento si raccomanda di monitorare i livelli di fattore VIII al fine di aggiustare la dose e la frequenza delle infusioni ripetute. In caso di intervento chirurgico maggiore, in particolare, è necessario un controllo preciso della terapia sostitutiva mediante la determinazione dell'attività del fattore VIII nel plasma. A seconda del fattore VIII, singoli pazienti possono presentare differenti emivite e tempi di recupero. Il regime posologico può essere aggiustato in base alla risposta del paziente.
La via di somministrazione è la stessa sia per adulti che per bambini e adolescenti; tuttavia, nei bambini e negli adolescenti potrebbero essere necessari intervalli più brevi tra le somministrazioni o dosi più elevate. Non sono disponibili dati sull'uso in bambini di età inferiore a 2 anni.
Modalità di somministrazione
Nuwiq® è destinato alla somministrazione endovenosa.
Si raccomanda di somministrare non più di 4 ml al minuto.
Istruzioni per la ricostituzione del medicinale prima della somministrazione.
Il prodotto in polvere deve essere ricostituito esclusivamente con il solvente fornito (2,5 ml di acqua per preparazioni iniettabili), utilizzando il set per la ricostituzione della soluzione iniettabile. Il flaconcino deve essere agitato delicatamente fino a completo scioglimento del contenuto. Dopo la ricostituzione, la soluzione deve essere aspirata nella siringa che precedentemente conteneva il solvente.
La soluzione ricostituita deve essere ispezionata visivamente per verificare la presenza di particelle insolubili e di eventuali alterazioni del colore prima della somministrazione. La soluzione ricostituita deve essere limpida, incolore, priva di particelle estranee e avere un pH compreso tra 6,5 e 7,5. Non devono essere utilizzate soluzioni torbide o contenenti sedimenti.
Istruzioni per la preparazione e la somministrazione del medicinale.
|
Fig. 1 |
|
Fig. 2 |
|
Fig. 3 |
|
Fig. 4 |
|
Fig. 5 |
|
Fig. 6 |
|
Fig. 7 |
|
Fig. 8 |
|
|
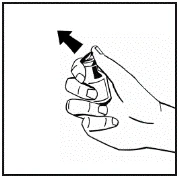
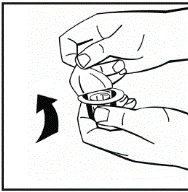
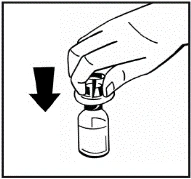
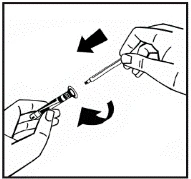
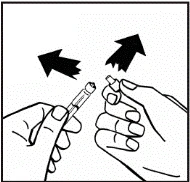
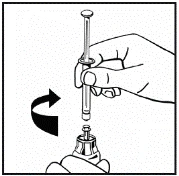
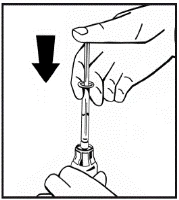
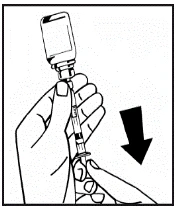
Se per una singola procedura si utilizzano più flaconcini di polvere, è possibile riutilizzare lo stesso ago per l'iniezione. L'adattatore per flaconcino e la siringa sono destinati a un solo utilizzo.
Il medicinale non utilizzato e i rifiuti devono essere smaltiti in conformità con i requisiti locali.
Bambini.
Nuwiq® può essere utilizzato in bambini di qualsiasi età, ma non sono disponibili dati sull'uso in bambini di età inferiore a 2 anni.
Sovradosaggio.
Non sono stati riportati casi di sovradosaggio.
Effetti indesiderati
Riassunto del profilo di sicurezza
Reazioni di ipersensibilità e allergiche (che possono includere angioedema, sensazione di calore e formicolio nel sito di infusione, brividi, iperemia, orticaria generalizzata, cefalea, orticaria, ipotensione arteriosa, sonnolenza, nausea, eruzioni cutanee, eccitazione, tachicardia, difficoltà respiratorie, formicolio, orticaria, orticaria generalizzata, vomito, raucedine/sibilanza) sono state osservate raramente con i preparati del fattore FVIII, ma in alcuni casi possono progredire verso una reazione anafilattica grave (incluso shock).
Nei pazienti con emofilia A trattati con fattore VIII o con il medicinale Nuwiq®, possono svilupparsi anticorpi neutralizzanti (inibitori) nei confronti del fattore VIII. In caso di comparsa di inibitori, la mancata risposta clinica, caratterizzata da sanguinamenti persistenti o ricorrenti nonostante un’adeguata profilassi, deve essere considerata un segnale d’allarme. In tali situazioni, si raccomanda di rivolgersi a un centro specializzato per l’emofilia.
Elenco degli effetti indesiderati in forma tabellare
La tabella 6 riportata di seguito è classificata secondo il sistema di organi MedDRA (Medical Dictionary for Regulatory Activities). La frequenza si basa sui dati degli studi clinici condotti su 355 pazienti con emofilia A grave, di cui 247 erano pazienti precedentemente trattati (PTP) e 108 erano pazienti mai trattati in precedenza (PUP).
La frequenza è definita secondo i seguenti criteri: molto comune (≥1/10); comune (≥1/100 a <1/10); non comune (≥1/1.000 a <1/100); raro (≥1/10.000 a <1/1.000); molto raro (<1/10.000); non noto (non può essere stimato sulla base dei dati disponibili).
All’interno di ciascuna categoria di frequenza, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Tabella 6.
| Classe sistemica MedDRA standard |
Reazioni avverse |
Frequenza |
| Alterazioni del sangue e del sistema linfatico |
Anemia Inibizione del fattore VIII Anemia emorragica |
non comune* non comune (PTPs)# molto comune (PUPs)# non comune* |
| Alterazioni del sistema immunitario |
Iper-sensibilità |
comune* |
| Alterazioni del sistema nervoso |
Vertigini Cefalea Pararestesia (disturbo della sensibilità) |
non comune* non comune* non comune* |
| Alterazioni dell'orecchio e del labirinto |
Vertigini |
non comune* |
| Alterazioni del sistema respiratorio, toracico e mediastinico |
Dispnea (difficoltà respiratoria) |
non comune* |
| Alterazioni gastrointestinali |
Secchezza orale |
non comune* |
| Alterazioni del sistema muscoloscheletrico e del tessuto connettivo |
Dolore alla schiena |
non comune* |
| Alterazioni sistemiche e alterazioni nel sito di somministrazione |
Pirosi (febbre) Dolore al petto Infiammazione nel sito di iniezione Dolore nel sito di iniezione Malessere |
comune* non comune* non comune* non comune* non comune* |
| Indagini |
Anticorpi positivi non neutralizzanti (nei PTPs) |
non comune* |
*Calcolato come numero di pazienti con reazioni avverse rispetto al numero totale di 355 pazienti studiati, di cui 247 pazienti precedentemente trattati (PTP) e 108 pazienti precedentemente non trattati (PUP).
La frequenza si basa su studi con tutti i farmaci del fattore VIII che includevano pazienti con emofilia A grave.
PTP = pazienti precedentemente trattati, PUP = pazienti precedentemente non trattati.
Descrizione delle singole reazioni avverse
Anticorpi contro gli antigeni del fattore VIII privi di attività neutralizzante sono stati rilevati in un paziente adulto (vedi tabella 6). Il risultato è risultato positivo solo con il diluente fattore 1 e il titolo degli anticorpi era molto basso. L'attività degli inibitori, misurata con il test di Bethesda modificato, non è stata rilevata in questo paziente. L'efficacia clinica e il recupero in vivo del medicinale Nuwiq® non sono stati influenzati in questo paziente.
Pediatria
Si prevede che la frequenza, il tipo e la gravità delle reazioni avverse nei bambini siano gli stessi degli adulti.
Segnalazione di potenziali reazioni avverse
La segnalazione di potenziali reazioni avverse dopo l'autorizzazione del medicinale è importante. Permette di continuare a monitorare il rapporto rischio/beneficio del farmaco. I professionisti sanitari devono segnalare qualsiasi potenziale reazione avversa.
Periodo di validità
Polvere: 2 anni.
Diluente (acqua per preparazioni iniettabili): 5 anni.
Per l'intero periodo di validità, il medicinale può essere conservato per un massimo di 1 mese a temperatura ambiente (fino a 25 °C). Se il medicinale è stato già tolto dal frigorifero, non deve essere rimesso al suo interno.
È necessario indicare la data di inizio del periodo di conservazione a temperatura ambiente sull'imballaggio del medicinale.
Dopo la ricostituzione, la stabilità chimica e fisica della soluzione iniettabile è stata dimostrata per 24 ore con conservazione a temperatura ambiente.
Dal punto di vista microbiologico, il medicinale deve essere utilizzato immediatamente dopo la ricostituzione. Se non viene utilizzato immediatamente, l'utilizzatore del medicinale è personalmente responsabile del periodo di validità e delle condizioni di utilizzo del prodotto.
Condizioni di conservazione.
Polvere
Conservare a una temperatura compresa tra 2 e 8 °C. Non congelare.
Conservare nella confezione di cartone per proteggere dalla luce.
Diluente (acqua per preparazioni iniettabili)
Conservare a una temperatura compresa tra 2 e 8 °C.
La soluzione ricostituita deve essere conservata a temperatura ambiente.
Non congelare la soluzione ricostituita.
Conservare fuori dalla portata dei bambini.
Incompatibilità.
A causa della mancanza di studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali.
Devono essere utilizzati esclusivamente i set per iniezione forniti, poiché il trattamento potrebbe risultare inefficace a causa dell'adsorbimento del fattore VIII della coagulazione del sangue sulle superfici interne di alcuni dispositivi per iniezione.
Confezione. 1500 UI, 2500 UI, 3000 UI o 4000 UI di polvere in un flaconcino; 1 flaconcino di polvere, 1 siringa preriempita con 2,5 ml di diluente (acqua per preparazioni iniettabili), insieme a un kit per ricostituzione e somministrazione endovenosa (1 adattatore per flaconcino, 1 ago farfalla, 2 tamponi alcolici) in una confezione di cartone.
Categoria di prescrizione.
Sotto prescrizione medica.
Produttore.
Octapharma AB.
Sede del produttore e indirizzo del luogo di attività.
Lars Forssells gata 23, Stoccolma, 11275, Svezia.