Novoeight®
UcrainaIndice
ISTRUZIONE PER L'USO MEDICO DEL MEDICINALE Novoeight® (NovoEight®)
Composizione:
sostanza attiva: turoctocog alfa;
1 flaconcino con polvere contiene 1500 UI o 2000 UI o 3000 UI di turoctocog alfa (fattore della coagulazione umano VIII (rDNA));
sostanze ausiliarie: cloruro di sodio; L-istidina; saccarosio; polisorbato 80; L-metionina; cloruro di calcio diidrato; idrossido di sodio; acido cloridrico.
Solvente: cloruro di sodio, acqua per preparazioni iniettabili.
Dopo ricostituzione, 1 ml del medicinale Novoeight® contiene circa 375 UI, 500 UI o 750 UI di turoctocog alfa (fattore della coagulazione umano VIII (rDNA)).
Forma farmaceutica. Polvere e solvente per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: polvere liofilizzata o massa friabile di colore bianco o leggermente giallastro. Solvente: soluzione iniettabile limpida e incolore.
Gruppo farmacoterapeutico. Agenti emostatici. Fattori della coagulazione. Fattore della coagulazione VIII.
Codice ATC B02B D02.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Il medicinale Novoeight® contiene turcoctocog alfa, un fattore della coagulazione VIII umano (rDNA) con dominio B troncato. Questa glicoproteina ha la stessa struttura del fattore VIII umano in forma attivata e presenta le stesse modifiche post-traduzionali della molecola isolata dal plasma sanguigno. È stato dimostrato che il sito di sulfatazione della tirosina localizzato in Tyr1680 (lunghezza completa del fattore nativo), importante per il legame con il fattore di von Willebrand, è completamente sulfatato nella molecola di turcoctocog alfa. Dopo somministrazione a pazienti con emofilia, il fattore VIII si lega al fattore di von Willebrand endogeno nel sangue dei pazienti. Il complesso fattore VIII/fattore di von Willebrand è costituito da due molecole (fattore VIII e fattore di von Willebrand) con diverse funzioni fisiologiche. Il fattore VIII attivato agisce come cofattore del fattore IX attivato, accelerando la conversione del fattore X in fattore X attivato. Il fattore X attivato converte la protrombina in trombina. La trombina a sua volta converte il fibrinogeno in fibrina, portando così alla formazione del trombo. L'emofilia A è una patologia ereditaria legata al sesso caratterizzata da un deficit del fattore VIII:C, che determina emorragie abbondanti nelle articolazioni, nei muscoli o negli organi interni, sia spontanee che conseguenti a traumi o interventi chirurgici. Grazie alla terapia sostitutiva, i livelli plasmatici di fattore VIII aumentano, determinando una correzione temporanea della carenza e riducendo la tendenza alle emorragie.
È importante notare che la frequenza annuale di emorragie (FAE) a diverse concentrazioni del fattore di coagulazione e nei diversi studi clinici non è comparabile.
Efficacia clinica
Sono stati condotti quattro studi clinici multicentrici, aperti e non controllati, per valutare la sicurezza ed efficacia del medicinale Novoeight® nella profilassi e nel trattamento delle emorragie e durante interventi chirurgici in pazienti con emofilia A grave (attività del fattore VIII ≤ 1%). Tre studi sono stati effettuati su pazienti precedentemente trattati, mentre uno è stato condotto su pazienti mai trattati in precedenza. Gli studi hanno coinvolto complessivamente 298 pazienti: 175 pazienti adulti e adolescenti di età pari o superiore a 12 anni (≥ 150 giorni di trattamento con il medicinale in studio), nei quali non sono stati rilevati inibitori; 63 pazienti pediatrici di età inferiore ai 12 anni (≥ 50 giorni di trattamento con il medicinale in studio), nei quali non sono stati rilevati inibitori; e 60 pazienti di età inferiore ai 6 anni, mai trattati in precedenza. 188 dei 238 pazienti precedentemente trattati hanno proseguito la partecipazione in uno studio aggiuntivo sulla sicurezza. È stato dimostrato che il trattamento con Novoeight® è sicuro e determina l'effetto emostatico e profilattico desiderato. Sono stati registrati 3293 episodi emorragici osservati nei 298 pazienti, dei quali 2902 (88,1%) si sono risolti dopo 1-2 infusioni di Novoeight®.
Tabella 1. Utilizzo del medicinale Novoeight® e parametri di efficacia nei pazienti mai trattati in precedenza e in quelli già trattati.
| Indicatore |
Bambini più giovani (da 0 a meno di 6 anni), mai trattati |
Bambini più giovani (da 0 a meno di 6 anni), già trattati |
Bambini più grandi (da 6 a meno di 12 anni), già trattati |
Adolescenti (da 12 a meno di 18 anni), già trattati |
Adulti (≥ 18 anni), già trattati |
Totale |
| Numero di pazienti |
60 |
31 |
32 |
24 |
151 |
298 |
| Dose utilizzata per la profilassi, per paziente (UI/kg PM) |
||||||
| Valore medio (DS) |
45,2 (14,4) |
41,5 (8,1) |
38,4 (9,4) |
28,5 (9,3) |
28,5 (8,3) |
32,8 (10,9) |
| Minimo; massimo |
4,5; 363,8 |
3,4; 196,3 |
3,2; 62,5 |
17,4; 73,9 |
12,0; 97,4 |
3,2; 363,8 |
| Dose utilizzata per il trattamento dell’emorragia (UI/kg PM) |
||||||
| Valore medio (DS) |
43,6 (15,2) |
44,0 (12,6) |
40,4 (10,5) |
29,3 (10,3) |
35,0 (12,3) |
37,5 (13,4) |
| Minimo; massimo |
11,9; 118,9 |
21,4; 193,8 |
24,0; 71,4 |
12,4; 76,8 |
6,4; 104,0 |
6,4; 193,8 |
| Indicatore di efficaciaa, % |
87,0 % |
92,2 % |
88,4 % |
85,1 % |
89,6 % |
88,9 % |
MT − massa corporea, SV − scarto quadratico medio.
a L'efficacia è definita come eccellente o buona.
I dati clinici ottenuti prima della registrazione sono stati confermati da uno studio di sicurezza post-marketing non interventistico, condotto allo scopo di fornire ulteriori informazioni sull'immunogenicità, efficacia e sicurezza del medicinale Novoeight® nella pratica clinica abituale. In totale, 68 pazienti già trattati (> 150 UI (unità efficaci)), di cui 14 pazienti di età inferiore a 12 anni e 54 pazienti di età pari o superiore a 12 anni, sono stati trattati su richiesta (N = 5) o con trattamento profilattico (N = 63) per un totale di 87,8 paziente-anni e 8967 UI.
Intervento chirurgico
Sono stati effettuati complessivamente 30 interventi chirurgici su 25 pazienti, di cui 26 interventi chirurgici maggiori e 4 minori. L'emostasi è stata efficace in tutti i casi di intervento chirurgico; non sono stati riportati casi di inefficacia del trattamento.
Sono stati raccolti dati riguardo all'induzione della tolleranza immunitaria (ITI) in pazienti con emofilia A nei quali sono stati riscontrati inibitori del fattore VIII. Durante gli studi clinici che hanno coinvolto pazienti precedentemente non trattati (in inglese: previously untreated patients (PUPs)), 21 pazienti hanno ricevuto trattamento ITI e 18 pazienti (86 %) hanno completato la terapia ITI con esito negativo del test per la ricerca di inibitori.
Farmacocinetica.
Tutti gli studi farmacocinetici con turcoctocog alfa sono stati condotti dopo somministrazione endovenosa di una dose di 50 UI/kg del medicinale Novoeight®, su pazienti con emofilia A grave (FVIII ≤ 1 %), già precedentemente trattati. L'analisi dei campioni di plasma è stata effettuata mediante saggio unistadio della attività coagulante e mediante saggio cromogeno.
L'attività del medicinale Novoeight® nel saggio FVIII:C è stata valutata e confrontata con un prodotto ricombinante di FVIII a lunghezza intera disponibile in commercio. Lo studio ha dimostrato che i risultati ottenuti sono comparabili e coerenti per entrambi i prodotti e che il medicinale Novoeight® può essere determinato in modo affidabile nel plasma senza la necessità di uno standard specifico per Novoeight®.
I parametri farmacocinetici del medicinale Novoeight® dopo somministrazione di una dose singola, determinati mediante analisi dell'attività coagulante del sangue, sono riportati nella Tabella 2, mentre quelli determinati mediante analisi cromogeno unistadio sono riportati nella Tabella 3.
Tabella 2. Farmacocinetica del turcoctocog alfa (50 UI/kg) in funzione dell'età – Unistadio, analisi dell'attività coagulante del sangue – Valore medio (scarto quadratico medio)
| Indicatore |
Da 0 a 6 anni |
Da 6 a 12 anni |
≥ 12 anni |
| n = 14 |
n = 14 |
n = 33 |
|
| Valore medio (DS) |
Valore medio (DS) |
Valore medio (DS) |
|
| Aumento del livello (MO/dl)/(MO/kg) |
1,8 (0,7) |
2,0 (0,4) |
2,2 (0,4) |
| AUC ((MO*ora)/dl) |
992 (411) |
1109 (374) |
1526 (577) |
| CL (ml/ora/kg) |
6,21 (3,66) |
5,02 (1,68) |
3,63 (1,09) |
| t½ (ore) |
7,65 (1,84) |
8,02 (1,89) |
11,00 (4,65) |
| Vss (ml/kg) |
56,68 (26,43) |
46,82 (10,63) |
47,40 (9,21) |
| Cmax (MO/dl) |
100 (58) |
107 (35) |
123 (41) |
| Durata media d'azione (ore) |
9,63 (2,50) |
9,91 (2,57) |
14,19 (5,08) |
AUC – area sotto la curva farmacocinetica, che descrive la dinamica temporale dell'attività del fattore VIII; CL – clearance; t1/2 – emivita terminale; Vss – volume di distribuzione allo stato stazionario; Cmax – attività massima del fattore VIII.
Tabella 3. Farmacocinetica di turcoctocog alfa (50 UI/kg) in base all'età – Saggio cromogenico – Valore medio (deviazione standard)
| Indicatore |
Da 0 a < 6 anni |
Da 6 a < 12 anni |
≥ 12 anni |
| n = 14 |
n = 14 |
n = 33 |
|
| Valore medio (DS) |
Valore medio (DS) |
Valore medio (DS) |
|
| Incremento del livello (MO/dl)/(MO/kg) |
2,2 (0,6) |
2,5 (0,6) |
2,9 (0,6) |
| AUC ((MO*ore)/dl) |
1223 (436) |
1437 (348) |
1963 (773) |
| CL (ml/ora/kg) |
4,59 (1,73) |
3,70 (1,00) |
2,86 (0,94) |
| t½ (ore) |
9,99 (1,71) |
9,42 (1,52) |
11,22 (6,86) |
| Vss (ml/kg) |
55,46 (23,53) |
41,23 (6,00) |
38,18 (10,24) |
| Cmax (MO/dl) |
112 (31) |
125 (27) |
163 (50) |
| Durata media dell'effetto (ore) |
12,06 (1,90) |
11,61 (2,32) |
14,54 (5,77) |
AUC – area sotto la curva farmacocinetica, che descrive la dinamica temporale dell'attività del fattore VIII; CL – clearance; t1/2 – emivita terminale; Vss – volume di distribuzione allo stato stazionario; Cmax – attività massima del fattore VIII.
I parametri farmacocinetici sono risultati comparabili nei pazienti di età inferiore ai 6 anni e tra i 6 e i 12 anni. Sono state osservate alcune differenze nei parametri farmacocinetici del medicinale Novoeight® nei bambini rispetto agli adulti affetti da emofilia A. Un clearance più elevato e un t½ più breve nei bambini rispetto agli adulti possono essere parzialmente spiegati dal maggiore volume di plasma per chilogrammo di peso corporeo nei pazienti più giovani.
È stato condotto uno studio sulla farmacocinetica dopo somministrazione singola di dose (50 UI/kg) su 35 pazienti emofilici (≥ 18 anni) appartenenti a diverse categorie di IMC. La concentrazione massima del farmaco nel plasma (Cmax) e la concentrazione totale del farmaco nel plasma (AUC) aumentano con l'aumentare dell'IMC, indicando che potrebbe essere necessaria una correzione della dose per i pazienti con peso corporeo insufficiente (IMC < 18,5 kg/m²) e per quelli con eccesso di peso (IMC ≥ 30 kg/m²); si prega di consultare la sezione «Modalità di somministrazione e dosi».
Tabella 4. Parametri farmacocinetici del medicinale Novoeight® dopo somministrazione di una dose singola (50 UI/kg) in base alla classe di IMCa – Analisi a stadio unico della coagulazione del sangue – Valore medio (deviazione standard)
| Indicatore farmacocinetico |
Insufficienza ponderale, N = 5 |
Peso corporeo normale, N = 7 |
Eccesso ponderale, N = 8 |
Obesità, classe I, N = 7 |
Obesità, classe II/III, N = 7 |
| Aumento del livello (MO/dl)/(MO/kg) |
1,7 (0,2) |
2,0 (0,2) |
2,4 (0,4) |
2,3 (0,3)b |
2,6 (0,3) |
| AUC ((MO*ore)/dl) |
1510 (360) |
1920 (610) |
1730 (610) |
2030 (840) |
2350 (590) |
| CL (ml/ora/kg) |
3,91 (0,94) |
3,20 (1,00) |
3,63 (1,24) |
3,37 (1,79) |
2,51 (0,63) |
| t½ (ore) |
11,3 (2,0) |
11,7 (3,5) |
9,4 (2,9) |
11,2 (3,5) |
11,1 (2,7) |
| Vss (ml/kg) |
56,8 (5,4) |
44,8 (6,5) |
39,6 (6,0) |
42,0 (9,0) |
35,0 (4,6) |
| Cmax (MO/dl) |
100 (11) |
121 (10) |
144 (26) |
140 (21) |
161 (32) |
| Durata media dell'effetto (ore) |
15,1 (3,0) |
15,3 (4,8) |
11,9 (3,7) |
14,4 (4,6) |
14,6 (3,7) |
a Classi di IMC: sottopeso, IMC < 18,5 kg/m²; peso normale, IMC 18,5–24,9 kg/m²; sovrappeso, IMC 25–29,9 kg/m²; obesità, classe I, IMC 30–34,9 kg/m²; obesità, classe II/III, IMC ≥ 35 kg/m².
b Dati disponibili solo per 6 pazienti.
Tabella 5. Parametri farmacocinetici del farmaco Novoeight® dopo somministrazione di una dose singola (50 UI/kg) in base alla classe di IMCa – Saggio cromogeno – Media (deviazione standard)
| Parametro farmacocinetico |
Massa corporea insufficiente, N = 5 |
Massa corporea normale, N = 7 |
Massa corporea eccessiva, N = 9 |
Obesità, classe I, N = 7 |
Obesità, classe II/III, N = 7 |
| Incremento del livello (MO/dl)/(MO/kg) |
2,2 (0,4) |
2,9 (0,3) |
3,0 (0,5) |
3,2 (0,5) |
3,5 (0,5) |
| AUC ((MO*ore)/dl) |
1860 (700) |
2730 (860) |
2310 (1020) |
2780 (1210) |
3050 (730) |
| CL (ml/ora/kg) |
3,28 (0,87) |
2,25 (0,73) |
2,84 (1,09) |
2,58 (1,56) |
1,94 (0,52) |
| t½ (ore) |
11,7 (2,4) |
11,5 (3,6) |
9,7 (3,4) |
10,4 (3,2) |
10,5 (2,5) |
| Vss (ml/kg) |
49,1 (10,4) |
31,2 (4,5) |
31,6 (5,8) |
28,9 (5,1) |
25,7 (4,0) |
| Cmax (MO/dl) |
138 (29) |
185 (24) |
194 (31) |
200 (33) |
227 (32) |
| Durata media dell'effetto (ore) |
15,5 (3,2) |
15,2 (4,9) |
12,6 (4,8) |
13,5 (4,6) |
13,9 (3,7) |
a Gruppi di IMC: massa corporea insufficiente, IMC < 18,5 kg/m²; massa corporea normale, IMC 18,5–24,9 kg/m²; massa corporea in eccesso, IMC 25–29,9 kg/m²; obesità, classe I, IMC 30–34,9 kg/m²; obesità, classe II/III, IMC ≥ 35 kg/m².
Dati preclinici di sicurezza
I dati preclinici indicano l'assenza di rischi particolari per l'uomo sulla base dei risultati degli studi standard di tossicologia di sicurezza e degli studi di tossicità da dosi ripetute.
Caratteristiche cliniche.
Indicazioni.
Trattamento e profilassi delle emorragie nei pazienti con emofilia A (deficit congenito del fattore VIII).
Il medicinale Novoeight® può essere utilizzato in pazienti di tutte le fasce d'età.
Controindicazioni.
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Reazione allergica nota alle proteine del criceto.
Interazioni con altri medicinali ed altre forme di interazione.
Non sono state riportate interazioni tra fattore della coagulazione VIII umano (rDNA) e altri medicinali.
Caratteristiche particolari di utilizzo.
Tracciabilità
Per migliorare il monitoraggio dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto del prodotto utilizzato.
Ipersensibilità
Nell’uso del medicinale Novoeight® possono verificarsi reazioni di ipersensibilità di tipo allergico. Il prodotto contiene tracce di proteine di criceto, che in alcuni pazienti possono causare reazioni allergiche. Se compaiono sintomi di ipersensibilità, si deve raccomandare ai pazienti di interrompere immediatamente l’uso del medicinale e di consultare un medico. I pazienti devono essere informati sui segni precoci di reazioni di ipersensibilità, tra cui eruzioni cutanee, orticaria generalizzata, sensazione di costrizione al torace, respiro sibilante, abbassamento della pressione sanguigna e anafilassi.
In caso di shock, si deve iniziare un trattamento farmacologico standard anti-shock.
Inibitori
La formazione di anticorpi neutralizzanti (inibitori) contro il fattore VIII è una complicanza nota nel trattamento di persone con emofilia A. Tali inibitori sono generalmente immunoglobuline IgG che interferiscono con l’attività procoagulante del fattore VIII; quantitativamente, vengono misurati in unità Bethesda (UB) per 1 ml di plasma mediante analisi modificato. Il rischio di formazione di inibitori è correlato all’esposizione al fattore VIII ed è massimo nei primi 50 giorni di esposizione, ma permane per tutta la vita del paziente, anche se tale rischio è raro.
L’importanza clinica della formazione di inibitori dipende dal loro titolo: un titolo basso comporta un rischio inferiore di risposta clinica inadeguata rispetto a un titolo elevato di inibitori.
In generale, tutti i pazienti in trattamento con medicinali contenenti fattore VIII della coagulazione devono essere attentamente monitorati per la comparsa di inibitori, attraverso un’appropriata osservazione clinica e analisi di laboratorio. Se non si raggiunge il livello atteso di attività del fattore VIII nel plasma o se non si riesce a controllare l’emorragia con un’appropriata dose, si deve effettuare un test per la ricerca di inibitori del fattore VIII. Nei pazienti con livelli elevati di inibitori, la terapia con fattore VIII può risultare inefficace e si devono considerare altre modalità di trattamento. La terapia di tali pazienti deve essere gestita sotto la supervisione di medici esperti nel trattamento di pazienti con emofilia e presenza di inibitori del fattore VIII.
Avvertenze relative agli eccipienti
Il medicinale contiene 30,5 mg di sodio per flaconcino con prodotto ricostituito, pari al 1,5% del consumo giornaliero massimo raccomandato di sodio dall’OMS per un adulto, pari a 2 g.
Complicazioni cardiovascolari
Nei pazienti con fattori di rischio cardiovascolari preesistenti, la terapia sostitutiva con fattore VIII può aumentare il rischio di complicazioni cardiovascolari.
Complicazioni legate al catetere
Se necessario l’inserimento di un catetere venoso centrale, si devono considerare i rischi di complicazioni legate al catetere, inclusi infezioni locali, batteriemia e trombosi nel sito di inserimento.
Al fine di mantenere un collegamento tra lo stato del paziente e il lotto del medicinale, si raccomanda vivamente di registrare il nome e il numero di lotto del prodotto ad ogni somministrazione di Novoeight® al paziente.
Bambini
Le avvertenze e le precauzioni sopra riportate si applicano sia agli adulti che ai bambini.
Uso durante la gravidanza o l’allattamento.
Non sono stati condotti studi sugli effetti di Novoeight® sulla funzione riproduttiva negli animali. Poiché l’emofilia A è rara nelle donne, non esiste esperienza clinica sull’uso del fattore VIII durante la gravidanza e l’allattamento. Pertanto, il fattore VIII deve essere somministrato alle donne in gravidanza e durante l’allattamento solo in caso di chiare indicazioni.
Capacità di guidare veicoli o di usare macchinari.
Il medicinale Novoeight® non altera la capacità di guidare veicoli né di usare macchinari.
Modalità e dosaggio di somministrazione
Il trattamento deve essere iniziato sotto la supervisione di un medico esperto nel trattamento dell'emofilia.
Monitoraggio del trattamento
Durante il corso del trattamento si raccomanda un'adeguata determinazione dei livelli del fattore VIII al fine di aggiustare la dose e la frequenza delle somministrazioni ripetute. I pazienti possono presentare risposte diverse al fattore VIII, nonché differenti emivite e tempi di recupero. La dose calcolata in base al peso corporeo potrebbe richiedere un aggiustamento nei pazienti con peso corporeo insufficiente o in eccesso. Studi di farmacocinetica del medicinale dopo somministrazione di dose singola ad adulti hanno dimostrato che la concentrazione massima del medicinale nel plasma (Cmax) e l'area sotto la curva concentrazione-tempo (AUC) aumentano all'aumentare dell'IMC. Ciò suggerisce che potrebbe essere necessario un aggiustamento della dose. I pazienti con peso corporeo insufficiente (IMC < 18,5 kg/m²) potrebbero richiedere un aumento della dose, mentre i pazienti con obesità (IMC ≥ 30 kg/m²) potrebbero richiedere una riduzione della dose. Tuttavia, attualmente non ci sono dati sufficienti per fornire raccomandazioni specifiche sull'aggiustamento della dose; si veda il paragrafo «Farmacocinetica».
In particolare, in caso di interventi chirurgici importanti è obbligatorio un accurato monitoraggio della terapia sostitutiva mediante analisi della coagulazione (attività del fattore VIII nel plasma).
Nel caso dell'analisi in un solo stadio della coagulazione basata sul tempo di tromboplastina parziale attivato in vitro (aPTT), sia il tipo di reagente aPTT utilizzato sia il preparato di riferimento impiegato nell'analisi possono influenzare in modo significativo i risultati della determinazione dell'attività del fattore VIII nel plasma. Inoltre, possono verificarsi notevoli discrepanze tra i risultati ottenuti con l'analisi in un solo stadio basata sull'aPTT e quelli ottenuti con l'analisi cromogenica secondo la Farmacopea Europea. Ciò è particolarmente importante in caso di cambio di laboratorio e/o di reagenti utilizzati per l'analisi.
Dosaggio
Il dosaggio e la durata della terapia sostitutiva dipendono dal grado di carenza del fattore VIII, dalla localizzazione e dalla gravità dell'emorragia, nonché dallo stato clinico del paziente.
La quantità di unità del fattore VIII è espressa in unità internazionali (UI), in riferimento allo standard vigente dell'OMS per i preparati del fattore VIII. L'attività del fattore VIII nel plasma è espressa in percentuale (rispetto al livello normale nel plasma umano) oppure in unità internazionali (rispetto allo standard internazionale per il fattore VIII nel plasma).
Un'unità internazionale (UI) di attività del fattore VIII equivale alla quantità di fattore VIII presente in 1 ml di plasma di un soggetto normale.
Trattamento su richiesta
Il calcolo della dose necessaria di fattore VIII si basa su un dato empirico secondo cui 1 unità internazionale (UI) di fattore VIII per kg di peso corporeo aumenta l'attività del fattore VIII nel plasma di 2 UI/dl. La dose necessaria può essere calcolata con la seguente formula:
Quantità necessaria di unità = peso corporeo (kg) × incremento desiderato del livello di fattore VIII (%) (UI/dl) × 0,5 (UI/kg per 1 UI/dl).
La quantità da somministrare e la frequenza di somministrazione devono sempre essere determinate caso per caso, in base all'efficacia clinica osservata.
In caso di manifestazioni emorragiche come quelle indicate di seguito, l'attività del fattore VIII non deve scendere al di sotto dei livelli indicati nel plasma (in % rispetto alla norma o in UI/dl) per il periodo corrispondente. La Tabella 6 può essere utilizzata come guida per il dosaggio in caso di emorragie e interventi chirurgici.
Tabella 6. Guida al dosaggio in caso di emorragie e interventi chirurgici
| Gravità dell’emorragia/tipo di procedura chirurgica |
Livello necessario di fattore VIII (%) (UI/ml) |
Frequenza di somministrazione (ore)/durata della terapia (giorni) |
| Emorragia |
||
| Primi segni di emartro, ematoma intramuscolare o emorragia orale |
20−40 |
Ripetere ogni 12−24 ore almeno per 1 giorno fino alla cessazione dell’emorragia, determinata dall’assenza di dolore o fino alla guarigione |
| Emartro più grave, ematoma intramuscolare o ematoma |
30−60 |
Ripetere l’infusione ogni 12−24 ore per 3−4 giorni o più a lungo, fino alla scomparsa del dolore e al ripristino della funzionalità |
| Emorragie potenzialmente letali |
60−100 |
Ripetere l’infusione ogni 8−24 ore fino alla scomparsa del pericolo per la vita |
| Interventi chirurgici |
||
| Interventi chirurgici minori, inclusa l’estrazione dentale |
30−60 |
Ogni 24 ore almeno per 1 giorno, fino alla guarigione |
| Interventi chirurgici maggiori |
80−100 (prima e dopo l’intervento) |
Ripetere l’infusione ogni 8−24 ore fino a un adeguato rimarginamento della ferita; successivamente continuare la terapia per almeno ulteriori 7 giorni al fine di mantenere l’attività del fattore VIII tra il 30 e il 60% (UI/ml) |
Prevenzione
Per la profilassi a lungo termine delle emorragie nei pazienti con emofilia A grave, la dose raccomandata abituale è di 20-40 UI di fattore VIII per kg di peso corporeo ogni due giorni oppure di 20-50 UI di fattore VIII per kg di peso corporeo tre volte alla settimana. Negli adulti e negli adolescenti (di età superiore ai 12 anni) può essere prevista una somministrazione meno frequente del medicinale (40-60 UI/kg ogni terzo giorno oppure due volte alla settimana). Talvolta, specialmente nei pazienti più giovani, potrebbero essere necessari intervalli più brevi tra le somministrazioni o dosi più elevate.
Intervento chirurgico
L'esperienza sull'uso del medicinale nei bambini in caso di interventi chirurgici è limitata.
Anziani
Non esiste esperienza sull'uso del medicinale nei pazienti di età superiore ai 65 anni.
Bambini
Per la profilassi a lungo termine delle emorragie nei pazienti di età inferiore ai 12 anni, si raccomandano dosi di 25-50 UI di fattore VIII per kg di peso corporeo ogni due giorni oppure di 25-60 UI di fattore VIII per kg di peso corporeo tre volte alla settimana. Le raccomandazioni sul dosaggio per i bambini di età pari o superiore ai 12 anni sono le stesse previste per i pazienti adulti.
Modalità di somministrazione
Somministrazione endovenosa.
La velocità raccomandata di infusione del medicinale Novoeight® è di 1-2 ml/min. La velocità deve essere determinata in base al livello di comfort del paziente.
Le istruzioni per la ricostituzione del medicinale prima della somministrazione sono riportate nelle Istruzioni per l'uso del medicinale Novoeight®.
Conservazione dopo ricostituzione
È stata dimostrata la stabilità chimica e fisica del medicinale ricostituito per 24 ore a una temperatura di 2-8 °C; per 4 ore a una temperatura non superiore a 30 °C, a condizione che il medicinale sia stato conservato per non più di 9 mesi a temperatura ambiente non superiore a 30 °C dal momento della produzione fino all'inizio della ricostituzione. Conservare per 4 ore a una temperatura non superiore a 40 °C, a condizione che il medicinale sia stato conservato per non più di 3 mesi a temperatura ambiente di 30-40 °C dal momento della produzione fino all'inizio della ricostituzione. Dal punto di vista microbiologico, il medicinale ricostituito deve essere utilizzato immediatamente. Se il medicinale non viene utilizzato immediatamente, l'utente è responsabile del periodo e delle condizioni di conservazione. In caso di ricostituzione effettuata in condizioni asettiche controllate e validate, il periodo di conservazione del medicinale ricostituito non deve superare il tempo indicato sopra.
Qualsiasi medicinale ricostituito non utilizzato e conservato per più di 4 ore a temperatura ambiente fino a 40 °C deve essere eliminato.
Istruzioni per l'uso del medicinale Novoeight®
LEGGERE ATTENTAMENTE QUESTE ISTRUZIONI PRIMA DELL'USO DEL MEDICINALE NOVOEIGHT®.
Il medicinale Novoeight® è fornito sotto forma di polvere. Prima dell'iniezione (somministrazione) deve essere ricostituito con il solvente fornito nella siringa. Il solvente è una soluzione di sodio cloruro 0,9% (9 mg/ml). Il medicinale Novoeight® viene somministrato per via endovenosa (iniezione endovenosa). Il contenuto della confezione (vedi sotto) è destinato alla ricostituzione e alla somministrazione del medicinale Novoeight®.
Per la somministrazione del medicinale sono inoltre necessari un set per infusione (tubi e ago a farfalla), tamponi sterili imbevuti di alcol, garze e cerotti. Questi dispositivi e materiali non sono inclusi nella confezione del medicinale Novoeight®.
Non utilizzare l'apparecchiatura senza un'adeguata formazione da parte del medico o dell'infermiere.
Prima dell'uso lavarsi sempre le mani e assicurarsi che lo spazio circostante sia pulito.
Durante la preparazione e la somministrazione del medicinale direttamente in vena è molto importante rispettare le norme di asepsi e antisepsi. Un'iniezione eseguita con tecnica inadeguata può provocare un'infezione del sangue.
Non aprire l'apparecchiatura finché non si è pronti a utilizzarla.
Non utilizzare l'apparecchiatura se è caduta o danneggiata. In tal caso, utilizzare una nuova confezione.
Non utilizzare l'apparecchiatura se è scaduta. In tal caso, utilizzare una nuova confezione. La data di scadenza è riportata dopo le parole "Scad." sulla scatola di cartone, sul flacone, sul connettore per flacone e sulla siringa pre-riempita.
Non utilizzare l'apparecchiatura se si sospetta che sia contaminata. In tal caso, utilizzare una nuova confezione.
Non gettare nessuna parte del kit finché non si è somministrata la soluzione preparata.
L'apparecchiatura è destinata a un uso singolo.
Contenuto della confezione:
1 flacone con polvere Novoeight®
1 connettore per flacone
1 siringa pre-riempita con solvente
1 stantuffo (posizionato sotto la siringa)
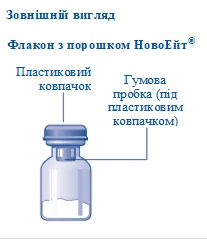
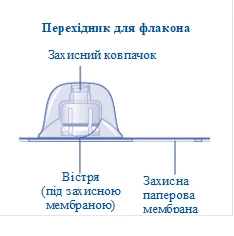
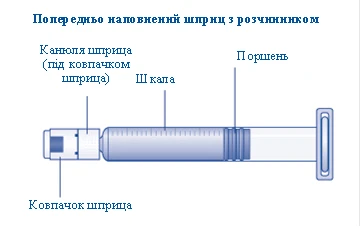

Fig. A
|
|
||
| Fig. B
|
|
||
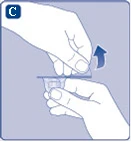
Fig. C
Se l’integrità della membrana protettiva di carta è compromessa o se è danneggiata, non utilizzare il connettore per fiala. Non estrarre il connettore per fiala dalla confezione con le dita. Se si tocca l’estremità del connettore, ciò potrebbe causare il trasferimento di microrganismi dalle dita. |
|
||
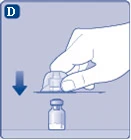
| Fig. D
Dopo l’applicazione, non rimuovere il connettore dalla fiala. |
|
||
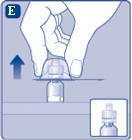
| Fig. E
Rimuovere il tappo protettivo dal connettore per fiala. Non rimuovere il connettore per fiala dalla fiala quando si rimuove il tappo protettivo. |
|
||
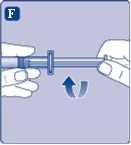
Fig. F
|
|
||
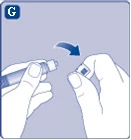
| Fig. G
Se il tappo della siringa è allentato o mancante, non utilizzare questa siringa pre-riempita. |
|
||
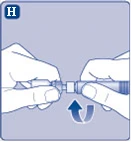
| Fig. H
|
|
||
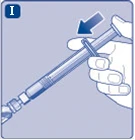
Fig. I
|
|
||
| Fig. J
Non agitare la fiala, poiché ciò potrebbe causare la formazione di schiuma.
|
Se la dose richiesta è maggiore di quella contenuta in una singola fiala, ripetere i passaggi da A a J con fiale aggiuntive, connettori per fiala e siringhe pre-riempite, fino a ottenere la dose necessaria. |
||
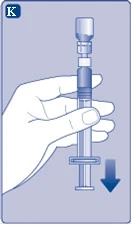
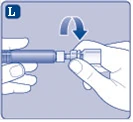
| Fig. K
Se accidentalmente entra aria nella siringa, espellerla nuovamente nella fiala.
|
|
||
| Fig. L
|
|
||
Il medicinale Novoeight® è ora pronto per la somministrazione endovenosa.
Iniezione del medicinale Novoeight® attraverso adattatori senza ago per cateteri endovenosi (e.v.) Attenzione! La siringa pre-riempita è in vetro ed è progettata per l’uso con il sistema di connessione standard Luer-Lok. Alcuni adattatori senza ago con ago interno non sono compatibili con questa siringa pre-riempita. Tale incompatibilità potrebbe impedire la somministrazione del farmaco e/o causare danni all’adattatore senza ago. Iniezione della soluzione attraverso un dispositivo di accesso venoso centrale (CVC), come un catetere venoso centrale (CVC) o una camera sottocutanea:
|
|||
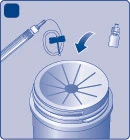
| Smaltimento Fig. M
Non gettare questi materiali nei rifiuti domestici. |
|
||
| Non smontare l'apparecchio prima dello smaltimento. Non riutilizzare l'apparecchio. |
|||

Bambini.
Il medicinale può essere somministrato ai bambini secondo le istruzioni riportate nella sezione «Modalità di somministrazione e posologia».
Overdose.
Non sono stati riportati sintomi di sovradosaggio con il fattore della coagulazione VIII ricombinante.
Effetti indesiderati.
Riassunto del profilo di sicurezza
Sono state osservate reazioni di ipersensibilità o allergiche (che possono includere angioedema, sensazione di calore e formicolio nel sito di infusione, brividi, vampate di calore, orticaria generalizzata, cefalea, eruzioni cutanee, ipotensione arteriosa, letargia, nausea, agitazione, tachicardia, sensazione di costrizione al petto, formicolio, vomito, respiro sibilante), raramente, e in alcuni casi possono evolvere verso una grave anafilassi (incluso shock).
È stato osservato molto raramente lo sviluppo di anticorpi contro le proteine del criceto e le correlate reazioni di ipersensibilità.
Nei pazienti con emofilia A possono svilupparsi anticorpi neutralizzanti (inibitori) contro il fattore VIII. Nel caso in cui si formassero tali inibitori, la risposta clinica sarebbe inadeguata. In tali casi si raccomanda di rivolgersi a un centro specializzato nel trattamento dell'emofilia.
Elenco degli effetti indesiderati
Nella Tabella 7 gli effetti indesiderati sono riportati secondo la classificazione per sistemi e organi (SOC) con l'indicazione dei termini preferenziali del Medical Dictionary for Regulatory Activities (MedDRA).
La frequenza è definita come segue: molto comune (≥ 1/10), comune (≥ 1/100, < 1/10), non comune (≥ 1/1000, < 1/100), raro (≥ 1/10000, < 1/1000), molto raro (< 1/10000); frequenza non nota (non può essere stimata sulla base dei dati disponibili).
All'interno di ciascun gruppo per frequenza, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Tabella 7. Frequenza degli effetti indesiderati del medicinale negli studi clinici
| Classi di sistemi e organi |
Frequenza nei pazienti precedentemente trattati (PTPs) |
Frequenza nei pazienti mai trattati in precedenza (PUPs) |
Reazione avversa |
| Disturbi del sistema emolinfopoietico |
Non comune |
Molto comune |
Inibizione del fattore VIII |
| Disturbi psichiatrici |
Non comune |
Insonnia |
|
| Disturbi del sistema nervoso |
Non comune |
Cefalea, capogiro, sensazione di bruciore |
|
| Disturbi cardiaci |
Non comune |
Tachicardia sinusale, infarto miocardico acuto |
|
| Disturbi vascolari |
Non comune |
Ipertensione arteriosa, linfedema, iperemia |
|
| Comune |
Calori improvvisi, tromboflebite delle vene superficiali |
||
| Disturbi della cute e del tessuto sottocutaneo |
Comune |
Eruzione cutanea, eruzione eritematosa |
|
| Non comune |
Eruzione cutanea, cheratosi lichenoide, sensazione di bruciore della cute |
||
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Non comune |
Rigidità muscolare e articolare, artropatia, dolore agli arti, dolore muscolare e osseo |
|
| Comune |
Emartro, emorragia muscolare |
||
| Disturbi del sistema respiratorio, toracico e mediastinico |
Comune |
Tosse |
|
| Disturbi generali e condizioni in rapporto con il sito di somministrazione |
Comune |
Reazioni nel sito di iniezione |
|
| Comune |
Ipertermia, eritema nel sito di cateterizzazione |
||
| Non comune |
Stanchezza, sensazione di calore, edema periferico, ipertermia |
||
| Anomalie nei test di laboratorio |
Comune |
Aumento dei livelli degli enzimi epaticig |
|
| Comune |
Test positivo per anticorpi contro il fattore VIII |
||
| Non comune |
Aumento della frequenza cardiaca |
||
| Disturbi del sistema gastrointestinale |
Comune |
Vomito |
|
| Lesioni, avvelenamenti e complicanze da procedure |
Comune |
Dosaggio errato |
|
| Comune |
Reazione correlata alla procedura di infusione |
||
| Non comune |
Contusione |
||
| Guasto tecnico |
Comune |
Ostruzione del dispositivo |
a Calcolato sulla base del numero totale di pazienti arruolati in tutti gli studi clinici (301), di cui 242 precedentemente trattati (in inglese: previously treated patients (PTPs)) e 60 mai trattati precedentemente (in inglese: previously untreated patients (PUPs)).
b Le frequenze si basano sui dati degli studi con tutti i farmaci del fattore VIII in pazienti con emofilia A grave.
c Le reazioni nel sito di iniezione comprendono eritema nel sito di iniezione, extravasazione nel sito di iniezione e prurito nel sito di iniezione.
d Gli enzimi epatici con livelli aumentati comprendono alanina aminotransferasi, aspartato aminotransferasi, gamma-glutamil transferasi e bilirubina.
Descrizione delle singole reazioni avverse
Negli studi clinici con Novoeight® in pazienti precedentemente trattati, sono state complessivamente osservate 35 reazioni avverse in 23 su 242 pazienti. Le reazioni avverse più comuni sono state quelle nel sito di iniezione, complicanze legate all’infusione di una dose errata e aumento dei livelli degli enzimi epatici. Due delle 35 reazioni avverse sono state osservate in 1 su 31 pazienti di età inferiore a 6 anni, nessuna in pazienti di età compresa tra 6 e 12 anni, un evento in 1 su 24 pazienti di età compresa tra 12 e 18 anni e 32 eventi in 21 su 155 pazienti adulti (di età pari o superiore a 18 anni).
Bambini
Negli studi clinici condotti in 63 bambini di età compresa tra 0 e 12 anni e in 24 adolescenti di età compresa tra 12 e 18 anni con emofilia A grave, non sono state osservate differenze nel profilo di sicurezza di Novoeight® tra bambini e adulti.
Nello studio condotto in pazienti di età compresa tra 0 e 6 anni mai trattati precedentemente, sono state complessivamente osservate 46 reazioni avverse in 33 su 60 pazienti trattati con Novoeight®. La reazione avversa più comune è stata l’inibizione del fattore VIII (vedere sezione «Informazioni importanti sull’uso»). Nel 92,3% del totale dei pazienti e nel 93,8% dei pazienti con titolo confermato alto di inibitori è stato osservato un alto rischio di mutazioni genetiche. Nessun altro fattore è risultato significativamente associato alla formazione di inibitori.
Segnalazione di reazioni avverse e mancata efficacia del medicinale
La segnalazione delle reazioni avverse dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette di monitorare continuamente il rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare qualsiasi sospetta reazione avversa o mancata efficacia del medicinale attraverso il Sistema informatizzato automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della conservazione.
La durata di conservazione del medicinale finito è di 30 mesi.
Non utilizzare il medicinale dopo la data di scadenza indicata sulla scatola, sul flacone e sull’etichetta della siringa. La data finale di utilizzo corrisponde all’ultimo giorno del mese indicato.
Condizioni di conservazione.
Conservare il medicinale a una temperatura compresa tra 2 e 8 °C. Non congelare. Tenere fuori dalla portata dei bambini. Durante il periodo di validità, il medicinale può essere conservato a temperatura ambiente (non superiore a 30 °C) per un massimo di 9 mesi, dal momento della produzione fino all’inizio della ricostituzione; il medicinale può essere conservato a temperatura ambiente (30–40 °C) per un massimo di 3 mesi, dal momento della produzione fino all’inizio della ricostituzione. Il medicinale non deve essere riportato in frigorifero. Sulla scatola va indicata la data e la temperatura di rimozione del medicinale dal frigorifero.
Conservare nell’imballaggio esterno per proteggerlo dalla luce.
Incompatibilità.
Poiché non sono stati effettuati studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali.
Confezione.
Un flacone di vetro (tipo I) da 5 ml contenente polvere, chiuso con tappo di gomma clorobutilica e sigillato con capsula d’alluminio con tappo plastico rimovibile, fornito con solvente (soluzione di sodio cloruro 0,9%) da 4 ml in una siringa pre-riempita da 5 ml con stantuffo in polipropilene con limitatore di corsa, stantuffo in gomma bromobutilica, capsula per ago con tappo in gomma bromobutilica, stantuffo in polipropilene, nonché un connettore sterile per flacone in confezione singola, il tutto contenuto in una scatola di cartone.
Categoria di prescrivibilità.
Sotto prescrizione medica.
Richiedente/Produttore.
A/T Novo Nordisk.
Sede del produttore e indirizzo del luogo in cui esercita la sua attività.
Novo Allé, Bagsværd, 2880, Danimarca.